
Epigenetics in a Spectrum of Myeloid Diseases and Its Exploitation for Therapy

 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
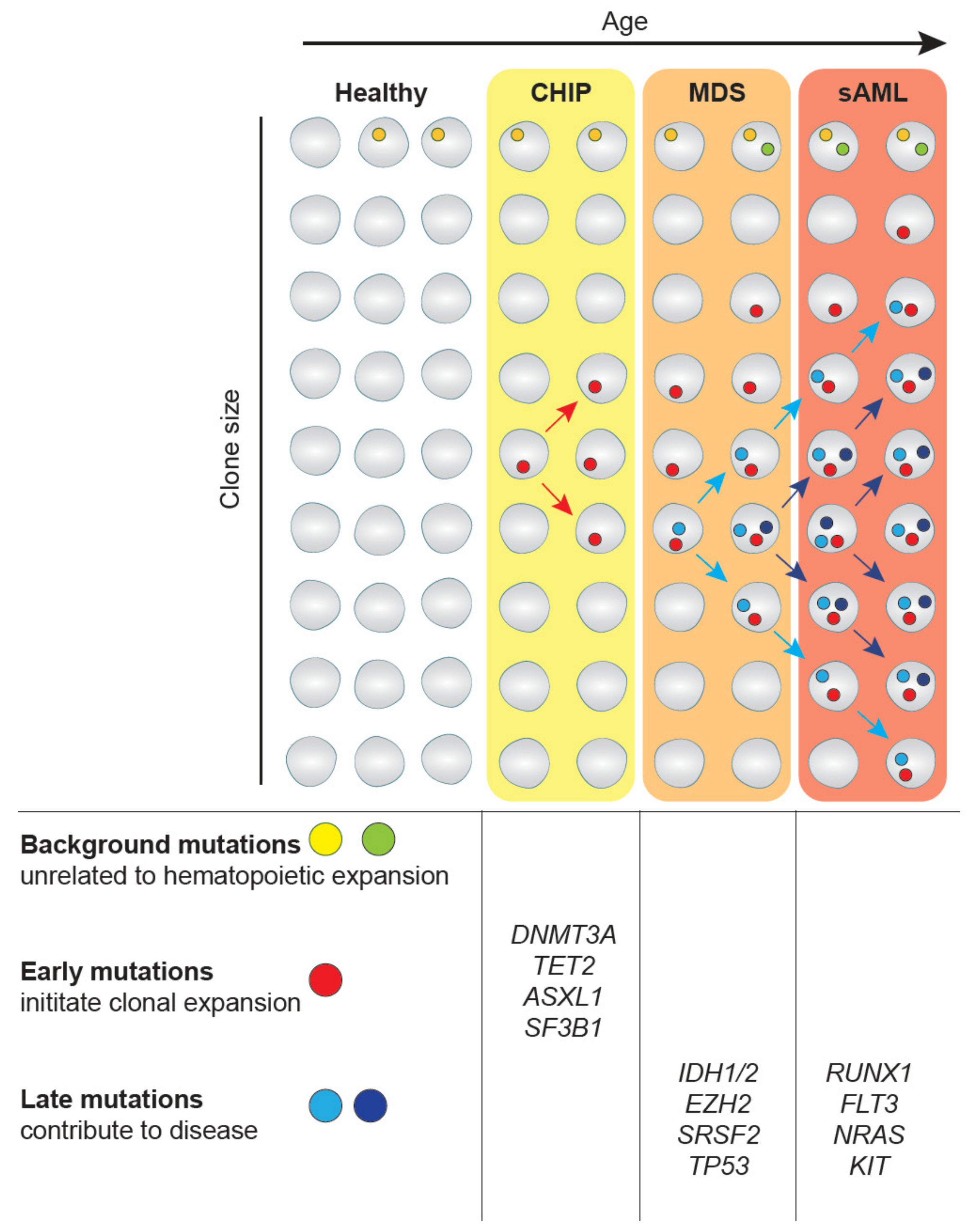
2. CHIP-MDS-sAML—A Spectrum Myeloid Diseases
3. Modifications of Chromatin Are the Molecular Basis of Epigenetic Regulation
4. Epigenetic Regulators Frequently Mutated in Myeloid Diseases and Their Function
4.1. Mutations Causing Aberrant DNA Methylation—TET2, DNMT3A, IDH
4.2. Dysregulation of Histone Modifications—EZH2, RUNX1, BCOR, ASXL1
4.3. Altering Chromatin Structure—The Cohesin Complex
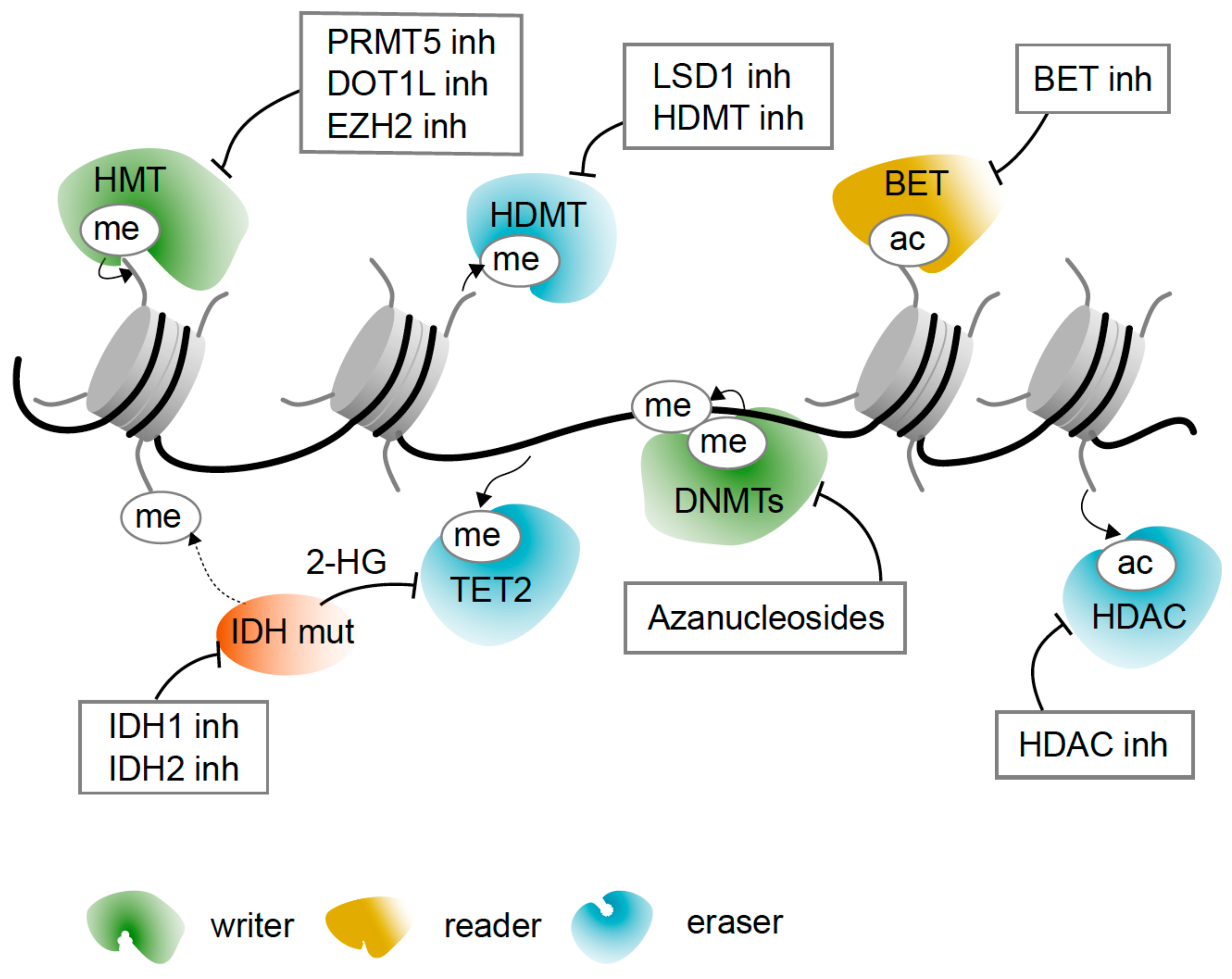
5. Epigenetic Drugs
5.1. Azanucleosides Are DNA Hypomethylating Agents and More
5.2. Targeting Histone Acetylation and Active Transcription
5.3. Targeting Histone Deacetylation and Gene Repression
5.4. Reversing Metabolic Change with IDH Inhibitors
5.5. Targeting Histone Methylation with Inhibitors of Histone Methylases and Demethylases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The Genetics of Myelodysplastic Syndrome: From Clonal Haematopoiesis to Secondary Leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The Role of Mutations in Epigenetic Regulators in Myeloid Malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P.J. The Myelodysplastic Syndrome as a Prototypical Epigenetic Disease. Blood 2013, 121. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Garcia-Manero, G. Myelodysplastic Syndromes: 2018 Update on Diagnosis, Risk-Stratification and Management. Am. J. Hematol. 2018, 93, 129–147. [Google Scholar] [CrossRef]
- Shlush, L.I. Age-Related Clonal Hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef]
- Wouters, B.J.; Delwel, R. Epigenetics and Approaches to Targeted Epigenetic Therapy in Acute Myeloid Leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical Consequences of Clonal Hematopoiesis of Indeterminate Potential. Blood Adv. 2018, 2, 3404–3410. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Kuendgen, A.; Strupp, C.; Aivado, M.; Hildebrandt, B.; Haas, R.; Gattermann, N.; Germing, U. Myelodysplastic Syndromes in Patients Younger Than Age 50. J. Clin. Oncol. 2006, 24, 5358–5365. [Google Scholar] [CrossRef]
- Sekeres, M.A. Epidemiology, Natural History, and Practice Patterns of Patients with Myelodysplastic Syndromes in 2010. J. Natl. Compr. Cancer Netw. 2011, 9, 57–63. [Google Scholar] [CrossRef]
- Cogle, C.R.; Craig, B.M.; Rollison, D.E.; List, A.F. Incidence of the Myelodysplastic Syndromes Using a Novel Claims-Based Algorithm: High Number of Uncaptured Cases by Cancer Registries. Blood 2011, 117, 7121–7125. [Google Scholar] [CrossRef]
- Germing, U.; Aul, C.; Niemeyer, C.M.; Haas, R.; Bennett, J.M. Epidemiology, Classification and Prognosis of Adults and Children with Myelodysplastic Syndromes. Ann. Hematol. 2008, 87, 691–699. [Google Scholar] [CrossRef]
- Efficace, F.; Gaidano, G.; Breccia, M.; Criscuolo, M.; Cottone, F.; Caocci, G.; Bowen, D.; Lübbert, M.; Angelucci, E.; Stauder, R.; et al. Prevalence, Severity and Correlates of Fatigue in Newly Diagnosed Patients with Myelodysplastic Syndromes. Br. J. Haematol. 2015, 168, 361–370. [Google Scholar] [CrossRef]
- Pomeroy, C.; Oken, M.; Rydell, R.E.; Filice, G.A. Infection in the Myelodysplastic Syndromes. Am. J. Med. 1991, 90, 338–344. [Google Scholar] [CrossRef]
- Anderson, L.A.; Pfeiffer, R.M.; Landgren, O.; Gadalla, S.; Berndt, S.I.; Engels, E.A. Risks of Myeloid Malignancies in Patients with Autoimmune Conditions. Br. J. Cancer 2009, 100, 822–828. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bennett, J.M. The Myelodysplastic Syndromes: Diagnosis and Treatment. Mayo Clin. Proc. 2006, 81, 104–130. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2, p. 188. [Google Scholar]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute Myeloid Leukemia Ontogeny Is Defined by Distinct Somatic Mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef]
- Lane, S.W.; Scadden, D.T.; Gilliland, D.G. The Leukemic Stem Cell Niche: Current Concepts and Therapeutic Opportunities. Blood 2009, 114, 1150–1157. [Google Scholar] [CrossRef]
- Pronk, E.; Raaijmakers, M.H.G.P. The Mesenchymal Niche in MDS. Blood 2019, 133, 1031–1038. [Google Scholar] [CrossRef]
- Hu, Z.; Tee, W.-W. Enhancers and Chromatin Structures: Regulatory Hubs in Gene Expression and Diseases. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- McGinty, R.K.; Tan, S. Nucleosome Structure and Function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21. [Google Scholar] [CrossRef]
- Stadhouders, R.; Filion, G.J.; Graf, T. Transcription Factors and 3D Genome Conformation in Cell-Fate Decisions. Nature 2019, 569, 345–354. [Google Scholar] [CrossRef]
- Eberharter, A.; Becker, P.B. Histone Acetylation: A Switch between Repressive and Permissive Chromatin: Second in Review Series on Chromatin Dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef]
- Marmorstein, R.; Zhou, M.-M. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of Histone Modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Lalonde, M.-E.; Côté, J.; Kutateladze, T.G. Perceiving the Epigenetic Landscape through Histone Readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic Regulation of Gene Expression: How the Genome Integrates Intrinsic and Environmental Signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET Enzymes, TDG and the Dynamics of DNA Demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Sashida, G.; Iwama, A. Epigenetic Regulation of Hematopoiesis. Int. J. Hematol. 2012, 96, 405–412. [Google Scholar] [CrossRef]
- Antoniani, C.; Romano, O.; Miccio, A. Concise Review: Epigenetic Regulation of Hematopoiesis: Biological Insights and Therapeutic Applications. Stem Cells Transl. Med. 2017, 6, 2106–2114. [Google Scholar] [CrossRef]
- Di Carlo, V.; Mocavini, I.; Di Croce, L. Polycomb Complexes in Normal and Malignant Hematopoiesis. J. Cell Biol. 2019, 218, 55–69. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 2013, 122, 3616–3627, quiz 3699. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Schnittger, S.; Eder, C.; Jeromin, S.; Alpermann, T.; Fasan, A.; Grossmann, V.; Kohlmann, A.; Illig, T.; Klopp, N.; Wichmann, H.-E.; et al. ASXL1 Exon 12 Mutations Are Frequent in AML with Intermediate Risk Karyotype and Are Independently Associated with an Adverse Outcome. Leukemia 2013, 27, 82–91. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Becker, H.; Maharry, K.; Radmacher, M.D.; Kohlschmidt, J.; Mrózek, K.; Nicolet, D.; Whitman, S.P.; Wu, Y.-Z.; Schwind, S.; et al. ASXL1 Mutations Identify a High-Risk Subgroup of Older Patients with Primary Cytogenetically Normal AML within the ELN Favorable Genetic Category. Blood 2011, 118, 6920–6929. [Google Scholar] [CrossRef]
- Boultwood, J.; Perry, J.; Pellagatti, A.; Fernandez-Mercado, M.; Fernandez-Santamaria, C.; Calasanz, M.J.; Larrayoz, M.J.; Garcia-Delgado, M.; Giagounidis, A.; Malcovati, L.; et al. Frequent Mutation of the Polycomb-Associated Gene ASXL1 in the Myelodysplastic Syndromes and in Acute Myeloid Leukemia. Leukemia 2010, 24, 1062–1065. [Google Scholar] [CrossRef]
- Sashida, G.; Oshima, M.; Iwama, A. Deregulated Polycomb Functions in Myeloproliferative Neoplasms. Int. J. Hematol. 2019, 110, 170–178. [Google Scholar] [CrossRef]
- Damm, F.; Chesnais, V.; Nagata, Y.; Yoshida, K.; Scourzic, L.; Okuno, Y.; Itzykson, R.; Sanada, M.; Shiraishi, Y.; Gelsi-Boyer, V.; et al. BCOR and BCORL1 Mutations in Myelodysplastic Syndromes and Related Disorders. Blood 2013, 122, 3169–3177. [Google Scholar] [CrossRef]
- Kelly, M.J.; So, J.; Rogers, A.J.; Gregory, G.; Li, J.; Zethoven, M.; Gearhart, M.D.; Bardwell, V.J.; Johnstone, R.W.; Vervoort, S.J.; et al. Bcor Loss Perturbs Myeloid Differentiation and Promotes Leukaemogenesis. Nat. Commun. 2019, 10, 1347. [Google Scholar] [CrossRef]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell 2014, 25. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a Is Essential for Hematopoietic Stem Cell Differentiation. Nat. Genet. 2012, 44. [Google Scholar] [CrossRef]
- Ettou, S.; Audureau, E.; Humbrecht, C.; Benet, B.; Jammes, H.; Clozel, T.; Bardet, V.; Lacombe, C.; Dreyfus, F.; Mayeux, P.; et al. Fas Expression at Diagnosis as a Biomarker of Azacitidine Activity in High-Risk MDS and Secondary AML. Leukemia 2012, 26, 2297–2299. [Google Scholar] [CrossRef][Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of Pre-Leukaemic Haematopoietic Stem Cells in Acute Leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; Mclellan, M.D.; et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 2012. [Google Scholar] [CrossRef]
- Herviou, L.; Cavalli, G.; Cartron, G.; Klein, B.; Moreaux, J. EZH2 in Normal Hematopoiesis and Hematological Malignancies. Oncotarget 2016, 7, 2284–2296. [Google Scholar] [CrossRef]
- Lund, K.; Adams, P.D.; Copland, M. EZH2 in Normal and Malignant Hematopoiesis. Leukemia 2014, 28, 44–49. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic Mutations of the Histone Methyltransferase Gene EZH2 in Myelodysplastic Syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 Loss Promotes Development of Myelodysplastic Syndrome but Attenuates Its Predisposition to Leukaemic Transformation. Nat. Commun. 2014, 5, 4177. [Google Scholar] [CrossRef]
- Neff, T.; Sinha, A.U.; Kluk, M.J.; Zhu, N.; Khattab, M.H.; Stein, L.; Xie, H.; Orkin, S.H.; Armstrong, S.A. Polycomb Repressive Complex 2 Is Required for MLL-AF9 Leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 5028–5033. [Google Scholar] [CrossRef]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate Dehydrogenase Mutations in Myeloid Malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jabbour, E.; Ravandi, F.; Takahashi, K.; Daver, N.; Routbort, M.; Patel, K.P.; Brandt, M.; Pierce, S.; Kantarjian, H.; et al. IDH1 and IDH2 Mutations in Myelodysplastic Syndromes and Role in Disease Progression. Leukemia 2016, 30, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- De Braekeleer, E.; Douet-Guilbert, N.; Morel, F.; Le Bris, M.-J.; Férec, C.; De Braekeleer, M. RUNX1 Translocations and Fusion Genes in Malignant Hemopathies. Future Oncol. Lond. Engl. 2011, 7, 77–91. [Google Scholar] [CrossRef]
- Osato, M. Point Mutations in the RUNX1/AML1 Gene: Another Actor in RUNX Leukemia. Oncogene 2004, 23, 4284–4296. [Google Scholar] [CrossRef]
- Harada, H.; Harada, Y.; Niimi, H.; Kyo, T.; Kimura, A.; Inaba, T. High Incidence of Somatic Mutations in the AML1/RUNX1 Gene in Myelodysplastic Syndrome and Low Blast Percentage Myeloid Leukemia with Myelodysplasia. Blood 2004, 103, 2316–2324. [Google Scholar] [CrossRef]
- Jacob, B.; Osato, M.; Yamashita, N.; Wang, C.Q.; Taniuchi, I.; Littman, D.R.; Asou, N.; Ito, Y. Stem Cell Exhaustion Due to Runx1 Deficiency Is Prevented by Evi5 Activation in Leukemogenesis. Blood 2010, 115, 1610–1620. [Google Scholar] [CrossRef]
- Huang, G.; Zhang, P.; Hirai, H.; Elf, S.; Yan, X.; Chen, Z.; Koschmieder, S.; Okuno, Y.; Dayaram, T.; Growney, J.D.; et al. PU.1 Is a Major Downstream Target of AML1 (RUNX1) in Adult Mouse Hematopoiesis. Nat. Genet. 2008, 40, 51–60. [Google Scholar] [CrossRef]
- Yu, M.; Mazor, T.; Huang, H.; Huang, H.-T.; Kathrein, K.L.; Woo, A.J.; Chouinard, C.R.; Labadorf, A.; Akie, T.E.; Moran, T.B.; et al. Direct Recruitment of Polycomb Repressive Complex 1 to Chromatin by Core Binding Transcription Factors. Mol. Cell 2012, 45, 330–343. [Google Scholar] [CrossRef]
- Nasmyth, K.; Haering, C.H. Cohesin: Its Roles and Mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef]
- Van Ruiten, M.S.; Rowland, B.D. On the Choreography of Genome Folding: A Grand Pas de Deux of Cohesin and CTCF. Curr. Opin. Cell Biol. 2021, 70, 84–90. [Google Scholar] [CrossRef]
- Thota, S.; Viny, A.D.; Makishima, H.; Spitzer, B.; Radivoyevitch, T.; Przychodzen, B.; Sekeres, M.A.; Levine, R.L.; Maciejewski, J.P. Genetic Alterations of the Cohesin Complex Genes in Myeloid Malignancies. Blood 2014, 124, 1790–1798. [Google Scholar] [CrossRef]
- Smith, J.S.; Lappin, K.M.; Craig, S.G.; Liberante, F.G.; Crean, C.M.; McDade, S.S.; Thompson, A.; Mills, K.I.; Savage, K.I. Chronic Loss of STAG2 Leads to Altered Chromatin Structure Contributing to De-Regulated Transcription in AML. J. Transl. Med. 2020, 18, 339. [Google Scholar] [CrossRef]
- Cuartero, S.; Weiss, F.D.; Dharmalingam, G.; Guo, Y.; Ing-Simmons, E.; Masella, S.; Robles-Rebollo, I.; Xiao, X.; Wang, Y.-F.; Barozzi, I.; et al. Control of Inducible Gene Expression Links Cohesin to Hematopoietic Progenitor Self-Renewal and Differentiation. Nat. Immunol. 2018, 19, 932–941. [Google Scholar] [CrossRef]
- Tothova, Z.; Valton, A.-L.; Gorelov, R.A.; Vallurupalli, M.; Krill-Burger, J.M.; Holmes, A.; Landers, C.C.; Haydu, J.E.; Malolepsza, E.; Hartigan, C.; et al. Cohesin Mutations Alter DNA Damage Repair and Chromatin Structure and Create Therapeutic Vulnerabilities in MDS/AML. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- López-Moyado, I.F.; Rao, A. DNMT3A and TET2 Mutations Reshape Hematopoiesis in Opposing Ways. Nat. Genet. 2020, 52. [Google Scholar] [CrossRef]
- Nakajima, H.; Kunimoto, H. TET2 as an Epigenetic Master Regulator for Normal and Malignant Hematopoiesis. Cancer Sci. 2014, 105, 1093–1099. [Google Scholar] [CrossRef]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of Heterozygosity 4q24 and TET2 Mutations Associated with Myelodysplastic/Myeloproliferative Neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in Hematopoietic Cells Leads to DNA Hypermethylation of Active Enhancers and Induction of Leukemogenesis. Genes Dev. 2015, 29. [Google Scholar] [CrossRef]
- Itzykson, R.; Kosmider, O.; Renneville, A.; Morabito, M.; Preudhomme, C.; Berthon, C.; Adès, L.; Fenaux, P.; Platzbecker, U.; Gagey, O.; et al. Clonal Architecture of Chronic Myelomonocytic Leukemias. Blood 2013, 121. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic Characterization of TET1, TET2, and TET3 Alterations in Myeloid Malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, O.; Gelsi-Boyer, V.; Cheok, M.; Grabar, S.; Della-Valle, V.; Picard, F.; Viguié, F.; Quesnel, B.; Beyne-Rauzy, O.; Solary, E.; et al. TET2 Mutation Is an Independent Favorable Prognostic Factor in Myelodysplastic Syndromes (MDSs). Blood 2009, 114, 3285–3291. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Levine, R.L.; Lim, K.-H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Finke, C.M.; Mullally, A.; Li, C.-Y.; Pardanani, A.; et al. Frequent TET2 Mutations in Systemic Mastocytosis: Clinical, KITD816V and FIP1L1-PDGFRA Correlates. Leukemia 2009, 23, 900–904. [Google Scholar] [CrossRef]
- Ji, H.; Ehrlich, L.I.R.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive Methylome Map of Lineage Commitment from Haematopoietic Progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA Methyltransferase 1 Is Essential for and Uniquely Regulates Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef]
- Bröske, A.-M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA Methylation Protects Hematopoietic Stem Cell Multipotency from Myeloerythroid Restriction. Nat. Genet. 2009, 41, 1207–1215. [Google Scholar] [CrossRef]
- Jiang, Y.; Dunbar, A.; Gondek, L.P.; Mohan, S.; Rataul, M.; O’Keefe, C.; Sekeres, M.; Saunthararajah, Y.; Maciejewski, J.P. Aberrant DNA Methylation Is a Dominant Mechanism in MDS Progression to AML. Blood 2009, 113. [Google Scholar] [CrossRef]
- Yang, X.; Wong, M.P.M.; Ng, R.K. Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications. Int. J. Mol. Sci. 2019, 20, 4576. [Google Scholar] [CrossRef]
- Valk-Lingbeek, M.E.; Bruggeman, S.W.M.; van Lohuizen, M. Stem Cells and Cancer. Cell 2004, 118, 409–418. [Google Scholar] [CrossRef]
- Trojer, P. Chapter 37: Histone Methylation Modifiers in Medical Therapeutics. In Medical Epigenetics; Academic Press: Cambridge, MA, USA, 2016; pp. 705–729. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Epigenetics of Haematopoietic Cell Development. Nat. Rev. Immunol. 2011, 11, 478–488. [Google Scholar] [CrossRef]
- Weishaupt, H.; Sigvardsson, M.; Attema, J.L. Epigenetic Chromatin States Uniquely Define the Developmental Plasticity of Murine Hematopoietic Stem Cells. Blood 2010, 115, 247–256. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Wang, Z.; Gearhart, M.D.; Lee, Y.-W.; Kumar, I.; Ramazanov, B.; Zhang, Y.; Hernandez, C.; Lu, A.Y.; Neuenkirchen, N.; Deng, J.; et al. A Non-Canonical BCOR-PRC1.1 Complex Represses Differentiation Programs in Human ESCs. Cell Stem Cell 2018, 22. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Diesch, J.; Zwick, A.; Garz, A.-K.; Palau, A.; Buschbeck, M.; Götze, K.S. A Clinical-Molecular Update on Azanucleoside-Based Therapy for the Treatment of Hematologic Cancers. Clin. Epigenet. 2016, 8, 71. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Santos, F.P.S.; Garcia-Manero, G. Therapy with Azanucleosides for Myelodysplastic Syndromes. Nat. Rev. Clin. Oncol. 2010, 7, 433–444. [Google Scholar] [CrossRef]
- Christman, J.; Mendelsohn, N.; Herzog, D.; Schneiderman, N. Effect of 5-Azacytidine on Differentiation and DNA Methylation in Human Promyelocytic Leukemia Cells (HL-60). Cancer Res. 1983, 43, 763–769. [Google Scholar]
- Jones, P.A.; Taylor, S.M. Cellular Differentiation, Cytidine Analogs and DNA Methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Lübbert, M.; Suciu, S.; Hagemeijer, A.; Rüter, B.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; et al. Decitabine Improves Progression-Free Survival in Older High-Risk MDS Patients with Multiple Autosomal Monosomies: Results of a Subgroup Analysis of the Randomized Phase III Study 06011 of the EORTC Leukemia Cooperative Group and German MDS Study Group. Ann. Hematol. 2016, 95. [Google Scholar] [CrossRef]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized Controlled Trial of Azacitidine in Patients with the Myelodysplastic Syndrome: A Study of the Cancer and Leukemia Group, B.J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of Azacitidine Compared with That of Conventional Care Regimens in the Treatment of Higher-Risk Myelodysplastic Syndromes: A Randomised, Open-Label, Phase III Study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Kaminskas, E. Approval Summary: Azacitidine for Treatment of Myelodysplastic Syndrome Subtypes. Clin. Cancer Res. 2005, 11, 3604–3608. [Google Scholar] [CrossRef]
- Goetze, K. The Role of Azacitidine in the Management of Myelodysplastic Syndromes (MDS). Cancer Manag. Res. 2009. [Google Scholar] [CrossRef][Green Version]
- Gore, S.D.; Fenaux, P.; Santini, V.; Bennett, J.M.; Silverman, L.R.; Seymour, J.F.; Hellstrom-Lindberg, E.; Swern, A.S.; Beach, C.L.; List, A.F. A Multivariate Analysis of the Relationship between Response and Survival among Patients with Higher-Risk Myelodysplastic Syndromes Treated within Azacitidine or Conventional Care Regimens in the Randomized AZA-001 Trial. Haematologica 2013, 98, 1067–1072. [Google Scholar] [CrossRef]
- Prébet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Adès, L.; et al. Outcome of High-Risk Myelodysplastic Syndrome After Azacitidine Treatment Failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef]
- Fenaux, P.; Ades, L. Review of Azacitidine Trials in Intermediate-2-and High-Risk Myelodysplastic Syndromes. Leuk. Res. 2009, 33, 7–11. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Stahl, M.; Sekeres, M.A.; Steensma, D.P.; Komrokji, R.S.; Gore, S.D. A Call for Action: Increasing Enrollment of Untreated Patients with Higher-Risk Myelodysplastic Syndromes in First-Line Clinical Trials. Cancer 2017, 123, 3662–3672. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellström-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine Prolongs Overall Survival Compared with Conventional Care Regimens in Elderly Patients with Low Bone Marrow Blast Count Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine versus Patient Choice, with Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients with Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Ravandi, F.; Liu-Dumlao, T.; Brandt, M.; Faderl, S.; Pierce, S.; Borthakur, G.; Garcia-Manero, G.; Cortes, J.; Kantarjian, H. Epigenetic Therapy Is Associated with Similar Survival Compared with Intensive Chemotherapy in Older Patients with Newly Diagnosed Acute Myeloid Leukemia. Blood 2012, 120, 4840–4845. [Google Scholar] [CrossRef] [PubMed]
- Huls, G.; Chitu, D.A.; Havelange, V.; Jongen-Lavrencic, M.; van de Loosdrecht, A.A.; Biemond, B.J.; Sinnige, H.; Hodossy, B.; Graux, C.; van Kooy, R.M.; et al. Azacitidine Maintenance after Intensive Chemotherapy Improves DFS in Older AML Patients. Blood 2019, 133, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.H.; Porkka, K.; et al. The QUAZAR AML-001 Maintenance Trial: Results of a Phase III International, Randomized, Double-Blind, Placebo-Controlled Study of CC-486 (Oral Formulation of Azacitidine) in Patients with Acute Myeloid Leukemia (AML) in First Remission. Blood 2019, 134. [Google Scholar] [CrossRef]
- Blum, W.; Klisovic, R.B.; Hackanson, B.; Liu, Z.; Liu, S.; Devine, H.; Vukosavljevic, T.; Huynh, L.; Lozanski, G.; Kefauver, C.; et al. Phase I Study of Decitabine Alone or in Combination with Valproic Acid in Acute Myeloid Leukemia. J. Clin. Oncol. 2007, 25, 3884–3891. [Google Scholar] [CrossRef]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 Mutations Predict Response to Hypomethylating Agents in Myelodysplastic Syndrome Patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef]
- Kuendgen, A.; Müller-Thomas, C.; Lauseker, M.; Haferlach, T.; Urbaniak, P.; Schroeder, T.; Brings, C.; Wulfert, M.; Meggendorfer, M.; Hildebrandt, B.; et al. Efficacy of Azacitidine Is Independent of Molecular and Clinical Characteristics—An Analysis of 128 Patients with Myelodysplastic Syndromes or Acute Myeloid Leukemia and a Review of the Literature. Oncotarget 2018, 9, 27882–27894. [Google Scholar] [CrossRef]
- Bristol Myers Squibb. U.S. Food and Drug Administration Approves Onureg® (Azacitidine Tablets), a New Oral Therapy, as Continued Treatment for Adults in First Remission with Acute Myeloid Leukemia. Press Release, 09/01/2020. Available online: https://news.bms.com/news/corporate-financial/2020/U.S.-Food-and-Drug-Administration-Approves-Onureg-azacitidine-tablets-a-New-Oral-Therapy-as-Continued-Treatment-for-Adults-in-First-Remission-with-Acute-Myeloid-Leukemia/default.aspx (accessed on 6 April 2021).
- Garcia-Manero, G.; Roboz, G.; Walsh, K.; Kantarjian, H.; Ritchie, E.; Kropf, P.; O’Connell, C.; Tibes, R.; Lunin, S.; Rosenblat, T.; et al. Guadecitabine (SGI-110) in Patients with Intermediate or High-Risk Myelodysplastic Syndromes: Phase 2 Results from a Multicentre, Open-Label, Randomised, Phase 1/2 Trial. Lancet Haematol. 2019, 6, 317–327. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Choy, G.; Redkar, S.; Taverna, P.; Azab, M.; Karpf, A.R. SGI-110: DNA Methyltransferase Inhibitor Oncolytic. Drugs Future 2013, 38, 535–543. [Google Scholar]
- Yoo, C.B.; Jeong, S.; Egger, G.; Liang, G.; Phiasivongsa, P.; Tang, C.; Redkar, S.; Jones, P.A. Delivery of 5-Aza-2′-Deoxycytidine to Cells Using Oligodeoxynucleotides. Cancer Res. 2007, 67, 6400. [Google Scholar] [CrossRef]
- Ball, B.J.; Famulare, C.A.; Stein, E.M.; Tallman, M.S.; Derkach, A.; Roshal, M.; Gill, S.I.; Manning, B.M.; Koprivnikar, J.; McCloskey, J.; et al. Venetoclax and Hypomethylating Agents (HMAs) Induce High Response Rates in MDS, Including Patients after HMA Therapy Failure. Blood Adv. 2020, 4, 2866–2870. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Walter-petrich, A.; Lehmann che, J.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. APR-246 Combined with Azacitidine (AZA) in TP53 Mutated Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). a Phase 2 Study by the Groupe Francophone Des Myélodysplasies (GFM). Blood 2019, 134, 677. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.F.; McGraw, K.; et al. Phase 2 Results of APR-246 and Azacitidine (AZA) in Patients with TP53 Mutant Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia (AML). Blood 2019, 134, 676. [Google Scholar] [CrossRef]
- Santini, V.; Prebet, T.; Fenaux, P.; Gattermann, N.; Nilsson, L.; Pfeilstöcker, M.; Vyas, P.; List, A.F. Minimizing Risk of Hypomethylating Agent Failure in Patients with Higher-Risk MDS and Practical Management Recommendations. Leuk. Res. 2014, 38, 1381–1391. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Nature 2011, 478. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Wroblewski, M.; Scheller-Wendorff, M.; Udonta, F.; Bauer, R.; Schlichting, J.; Zhao, L.; Ben Batalla, I.; Gensch, V.; Päsler, S.; Wu, L.; et al. BET-Inhibition by JQ1 Promotes Proliferation and Self-Renewal Capacity of Hematopoietic Stem Cells. Haematologica 2018, 103, 939–948. [Google Scholar] [CrossRef]
- Roe, J.-S.; Vakoc, C.R. The Essential Transcriptional Function of BRD4 in Acute Myeloid Leukemia. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Gudgin, E.J.; Horton, S.J.; Giotopoulos, G.; Meduri, E.; Robson, S.; Cannizzaro, E.; Osaki, H.; Wiese, M.; Putwain, S.; et al. Recurrent Mutations, Including NPM1c, Activate a BRD4-Dependent Core Transcriptional Program in Acute Myeloid Leukemia. Leukemia 2014, 28, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Garau, D.; Ribeiro, M.L.; Roué, G. Pharmacological Targeting of BET Bromodomain Proteins in Acute Myeloid Leukemia and Malignant Lymphomas: From Molecular Characterization to Clinical Applications. Cancers 2019, 11, 1483. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain Inhibitor OTX015 in Patients with Acute Leukaemia: A Dose-Escalation, Phase 1 Study. Lancet Haematol. 2016, 3, 186–195. [Google Scholar] [CrossRef]
- Dawson, M.; Stein, E.M.; Huntly, B.J.P.; Karadimitris, A.; Kamdar, M.; Fernandez de Larrea, C.; Dickinson, M.J.; Yeh, P.S.-H.; Daver, N.; Chaidos, A.; et al. A Phase I Study of GSK525762, a Selective Bromodomain (BRD) and Extra Terminal Protein (BET) Inhibitor: Results from Part 1 of Phase I/II Open Label Single Agent Study in Patients with Acute Myeloid Leukemia (AML). Blood 2017, 130, 1377. [Google Scholar] [CrossRef]
- Borthakur, G.; Wolff, J.E.; Aldoss, I.; Hu, B.; Dinh, M.; Torres, A.; Chen, X.; Rizzieri, D.; Sood, A.; Odenike, O.; et al. First-in-Human Study of ABBV-075 (Mivebresib), a Pan-Inhibitor of Bromodomain and Extra Terminal (BET) Proteins, in Patients (Pts) with Relapsed/Refractory (RR) Acute Myeloid Leukemia (AML): Preliminary Data. J. Clin. Oncol. 2018, 36, 7019. [Google Scholar] [CrossRef]
- Sun, Y.; Han, J.; Wang, Z.; Li, X.; Sun, Y.; Hu, Z. Safety and Efficacy of Bromodomain and Extra-Terminal Inhibitors for the Treatment of Hematological Malignancies and Solid Tumors: A Systematic Study of Clinical Trials. Front. Pharmacol. 2021, 11, 2440. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, Q. Histone Deacetylase Inhibitors and Cell Death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in Normal and Malignant Hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Santoro, F.; Botrugno, O.A.; Dal Zuffo, R.; Pallavicini, I.; Matthews, G.M.; Cluse, L.; Barozzi, I.; Senese, S.; Fornasari, L.; Moretti, S.; et al. A Dual Role for Hdac1: Oncosuppressor in Tumorigenesis, Oncogene in Tumor Maintenance. Blood 2013, 121, 3459–3468. [Google Scholar] [CrossRef]
- Chandhok, N.S.; Prebet, T. Insights into Novel Emerging Epigenetic Drugs in Myeloid Malignancies. Ther. Adv. Hematol. 2019, 10, 204062071986608. [Google Scholar] [CrossRef]
- Pan, D.; Rampal, R.; Mascarenhas, J. Clinical Developments in Epigenetic-Directed Therapies in Acute Myeloid Leukemia. Blood Adv. 2020, 4, 970–982. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-Mutant-IDH1 Inhibitor BAY1436032 Is Highly Effective against Human IDH1 Mutant Acute Myeloid Leukemia in Vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-Based Design and Identification of FT-2102 (Olutasidenib), a Potent Mutant-Selective IDH1 Inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar] [CrossRef]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and Evaluation of Clinical Candidate IDH305, a Brain Penetrant Mutant IDH1 Inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef]
- Brooks, N.; DeWalt, R.; Boulet, S.; Lu, Z.; Kays, L.; Cavitt, R.; Gomez, S.; Strelow, J.; Milligan, P.; Roth, K.; et al. Abstract LB-274: Identification and Characterization of LY3410738, a Novel Covalent Inhibitor of Cancer-Associated Mutant Isocitrate Dehydrogenase 1 (IDH1). Cancer Res. 2019, 79, 8274. [Google Scholar] [CrossRef]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Kernytsky, A.; Wang, F.; Hansen, E.; Schalm, S.; Straley, K.; Gliser, C.; Yang, H.; Travins, J.; Murray, S.; Dorsch, M.; et al. IDH2 Mutation-Induced Histone and DNA Hypermethylation Is Progressively Reversed by Small-Molecule Inhibition. Blood 2015, 125, 296–303. [Google Scholar] [CrossRef]
- Galkin, M.; Jonas, B.A. Enasidenib in the Treatment of Relapsed/Refractory Acute Myeloid Leukemia: An Evidence-Based Review of Its Place in Therapy. Core Evid. 2019, 14, 3–17. [Google Scholar] [CrossRef]
- Abou Dalle, I.; DiNardo, C.D. The Role of Enasidenib in the Treatment of Mutant IDH2 Acute Myeloid Leukemia. Ther. Adv. Hematol. 2018, 9, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Wang, F.; Travins, J.; Chen, Y.; Yang, H.; Straley, K.; Choe, S.; Dorsch, M.; Agresta, S.; Schenkein, D.; et al. O11.2—AG-221 Offers a Survival Advantage in a Primary Human IDH2 Mutant AML Xenograft Model. Ann. Oncol. 2015, 26, 15. [Google Scholar] [CrossRef][Green Version]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib Induces Acute Myeloid Leukemia Cell Differentiation to Promote Clinical Response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Zhang, Y. The Diverse Functions of Dot1 and H3K79 Methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Choi, M.; Kim, J.-E. The Histone Methyltransferase Dot1/DOT1L as a Critical Regulator of the Cell Cycle. Cell Cycle 2014, 13, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. HDOT1L Links Histone Methylation to Leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Taranova, O.; He, J.; Zhang, Y. DOT1L, the H3K79 Methyltransferase, Is Required for MLL-AF9-Mediated Leukemogenesis. Blood 2011, 117, 6912–6922. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L Inhibitor Pinometostat Reduces H3K79 Methylation and Has Modest Clinical Activity in Adult Acute Leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Perner, F.; Gadrey, J.Y.; Xiong, Y.; Hatton, C.; Eschle, B.K.; Weiss, A.; Stauffer, F.; Gaul, C.; Tiedt, R.; Perry, J.A.; et al. Novel Inhibitors of the Histone Methyltransferase DOT1L Show Potent Antileukemic Activity in Patient-Derived Xenografts. Blood 2020, 136, 1983–1988. [Google Scholar] [CrossRef]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 Arginine Methyltransferase: Many Roles in Development, Cancer and Beyond. Cell. Mol. Life Sci. CMLS 2015, 72, 2041–2059. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein Arginine Methyltransferases and Cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Tarighat, S.S.; Santhanam, R.; Frankhouser, D.; Radomska, H.S.; Lai, H.; Anghelina, M.; Wang, H.; Huang, X.; Alinari, L.; Walker, A.; et al. The Dual Epigenetic Role of PRMT5 in Acute Myeloid Leukemia: Gene Activation and Repression via Histone Arginine Methylation. Leukemia 2016, 30, 789–799. [Google Scholar] [CrossRef]
- Radzisheuskaya, A.; Shliaha, P.V.; Grinev, V.; Lorenzini, E.; Kovalchuk, S.; Shlyueva, D.; Gorshkov, V.; Hendrickson, R.C.; Jensen, O.N.; Helin, K. PRMT5 Methylome Profiling Uncovers a Direct Link to Splicing Regulation in Acute Myeloid Leukemia. Nat. Struct. Mol. Biol. 2019, 26, 999–1012. [Google Scholar] [CrossRef]
- Kim, H.; Ronai, Z.A. PRMT5 Function and Targeting in Cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef]
- Hayami, S.; Kelly, J.D.; Cho, H.-S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.J.; et al. Overexpression of LSD1 Contributes to Human Carcinogenesis through Chromatin Regulation in Various Cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Ora, I.; Pajtler, K.; Klein-Hitpass, L.; Kuhfittig-Kulle, S.; et al. Lysine-Specific Demethylase 1 Is Strongly Expressed in Poorly Differentiated Neuroblastoma: Implications for Therapy. Cancer Res. 2009, 69, 2065–2071. [Google Scholar] [CrossRef]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The Histone Demethylase KDM1A Sustains the Oncogenic Potential of MLL-AF9 Leukemia Stem Cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef]
- Schenk, T.; Chen, W.C.; Göllner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.-U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) Demethylase Reactivates the All-Trans-Retinoic Acid Differentiation Pathway in Acute Myeloid Leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A Inhibitors in Clinical Trials: Advances and Prospects. J. Hematol. Oncol. 2019, 12, 129. [Google Scholar] [CrossRef] [PubMed]
- Salamero, O.; Montesinos, P.; Willekens, C.; Pérez-Simón, J.A.; Pigneux, A.; Récher, C.; Popat, R.; Carpio, C.; Molinero, C.; Mascaró, C.; et al. First-in-Human Phase I Study of Iadademstat (ORY-1001): A First-in-Class Lysine-Specific Histone Demethylase 1A Inhibitor, in Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2020, 38, 4260–4273. [Google Scholar] [CrossRef]
- Salamero, O.; Somervaille, T.C.; Molero, A.; Acuna, E.; Perez, A.; Cano, I.; Rodriguez-Veiga, R.; Gutierrez, S.; Bullock, R.; Buesa, C.; et al. Robust Efficacy Signals in Elderly Aml Patients Treated with Iadademstat in Combination with Azacitidine (ALICE Phase IIa Trial). In Proceedings of the 2020 ASH Annual Meeting, Atlanta, GA, USA, 5 December 2020; Volume 1916. [Google Scholar]
- Stresemann, C.; Lyko, F. Modes of Action of the DNA Methyltransferase Inhibitors Azacytidine and Decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic Therapy in Immune-Oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kiziltepe, T.; Hideshima, T.; Catley, L.; Raje, N.; Yasui, H.; Shiraishi, N.; Okawa, Y.; Ikeda, H.; Vallet, S.; Pozzi, S.; et al. 5-Azacytidine, a DNA Methyltransferase Inhibitor, Induces ATR-Mediated DNA Double-Strand Break Responses, Apoptosis, and Synergistic Cytotoxicity with Doxorubicin and Bortezomib against Multiple Myeloma Cells. Mol. Cancer Ther. 2007, 6, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Siegel, S.; Avila, F.; Lee, T.; Karon, M. Effect of TRNA from 5-Azacytidine-Treated Hamster Fibrosarcoma Cells on Protein Synthesis in Vitro in a Cell-Free System. Biochem. Pharmacol. 1976, 25, 389–392. [Google Scholar] [CrossRef]
- Lu, L.J.; Randerath, K. Mechanism of 5-Azacytidine-Induced Transfer RNA Cytosine-5-Methyltransferase Deficiency. Cancer Res. 1980, 40, 2701–2705. [Google Scholar]
- Schaefer, M.; Hagemann, S.; Hanna, K.; Lyko, F. Azacytidine Inhibits RNA Methylation at DNMT2 Target Sites in Human Cancer Cell Lines. Cancer Res. 2009, 69, 8127–8132. [Google Scholar] [CrossRef]
- Cheng, J.X.; Chen, L.; Li, Y.; Cloe, A.; Yue, M.; Wei, J.; Watanabe, K.A.; Shammo, J.M.; Anastasi, J.; Shen, Q.J.; et al. RNA Cytosine Methylation and Methyltransferases Mediate Chromatin Organization and 5-Azacytidine Response and Resistance in Leukaemia. Nat. Commun. 2018, 9, 1163. [Google Scholar] [CrossRef]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Stahl, M.; Hu, X.; Wang, R.; Huntington, S.F.; Podoltsev, N.A.; Gore, S.D.; Ma, X.; Davidoff, A.J. Long-Term Survival of Older Patients with MDS Treated with HMA Therapy without Subsequent Stem Cell Transplantation. Blood 2018, 131, 818–821. [Google Scholar] [CrossRef]
- Cabezón, M.; Malinverni, R.; Bargay, J.; Xicoy, B.; Marcé, S.; Garrido, A.; Tormo, M.; Arenillas, L.; Coll, R.; Borras, J.; et al. Different Methylation Signatures at Diagnosis in Patients with High-Risk Myelodysplastic Syndromes and Secondary Acute Myeloid Leukemia Predict Azacitidine Response and Longer Survival. Clin. Epigenet. 2021, 13, 9. [Google Scholar] [CrossRef]
- Voso, M.T.; Lo-Coco, F.; Fianchi, L. Epigenetic Therapy of Myelodysplastic Syndromes and Acute Myeloid Leukemia. Curr. Opin. Oncol. 2015, 27, 532–539. [Google Scholar] [CrossRef]
- Ribeiro, M.L.; Reyes-Garau, D.; Armengol, M.; Fernández-Serrano, M.; Roué, G. Recent Advances in the Targeting of Epigenetic Regulators in B-Cell Non-Hodgkin Lymphoma. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Shilatifard, A. The Super Elongation Complex (SEC) Family in Transcriptional Control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547. [Google Scholar] [CrossRef]
- Wang, F.; Marshall, C.B.; Ikura, M. Transcriptional/Epigenetic Regulator CBP/P300 in Tumorigenesis: Structural and Functional Versatility in Target Recognition. Cell. Mol. Life Sci. 2013, 70, 3989–4008. [Google Scholar] [CrossRef]
- Brooks, N.; Raja, M.; Young, B.W.; Spencer, G.J.; Somervaille, T.C.P.; Pegg, N.A. CCS1477: A Novel Small Molecule Inhibitor of P300/CBP Bromodomain for the Treatment of Acute Myeloid Leukaemia and Multiple Myeloma. Blood 2019, 134, 2560. [Google Scholar] [CrossRef]
- Pegg, N.; Brooks, N.; Worthington, J.; Young, B.; Prosser, A.; Lane, J.; Taddei, D.; Brown, R.; Harbottle, G.; Shannon, J.; et al. Characterisation of CCS1477: A Novel Small Molecule Inhibitor of P300/CBP for the Treatment of Castration Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 11590. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Licht, J.D. Histone Deacetylase Inhibitors in Cancer Therapy. Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone Deacetylases and Cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Luo, L.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.-L.; Sheth, C.M.; Charlab, R.; et al. FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia with an Isocitrate Dehydrogenase-1 Mutation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-Lysine 79 Is Mediated by a New Family of HMTases without a SET Domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in Cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.-U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the Histone Methyltransferase EZH2 Induces Resistance to Multiple Drugs in Acute Myeloid Leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Karkhanis, V.; Hu, Y.-J.; Baiocchi, R.A.; Imbalzano, A.N.; Sif, S. Versatility of PRMT5-Induced Methylation in Growth Control and Development. Trends Biochem. Sci. 2011, 36, 633–641. [Google Scholar] [CrossRef]
- Zhao, Q.; Rank, G.; Tan, Y.T.; Li, H.; Moritz, R.L.; Simpson, R.J.; Cerruti, L.; Curtis, D.J.; Patel, D.J.; Allis, C.D.; et al. PRMT5-Mediated Methylation of Histone H4R3 Recruits DNMT3A, Coupling Histone and DNA Methylation in Gene Silencing. Nat. Struct. Mol. Biol. 2009, 16, 304–311. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Maiques-Diaz, A.; Somervaille, T.C. LSD1: Biologic Roles and Therapeutic Targeting. Epigenomics 2016, 8, 1103–1116. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical Epigenetics: Seizing Opportunities for Translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef]
- Greenberg, P.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Saber, W.; Horowitz, M.M. Transplantation for Myelodysplastic Syndromes: Who, When, and Which Conditioning Regimens. Hematology 2016, 2016, 478–484. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Mutation Effect on Gene | Mutational Frequency | Characteristics |
|---|---|---|---|
| ASXL1 [42,43,44,45] | Loss-of-function mutation | 20% in MDS | Mutations enriched in elderly AML and sAML patients |
| 6–30% in AML | |||
| BCOR [46,47,48] | Loss-of-function mutation | 5% in MDS | Associated with poor prognosis |
| 9% in AML | |||
| DNMT3A [49,50,51,52,53,54] | Loss-of-function mutation | 13% in MDS | Thought to be initiating mutation during the pre-leukemic state |
| 20% in AML | Important for the balance of differentiation and self-renewal | ||
| EZH2 [55,56,57,58,59] | Loss-of-function mutation as well as gain of function mutations | 5% in MDS | Thought to regulate the balance between self-renewal and differentiation |
| 1–2% de novo AML | In MDS associated with poor prognosis | ||
| IDH1/2 [60,61,62,63,64,65] | Gain of function | 5% in MDS | Leads to the production of oncometabolite, which interferes with TET2 activity and histone demethylases |
| 20% in AML | IDH2 mutations are more common | ||
| RUNX1 [66,67,68,69,70,71] | Translocations | 10–20% in MDS | Significantly associated with EZH2 mutations |
| Loss-of-function mutation | 2–20% in AML | ||
| Cohesin [72,73,74,75,76,77] | Loss-of-function mutation | 10–15% in MDS, | Mutually exclusive |
| 10% in AML | often associated with mutations in NPM1, TET2, ASXL1 and EZH2 | ||
| TET2 [78,79,80,81,82,83,84,85] | Loss-of-function mutation | 30–50% in MDS | Important for myeloid differentiation and lineage commitment |
| 30% in sAML | Associated with poor prognosis in some studies |
| Targets/Agents | Characteristics/Mechanisms of Action |
|---|---|
| Azanucleosides [100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127] | Promote differentiation, activate the innate immune response and lead to DNA damage response causing cytotoxicity. |
| Through incorporation into RNA, AZA also reduces protein synthesis and impairs DNA synthesis and repair. | |
| Azacitidine and decitabine are FDA-approved for the treatment of MDS. | |
| Oral azacitidine CC-486 FDA approved as maintenance therapy in AML. | |
| BET [128,129,130,131,132,133,134,135,136,137,138,139,140] | Mainly BRD4 inhibitors. |
| Reduce expression of oncogenes, including MYC and BCL2, thus lead to reduced proliferation and increased apoptosis. | |
| In clinical trials, modest efficacy and adverse effects suggesting their use in combinatorial therapy. | |
| HDAC [141,142,143,144,145,146] | Inhibitors restore histone acetylation, promoting differentiation and apoptosis. |
| Often have dual roles making their use as monotherapies difficult. | |
| IDH1/IDH2 [147,148,149,150,151,152,153,154,155,156,157] | IDH inhibitors reduce the total serum 2-HG level and induce AML cell differentiation. |
| IDH1 inhibitor Ivosidenib and IDH2 inhibitor enasidenib are FDA approved for the treatment of adult relapsed or refractory AML with IDH1 or IDH2 mutations, respectively. | |
| EZH2 [158,159,160] | S-adenosyl methionine-competitive EZH2 inhibitor tazemetostat is FDA approved for the treatment of epithelioid sarcoma. |
| DOT1L [160,161,162,163,164,165] | DOT1L inhibitor pinometostat selectively kills MLL-rearranged AML cells and is in phase I clinical trial in patients with MLL translocation. |
| Pinometostat has limited pharmacokinetics (requires continuous intravenous administration); thus, new DOT1L inhibitors are currently being assessed in vitro and in PDX models. | |
| PRMT5 [166,167,168,169,170] | PRMT5 inhibition has anti-leukemic effects in AML due to the downregulation of FLT3 expression. |
| PRMT5 inhibition induces alternative splicing and downregulation of proteins required for proliferation. | |
| LSD1 [171,172,173,174,175,176,177] | LSD1 inhibition abrogates the clonogenic potential and induces differentiation of MLL-rearranged AML as well as sensitizes AML cells to differentiation induced by all-trans-retinoic acid. |
| Targets/Agents | Drug Name | Diseases | NCT Number | Phase Trial | Combination | Completion Date |
|---|---|---|---|---|---|---|
| HMAs | Azacitidine | IDH1-mutant AML and MDS | NCT03471260 | Phase I/II | Venetoclax and ivosidenib | 2021 |
| TP53-mutant AML and MDS | NCT03588078 | Phase I/II | APR-246 | 2021 | ||
| TP53-mutant MDS | NCT03745716 | Phase III | APR-246 | 2021 | ||
| TP53-mutant myeloid malignancies | NCT04214860 | Phase I | Venetoclax and APR-246 | 2021 | ||
| AML, MDS | NCT02775903 | Phase II | PD-L1 inhibitor durvalumab (MEDI4736) | 2021 | ||
| AML, MDS | NCT03030612 | Phase I/II | Anti-CD70 antibody ARGX-110 | 2021 | ||
| R/R AML, MDS | NCT01869114 | Phase II | mTOR inhibitor sirolimus | 2021 | ||
| Treatment-naïve MDS | NCT02942290 | Phase I | Venetoclax | 2022 | ||
| AML, MDS, CMML | NCT02397720 | Phase II | PD-1 inhibitor nivolumab | 2022 | ||
| AML, MDS | NCT04275518 | Phase I | MDM2 inhibitor APG-115 | 2022 | ||
| AML, MDS | NCT02319369 | Phase I | MDM2 inhibitor milademetan | 2022 | ||
| AML, MDS, CMML | NCT04256317 | Phase II/III | Cytidine deaminase inhibitor ASTX727 | 2023 | ||
| AML, MDS | NCT04609826 | Phase I | Dihydroorotate dehydrogenase inhibitor JNJ-74856665 | 2023 | ||
| AML, MDS | NCT03113643 | Phase I | Venetoclax and SL-401 | 2024 | ||
| AML, MDS, CMML, MPN | NCT03862157 | Phase I/II | Venetoclax and pevonedistat | 2024 | ||
| R/R AML, MDS | NCT04487106 | Phase II | Venetoclax and trametinib | 2024 | ||
| R/R FLT3-mutant AML, R/R MDS, R/R CMML, R/R MPN | NCT04140487 | Phase I/II | Venetoclax and gilteritinib | 2024 | ||
| AML, MDS, CMML | NCT04730258 | Phase I/II | PLK4 inhibitor CFI-400945 | 2024 | ||
| AML, MDS, MPN | NCT04771130 | Phase I/II | BCL2 inhibitor BGB-11417 | 2024 | ||
| AML, MDS with impending relapse | NCT04712942 | Phase II | NEDD8-inhibitor pevonedistat | 2026 | ||
| CC-486 | AML, MDS after allo-HSCT | NCT04173533 | Phase III | 2024 | ||
| Decitabine | AML, MDS | NCT03066648 | Phase I | PD-1 inhibitor PDR001 and checkpoint inhibitor MBG453 | 2021 | |
| Untreated AML or R/R AML | NCT02878785 | Phase I/II | PARP inhibitor talazoparib | 2022 | ||
| AML, MDS, CMML | NCT03306264 | Phase III | Cytidine deaminase inhibitor ASTX727 | 2022 | ||
| AML, MDS, CMML | NCT04730258 | Phase I/II | PLK4 inhibitor CFI-400945 | 2024 | ||
| R/R AML, R/R high-risk MDS | NCT03404193 | Phase II | Venetoclax | 2024 | ||
| R/R AML, MDS | NCT02190695 | Phase II | Carboplatin, arsenic trioxide | 2026 | ||
| Guadecitabine (SGI-110) | AML, MDS, CMML | NCT01261312 | Phase I/II | 2019 | ||
| AML, MDS | NCT03603964 | Phase II | 2021 | |||
| AML and MDS after allo-HSCT | NCT03454984 | Phase II | 2022 | |||
| AML, MDS, CMML | NCT02935361 | Phase I/II | PD-L1 inhibitor atezolizumab | 2021 | ||
| NTX-301 (DNMT1 inhibitor) | AML, MDS, CMML | NCT04167917 | Phase I | 2025 | ||
| BET | Birabresib (OTX015, MK-8628) | AML, sAML, DLBCL | NCT02698189 | Phase I | 2021 | |
| CPI0610 | AML, MDS, MPN | NCT02158858 | Phase I/II | JAK1/2 inhibitor ruxolitinib | 2021 | |
| ABBV-744 | R/R AML | NCT03360006 | Phase I | 2022 | ||
| FT-1101 | R/R AML, MDS, non-Hodgkin’s lymphoma | NCT02543879 | Phase I | Azacitidine | 2019 | |
| PLX2853 | R/R AML, MDS | NCT03787498 | Phase I | 2021 | ||
| PLX51107 | AML, MDS | NCT04022785 | Phase I | 2022 | ||
| HDAC | LBH589 (Panobinostat) | AML, MDS, CMML | NCT00946647 | Phase Ib/IIb | Azacitidine | 2019 |
| High-risk AML and MDS after allo-HSCT | NCT04326764 | Phase III | 2023 | |||
| Vorinostat | AML, MDS | NCT00948064 | Phase II | Azacitidine | 2017 | |
| AML and MDS after allo-HSCT | NCT03843528 | Phase I | Low-dose azacitidine | 2021 | ||
| Belinostat | R/R AML, R/R MDS | NCT03772925 | Phase I | NEDD8-inhibitor pevonedistat | 2021 | |
| IDH1 | Ivosidenib | IDH1-mutant AML and MDS | NCT03503409 | Phase II | 2025 | |
| IDH1-mutant AML | NCT03173248 | Phase III | Azacitidine | 2022 | ||
| BAY1436032 | IDH1-mutant AML | NCT03127735 | Phase I | 2019 | ||
| FT-2102 | IDH1-mutant AML and MDS | NCT02719574 | Phase I/II | Azacitidine or cytarabine | 2020 | |
| IDH305 | IDH1-R132 mutant AML and MDS | NCT02381886 | Phase I | 2022 | ||
| LY3410738 | IDH1- or IDH2-mutant AML, MDS, CMML, MPN | NCT04603001 | Phase I | 2023 | ||
| IDH2 | Enasidenib | IDH2-mutant AML and MDS | NCT03744390 | Phase II | 2023 | |
| IDH2-mutant AML and MDS | NCT03383575 | Phase II | Azacitidine | 2023 | ||
| IDH2-mutant AML, MDS, CMML after allo-HSCT | NCT04522895 | Phase II | 2024 | |||
| EZH2 | Tazemetostat | R/R Non-Hodgkin’s lymphoma | NCT03009344 | Phase I | 2020 | |
| B-cell lymphomas, advanced solid tumors, DLBCL | NCT01897571 | Phase I/II | 2021 | |||
| DOT1L | Pinometostat | R/R AML or AML with MLL-rearrangement | NCT03701295 | Phase I/II | Azacitidine | 2021 |
| AML with MLL-rearrangement | NCT03724084 | Phase I/II | Standard chemotherapy | 2021 | ||
| PRMT5 | GSK3326595 | AML, MDS | NCT03614728 | Phase I | Azacitidine | 2023 |
| JNJ-64619178 | Advanced solid tumors, non-Hodgkin’s lymphoma, low-risk MDS | NCT03573310 | Phase I | 2022 | ||
| PF-06939999 | Advanced or metastatic solid tumors | NCT03854227 | Phase I | 2024 | ||
| LSD1 | Tranylcypromine | AML, MDS | NCT02273102 | Phase I | ATRA | 2020 |
| GSK2879552 | AML, MDS | NCT02177812 | Phase I | ATRA | 2017 | |
| IMG-7289 | AML, MDS | NCT02842827 | Phase I | ATRA | 2018 | |
| INCB059872 | Solid tumors and AML, MDS | NCT02712905 | Phase I/II | ATRA, azacitidinecitidine and nivolumab | 2020 | |
| Seclidemstat (SP-2577) | CMML, MDS | NCT04734990 | Phase I/II | Azacitidine | 2022 | |
| CC-90011 | R/R AML, treatment-naïve AML not eligible for chemotherapy | NCT04748848 | Phase I/II | Venetoclax and azacitidine | 2025 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maher, M.; Diesch, J.; Le Pannérer, M.-M.; Buschbeck, M. Epigenetics in a Spectrum of Myeloid Diseases and Its Exploitation for Therapy. Cancers 2021, 13, 1746. https://doi.org/10.3390/cancers13071746
Maher M, Diesch J, Le Pannérer M-M, Buschbeck M. Epigenetics in a Spectrum of Myeloid Diseases and Its Exploitation for Therapy. Cancers. 2021; 13(7):1746. https://doi.org/10.3390/cancers13071746
Chicago/Turabian StyleMaher, Michael, Jeannine Diesch, Marguerite-Marie Le Pannérer, and Marcus Buschbeck. 2021. "Epigenetics in a Spectrum of Myeloid Diseases and Its Exploitation for Therapy" Cancers 13, no. 7: 1746. https://doi.org/10.3390/cancers13071746
APA StyleMaher, M., Diesch, J., Le Pannérer, M.-M., & Buschbeck, M. (2021). Epigenetics in a Spectrum of Myeloid Diseases and Its Exploitation for Therapy. Cancers, 13(7), 1746. https://doi.org/10.3390/cancers13071746