
Combination of PKCδ Inhibition with Conventional TKI Treatment to Target CML Models

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
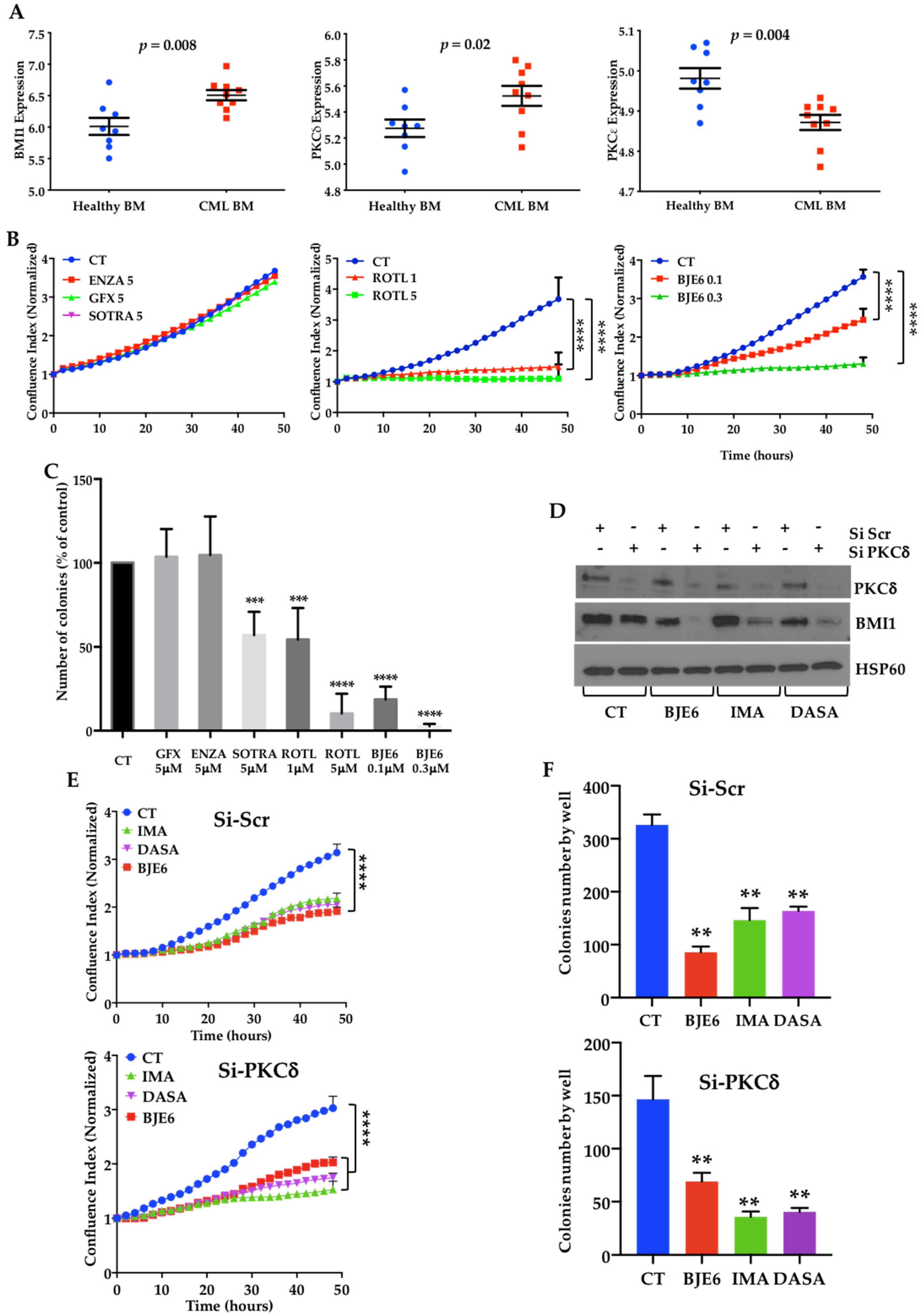
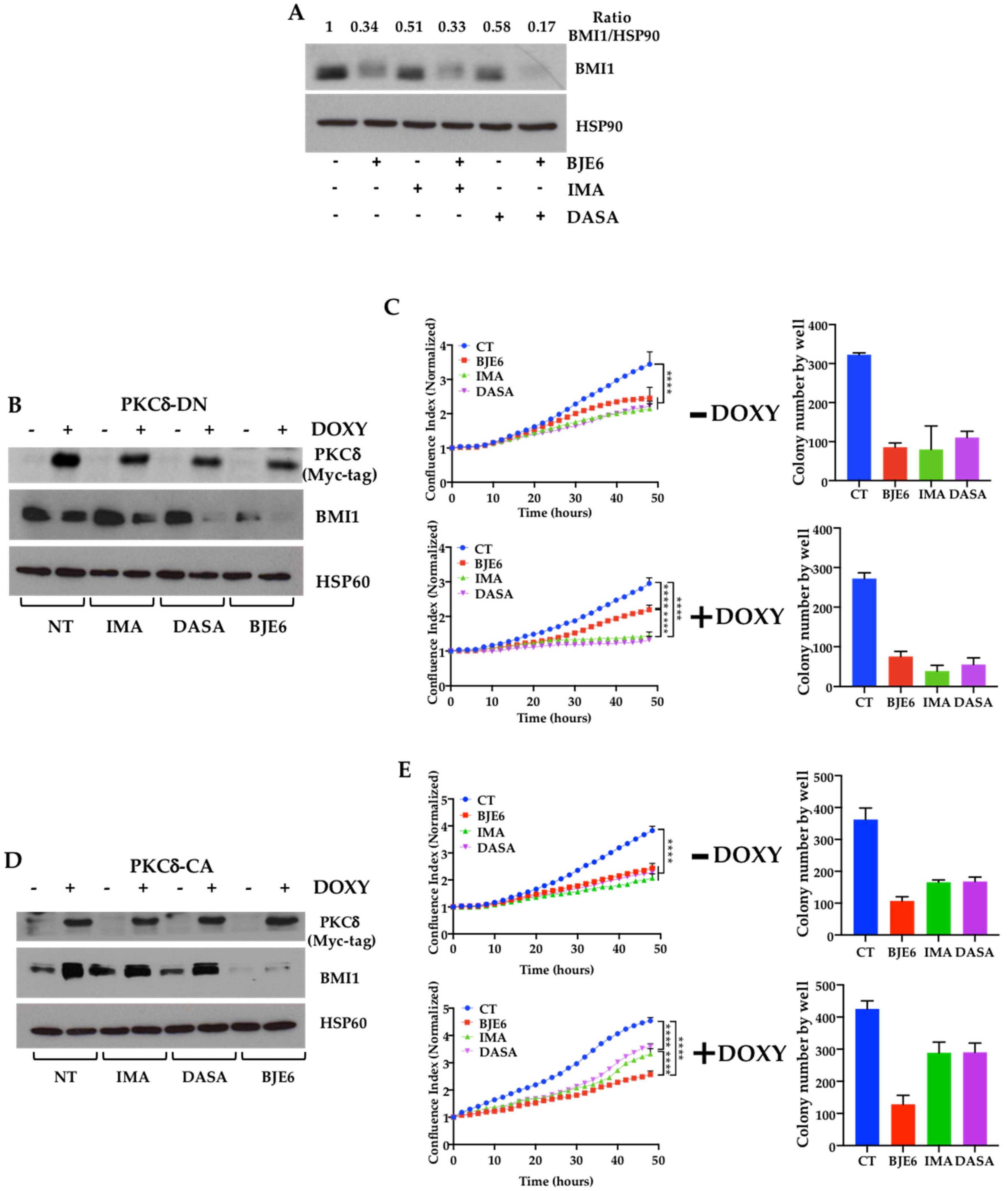
2.1. BCR-ABL Controls BMI1 Expression by Increasing PKCδ Levels in CML Cells
2.2. PKCδ Inhibition Synergizes with TKIs to Decrease Proliferation and Clonogenicity of CML Cells
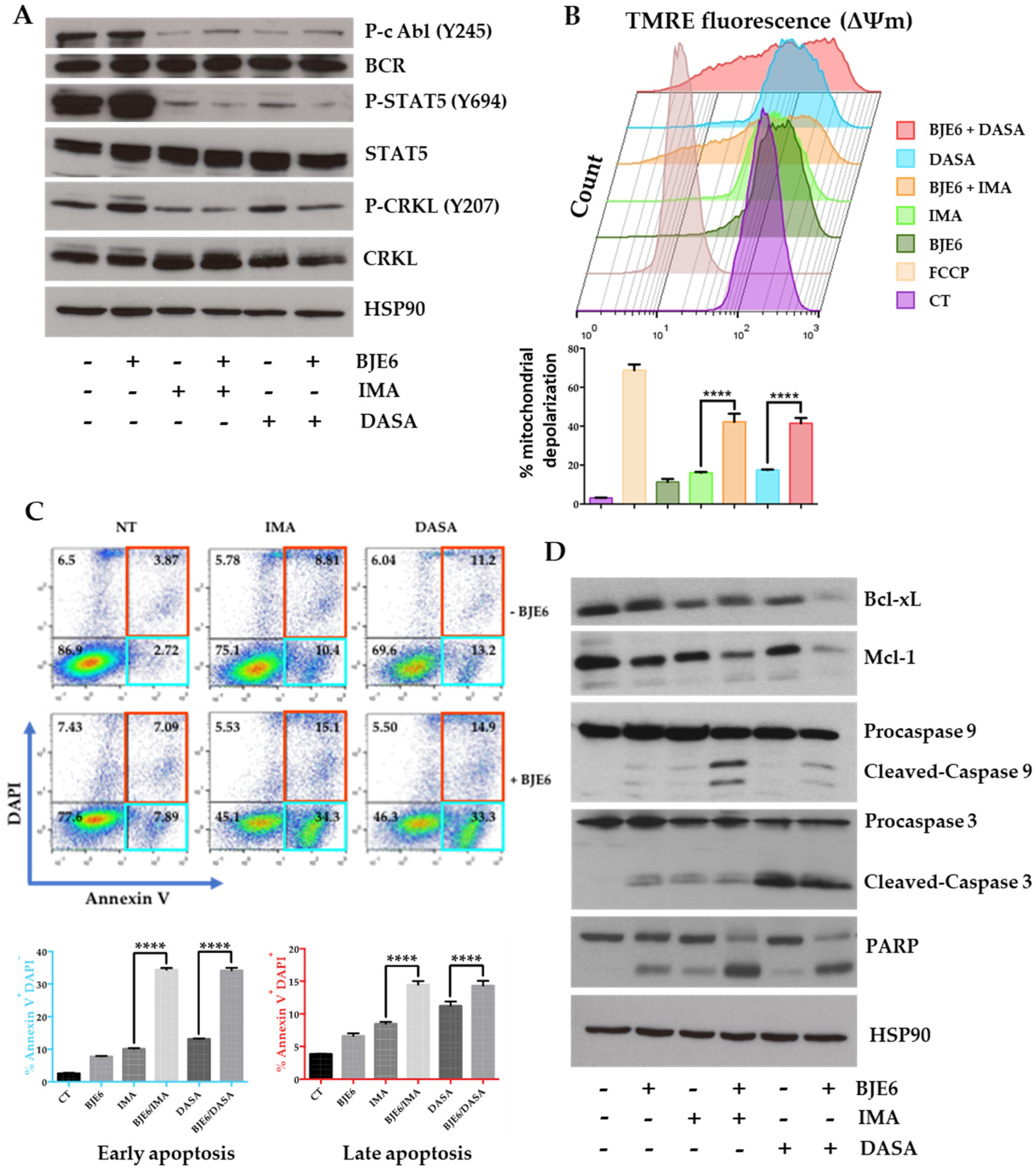
2.3. PKCδ Inhibition Synergistically Enhances TKI-Induced Mitochondrial Apoptotic Events
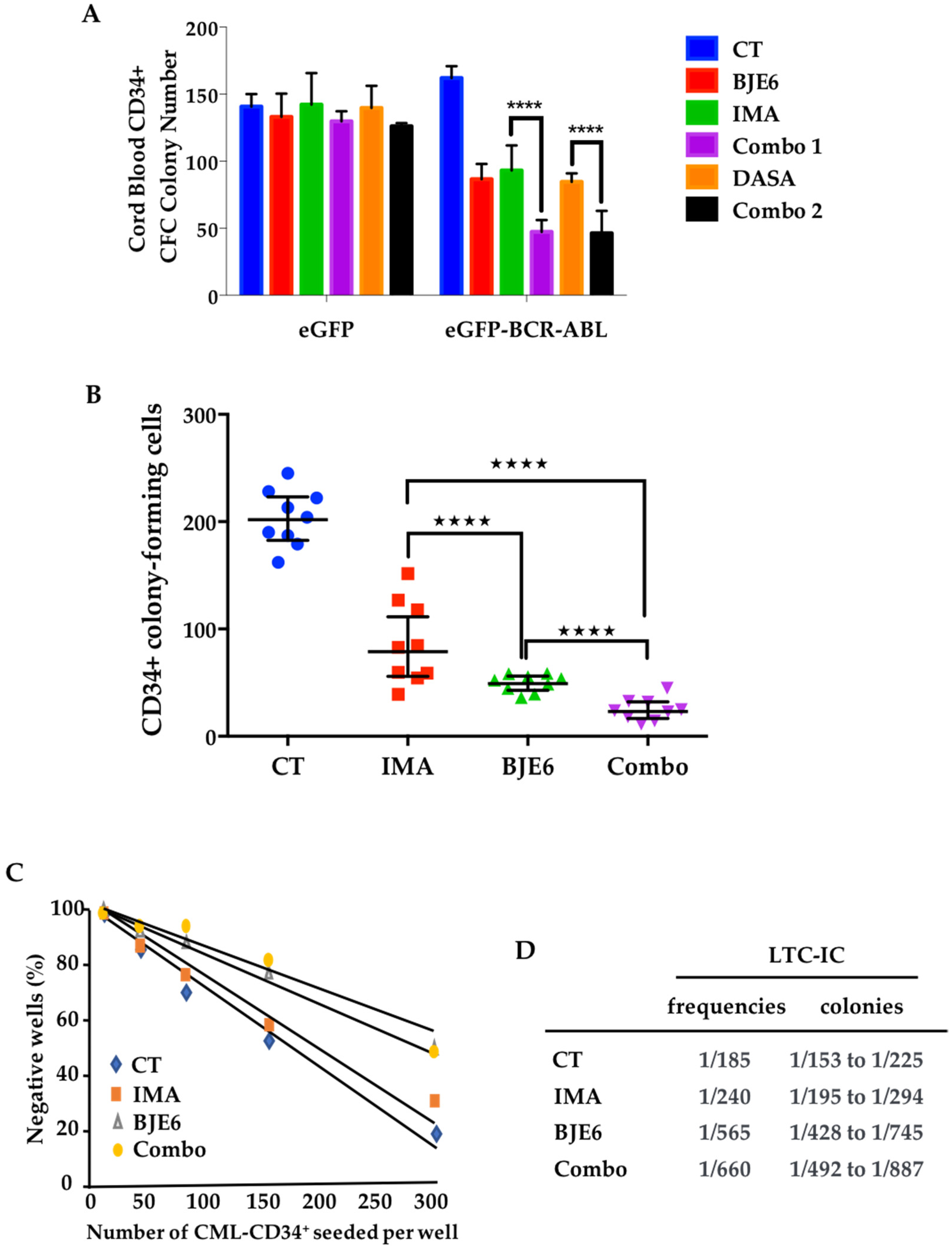
2.4. Combination of BCR-ABL and PKCδ Inhibitors Targeted Stem/Progenitor CML Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines
4.3. Proliferation Assay
4.4. Viability Assay and Synergy Score Calculation
4.5. Analysis of Apoptosis
4.6. Mitochondrial Membrane Potential (ΔΨm) Measurement
4.7. Primary Cell Isolation
4.8. Colony-Forming Cell (CFC) Assay
4.9. Long-Term Culture-Initiating Cell (LTC-IC) Experiments with Limiting Dilution Assay (LDA)
4.10. Plasmid, Transfection and Luciferase Reporter Assays
4.11. Western Blotting
4.12. Stastitical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma–extra large |
| BCR-ABL | Breakpoint cluster region–Abelson murine leukemia viral oncogene homolog 1 |
| BJE6 | BJE6-106 |
| BMI1 | B cell-specific Moloney murine leukemia virus integration site 1 |
| BOSU | Bosutinib |
| CA | Constitutively active |
| CFC | Colony-forming cell |
| CRKL | Crk-like protein |
| CSC | Cancer stem cell |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DASA | Dasatinib |
| DN | Dominant negative |
| DOXY | Doxycycline |
| EGFR | Epithelial growth factor receptor |
| ENZA | Enzastaurin |
| GFX | GF109203X |
| HSC | Hematopoietic stem cell |
| IMA | Imatinib |
| LIC | Leukemia-initiating cell |
| LSC | Leukemic stem cell |
| Mcl-1 | Induced myeloid leukemia cell differentiation protein |
| NILO | Nilotinib |
| PKCδ | Protein kinase Cδ |
| PONA | Ponatinib |
| PRC1 | Polycomb repressive complex 1 |
| SOTRA | Sotrastaurin |
| STAT5 | Signal transducer and activator of transcription 5 |
| TKI | Tyrosine kinase inhibitor |
| TMRE | Tetramethylrhodamine ethyl ester perchlorate |
References
- Dan, S.; Naito, M.; Tsuruo, T. Selective induction of apoptosis in Philadelphia chromosome-positive chronic myelogenous leukemia cells by an inhibitor of BCR-ABL tyrosine kinase, CGP 57148. Cell Death Differ. 1998, 5, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Kim, C.N.; Perkins, C.L.; Ramadevi, N.; Winton, E.; Wittmann, S.; Bhalla, K.N. CGP57148B (STI-571) induces differentiation and apoptosis and sensitizes Bcr-Abl-positive human leukemia cells to apoptosis due to antileukemic drugs. Blood 2000, 96, 2246–2253. [Google Scholar] [CrossRef]
- An, X.; Tiwari, A.K.; Sun, Y.; Ding, P.R.; Ashby, C.R., Jr.; Chen, Z.S. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: A review. Leuk. Res. 2010, 34, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Tipping, A.J.; Baluch, S.; Barnes, D.J.; Veach, D.R.; Clarkson, B.M.; Bornmann, W.G.; Mahon, F.X.; Goldman, J.M.; Melo, J.V. Efficacy of dual-specific Bcr-Abl and Src-family kinase inhibitors in cells sensitive and resistant to imatinib mesylate. Leukemia 2004, 18, 1352–1356. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.; Schafhausen, P.; Brummendorf, T.H. Bosutinib: A dual SRC/ABL kinase inhibitor for the treatment of chronic myeloid leukemia. Expert Rev. Hematol. 2009, 2, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Muselli, F.; Peyron, J.F.; Mary, D. Druggable Biochemical Pathways and Potential Therapeutic Alternatives to Target Leukemic Stem Cells and Eliminate the Residual Disease in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2019, 20, 5616. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.; Trotta, R.; Stramucci, L.; Ellis, J.J.; Harb, J.G.; Neviani, P.; Wang, S.; Eisfeld, A.K.; Walker, C.J.; Zhang, B.; et al. Persistence of Drug-Resistant Leukemic Stem Cells and Impaired NK Cell Immunity in CML Patients Depend on MIR300 Antiproliferative and PP2A-Activating Functions. Blood Cancer Discov. 2020, 1, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.L.; et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018, 24, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein kinase C in cancer: The top five unanswered questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef]
- Parker, P.J.; Brown, S.J.; Calleja, V.; Chakravarty, P.; Cobbaut, M.; Linch, M.; Marshall, J.J.T.; Martini, S.; McDonald, N.Q.; Soliman, T.; et al. Equivocal, explicit and emergent actions of PKC isoforms in cancer. Nat. Rev. Cancer. 2021, 21, 51–63. [Google Scholar] [CrossRef]
- Jackson, D.N.; Foster, D.A. The enigmatic protein kinase Cdelta: Complex roles in cell proliferation and survival. FASEB J. 2004, 18, 627–636. [Google Scholar] [CrossRef]
- Chu, W.K.; Dai, P.M.; Li, H.L.; Pao, C.C.; Chen, J.K. Nanog expression is negatively regulated by protein kinase C activities in human cancer cell lines. Carcinogenesis 2013, 34, 1497–1509. [Google Scholar] [CrossRef]
- Kahana, S.; Finniss, S.; Cazacu, S.; Xiang, C.; Lee, H.K.; Brodie, S.; Goldstein, R.S.; Roitman, V.; Slavin, S.; Mikkelsen, T.; et al. Proteasome inhibitors sensitize glioma cells and glioma stem cells to TRAIL-induced apoptosis by PKCepsilon-dependent downregulation of AKT and XIAP expressions. Cell. Signal. 2011, 23, 1348–1357. [Google Scholar] [CrossRef]
- Basu, A.; Pal, D. Two faces of protein kinase Cdelta: The contrasting roles of PKCdelta in cell survival and cell death. Sci. World J. 2010, 10, 2272–2284. [Google Scholar] [CrossRef]
- Chen, Z.; Forman, L.W.; Williams, R.M.; Faller, D.V. Protein kinase C-delta inactivation inhibits the proliferation and survival of cancer stem cells in culture and in vivo. BMC Cancer 2014, 14, 90. [Google Scholar] [CrossRef]
- Lee, P.C.; Fang, Y.F.; Yamaguchi, H.; Wang, W.J.; Chen, T.C.; Hong, X.; Ke, B.; Xia, W.; Wei, Y.; Zha, Z.; et al. Targeting PKCdelta as a Therapeutic Strategy against Heterogeneous Mechanisms of EGFR Inhibitor Resistance in EGFR-Mutant Lung Cancer. Cancer Cell 2018, 34, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Bazuine, M.; van der Zon, G.C.; van de Ven, R.; van den Broek, P.J.; Antonie Maassen, J. Rottlerin inhibits multiple steps involved in insulin-induced glucose uptake in 3T3-L1 adipocytes. Biochem. Pharmacol. 2004, 68, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P. Rottlerin: An inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol. Sci. 2007, 28, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Mourgues, L.; Imbert, V.; Nebout, M.; Colosetti, P.; Neffati, Z.; Lagadec, P.; Verhoeyen, E.; Peng, C.; Duprez, E.; Legros, L.; et al. The BMI1 polycomb protein represses cyclin G2-induced autophagy to support proliferation in chronic myeloid leukemia cells. Leukemia 2015, 29, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, T.; Tsuji, K.; Kida, A.; Koyama, T.; Yamamoto, M.; Miura, O. Rottlerin synergistically enhances imatinib-induced apoptosis of BCR/ABL-expressing cells through its mitochondrial uncoupling effect independent of protein kinase C-delta. Oncogene 2007, 26, 2975–2987. [Google Scholar] [CrossRef]
- Takashima, A.; English, B.; Chen, Z.; Cao, J.; Cui, R.; Williams, R.M.; Faller, D.V. Protein kinase Cdelta is a therapeutic target in malignant melanoma with NRAS mutation. ACS Chem. Biol. 2014, 9, 1003–1014. [Google Scholar] [CrossRef]
- Mauro, L.V.; Grossoni, V.C.; Urtreger, A.J.; Yang, C.; Colombo, L.L.; Morandi, A.; Pallotta, M.G.; Kazanietz, M.G.; Bal de Kier Joffe, E.D.; Puricelli, L.L. PKC Delta (PKCdelta) promotes tumoral progression of human ductal pancreatic cancer. Pancreas 2010, 39, e31–e41. [Google Scholar] [CrossRef]
- Yoon, C.H.; Kim, M.J.; Park, M.J.; Park, I.C.; Hwang, S.G.; An, S.; Choi, Y.H.; Yoon, G.; Lee, S.J. Claudin-1 acts through c-Abl-protein kinase Cdelta (PKCdelta) signaling and has a causal role in the acquisition of invasive capacity in human liver cells. J. Biol. Chem. 2010, 285, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef]
- Siddique, H.R.; Parray, A.; Tarapore, R.S.; Wang, L.; Mukhtar, H.; Karnes, R.J.; Deng, Y.; Konety, B.R.; Saleem, M. BMI1 polycomb group protein acts as a master switch for growth and death of tumor cells: Regulates TCF4-transcriptional factor-induced BCL2 signaling. PLoS ONE 2013, 8, e60664. [Google Scholar] [CrossRef]
- Kreso, A.; van Galen, P.; Pedley, N.M.; Lima-Fernandes, E.; Frelin, C.; Davis, T.; Cao, L.; Baiazitov, R.; Du, W.; Sydorenko, N.; et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef]
- Kong, Y.; Ai, C.; Dong, F.; Xia, X.; Zhao, X.; Yang, C.; Kang, C.; Zhou, Y.; Zhao, Q.; Sun, X.; et al. Targeting of BMI-1 with PTC-209 inhibits glioblastoma development. Cell Cycle 2018, 17, 1199–1211. [Google Scholar] [CrossRef]
- Shen, H.T.; Chien, P.J.; Chen, S.H.; Sheu, G.T.; Jan, M.S.; Wang, B.Y.; Chang, W.W. BMI1-Mediated Pemetrexed Resistance in Non-Small Cell Lung Cancer Cells Is Associated with Increased SP1 Activation and Cancer Stemness. Cancers 2020, 12, 2069. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, I.; Danis, E.; Pierce, A.; Madhavan, K.; Wang, D.; Dahl, N.; Sanford, B.; Birks, D.K.; Davidson, N.; Metselaar, D.S.; et al. Senescence Induced by BMI1 Inhibition Is a Therapeutic Vulnerability in H3K27M-Mutant DIPG. Cell Rep. 2020, 33, 108286. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’ Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Goff, D.J.; Court Recart, A.; Sadarangani, A.; Chun, H.J.; Barrett, C.L.; Krajewska, M.; Leu, H.; Low-Marchelli, J.; Ma, W.; Shih, A.Y.; et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 2013, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Beverly, L.J.; Varmus, H.E. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene 2009, 28, 1274–1279. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, S.; Dai, Y.; Harada, H.; Dent, P.; Grant, S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007, 67, 782–791. [Google Scholar] [CrossRef]
- Vogler, M.; Butterworth, M.; Majid, A.; Walewska, R.J.; Sun, X.M.; Dyer, M.J.; Cohen, G.M. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 2009, 113, 4403–4413. [Google Scholar] [CrossRef] [PubMed]
- Yecies, D.; Carlson, N.E.; Deng, J.; Letai, A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 2010, 115, 3304–3313. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Mochizuki, Y.; Ogahara, I.; Watanabe, T.; Hogdal, L.; Takagi, S.; Sato, K.; Kaneko, A.; Kajita, H.; Uchida, N.; et al. Overcoming mutational complexity in acute myeloid leukemia by inhibition of critical pathways. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Joseloff, E.; Cataisson, C.; Aamodt, H.; Ocheni, H.; Blumberg, P.; Kraker, A.J.; Yuspa, S.H. Src family kinases phosphorylate protein kinase C delta on tyrosine residues and modify the neoplastic phenotype of skin keratinocytes. J. Biol. Chem. 2002, 277, 12318–12323. [Google Scholar] [CrossRef]
- Verhoeyen, E.; Relouzat, F.; Cambot, M.; Costa, C.; Negre, D.; Legrand, F.; Joubert, C.; Le Grand, R.; Cosset, F.L.; Leboulch, P.; et al. Stem cell factor-displaying simian immunodeficiency viral vectors together with a low conditioning regimen allow for long-term engraftment of gene-marked autologous hematopoietic stem cells in macaques. Hum. Gene Ther. 2012, 23, 754–768. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BJE6 (µM) | IMA (µM) | % Cell Death | Relative Synergy | Standard Deviation |
|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 0 |
| 0.1 | 0 | 24.74 | 0 | 12.23 |
| 0.3 | 0 | 61.95 | 0 | 0.56 |
| 1 | 0 | 96.36 | 0 | 0.61 |
| 0 | 0.1 | 18.98 | 0 | 9.98 |
| 0.1 | 0.1 | 29.11 | 5.325283 | 0.73 |
| 0.3 | 0.1 | 71.88 | 12.1007 | 8.41 |
| 1 | 0.1 | 96.56 | 0.24372 | 0.39 |
| 0 | 0.3 | 39.48 | 0 | 3.64 |
| 0.1 | 0.3 | 55 | 18.91267 | 4.63 |
| 0.3 | 0.3 | 70.98 | 11.00396 | 1.84 |
| 1 | 0.3 | 95.71 | −0.79209 | 0.59 |
| 0 | 1 | 57.57 | 0 | 2.55 |
| 0.1 | 1 | 69.09 | 14.03827 | 1.36 |
| 0.3 | 1 | 81.28 | 23.55554 | 0.47 |
| 1 | 1 | 95.88 | −0.58493 | 0.18 |
| BJE6 (µM) | DASA (nM) | % Cell Death | Relative Synergy | Standard Deviation |
|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 0 |
| 0.1 | 0 | 22.45 | 0 | 7.18 |
| 0.3 | 0 | 65.86 | 0 | 4.01 |
| 1 | 0 | 95.74 | 0 | 0.55 |
| 0 | 0.1 | 13.97 | 0 | 7.27 |
| 0.1 | 0.1 | 35.04 | 14.92119 | 5.59 |
| 0.3 | 0.1 | 70.49 | 5.487299 | 0.86 |
| 1 | 0.1 | 96.15 | 0.485916 | 0.60 |
| 0 | 0.3 | 42.25 | 0 | 4.11 |
| 0.1 | 0.3 | 41.03 | −1.4459 | 2.99 |
| 0.3 | 0.3 | 72.96 | 8.414648 | 2.29 |
| 1 | 0.3 | 95.36 | −0.45036 | 0.68 |
| 0 | 1 | 52.48 | 0 | 10.10 |
| 0.1 | 1 | 54.95 | 2.92735 | 5.51 |
| 0.3 | 1 | 78.59 | 15.08711 | 4.65 |
| 1 | 1 | 95.1 | −0.7585 | 1.13 |
| 0 | 3 | 60.97 | 0 | 8.02 |
| 0.1 | 3 | 64.47 | 4.148066 | 4.41 |
| 0.3 | 3 | 83.4 | 20.78774 | 4.20 |
| 1 | 3 | 94.6 | −1.35108 | 0.89 |
| 0 | 10 | 65.76 | 0 | 6.63 |
| 0.1 | 10 | 60.09 | −6.71987 | 2.80 |
| 0.3 | 10 | 82.12 | 19.27073 | 2.25 |
| 1 | 10 | 94.66 | −1.27997 | 0.59 |
| Target | Antibody | Ref | Supplier |
|---|---|---|---|
| Actin | Actin (I-19) | sc-1616 | SCB |
| Bcl-2 | Bcl-2 (C 21) | sc-783 | SCB |
| Bcl-xL | Bcl-xL | 2762 | CST |
| Bmi1 | Bmi1 (F6) | 05-637 | Merck Millipore (Upstate) |
| BCR | BCR | 3902 S | CST |
| Caspase 3 | Caspase 3 | 9662 S | CST |
| Caspase 9 | Caspase 9 | 9502 T | CST |
| CRKL | CRKL (32H4) | 3182 | CST |
| HSP60 | HSP60 (K-19) | sc-1722 | SCB |
| HSP90 | HSP 90α/β Antibody (F-8) | sc-13119 | SCB |
| Mcl-1 | Mcl-1 (D35A5) | 5453 S | CST |
| Myc-tag | Myc-tag clone 4A6 | 05-724 | Merck Millipore (Upstate) |
| PARP | PARP | 9542 S | CST |
| P-c Abl | P-c Abl (Y245) (73E5) | 2868 S | CST |
| P-CRKL | P-CRKL (Tyr207) | 3181 S | CST |
| P-STAT5 | P-STAT5 (Tyr694) (C11C5) | 9359 S | CST |
| PKCδ | PKCδ | 2058 S | CST |
| P-KCδ | P-PKCδ (Thr505) | 9374S | CST |
| STAT5 | STAT5 (D2O6Y) | 94205 | CST |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muselli, F.; Mourgues, L.; Morcos, R.; Rochet, N.; Nebout, M.; Guerci-Bresler, A.; Faller, D.V.; William, R.M.; Mhaidly, R.; Verhoeyen, E.; et al. Combination of PKCδ Inhibition with Conventional TKI Treatment to Target CML Models. Cancers 2021, 13, 1693. https://doi.org/10.3390/cancers13071693
Muselli F, Mourgues L, Morcos R, Rochet N, Nebout M, Guerci-Bresler A, Faller DV, William RM, Mhaidly R, Verhoeyen E, et al. Combination of PKCδ Inhibition with Conventional TKI Treatment to Target CML Models. Cancers. 2021; 13(7):1693. https://doi.org/10.3390/cancers13071693
Chicago/Turabian StyleMuselli, Fabien, Lucas Mourgues, Rita Morcos, Nathalie Rochet, Marielle Nebout, Agnès Guerci-Bresler, Douglas V Faller, Robert M William, Rana Mhaidly, Els Verhoeyen, and et al. 2021. "Combination of PKCδ Inhibition with Conventional TKI Treatment to Target CML Models" Cancers 13, no. 7: 1693. https://doi.org/10.3390/cancers13071693
APA StyleMuselli, F., Mourgues, L., Morcos, R., Rochet, N., Nebout, M., Guerci-Bresler, A., Faller, D. V., William, R. M., Mhaidly, R., Verhoeyen, E., Legros, L., Peyron, J.-F., & Mary, D. (2021). Combination of PKCδ Inhibition with Conventional TKI Treatment to Target CML Models. Cancers, 13(7), 1693. https://doi.org/10.3390/cancers13071693