
Targeting Feedforward Loops Formed by Nuclear Receptor RORγ and Kinase PBK in mCRPC with Hyperactive AR Signaling

 , , , and
, , , and 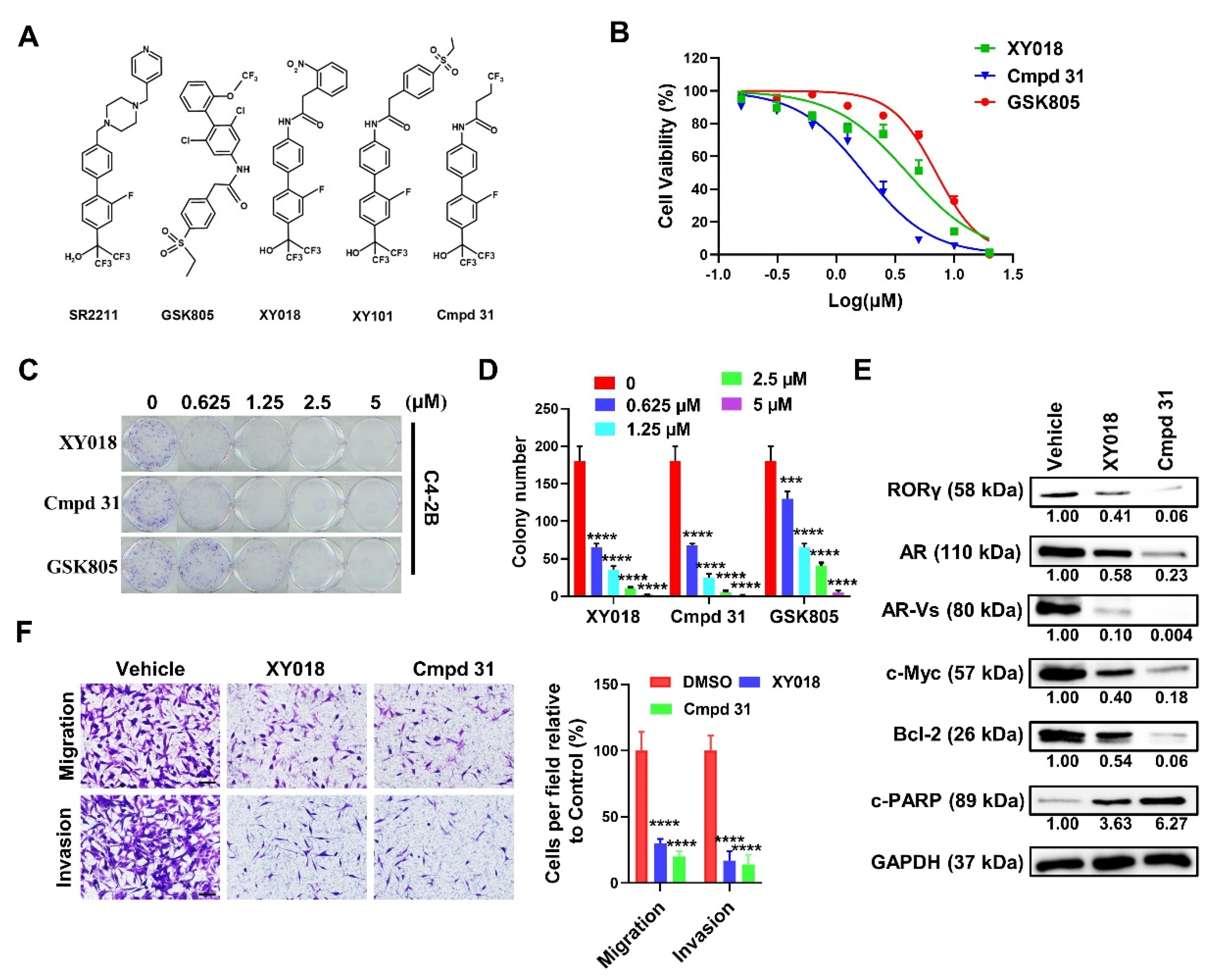{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. RORγ Antagonist/Inverse Agonist cmpd 31 Ootently Inhibits CRPC Cell Survival, Migration, and Invasion
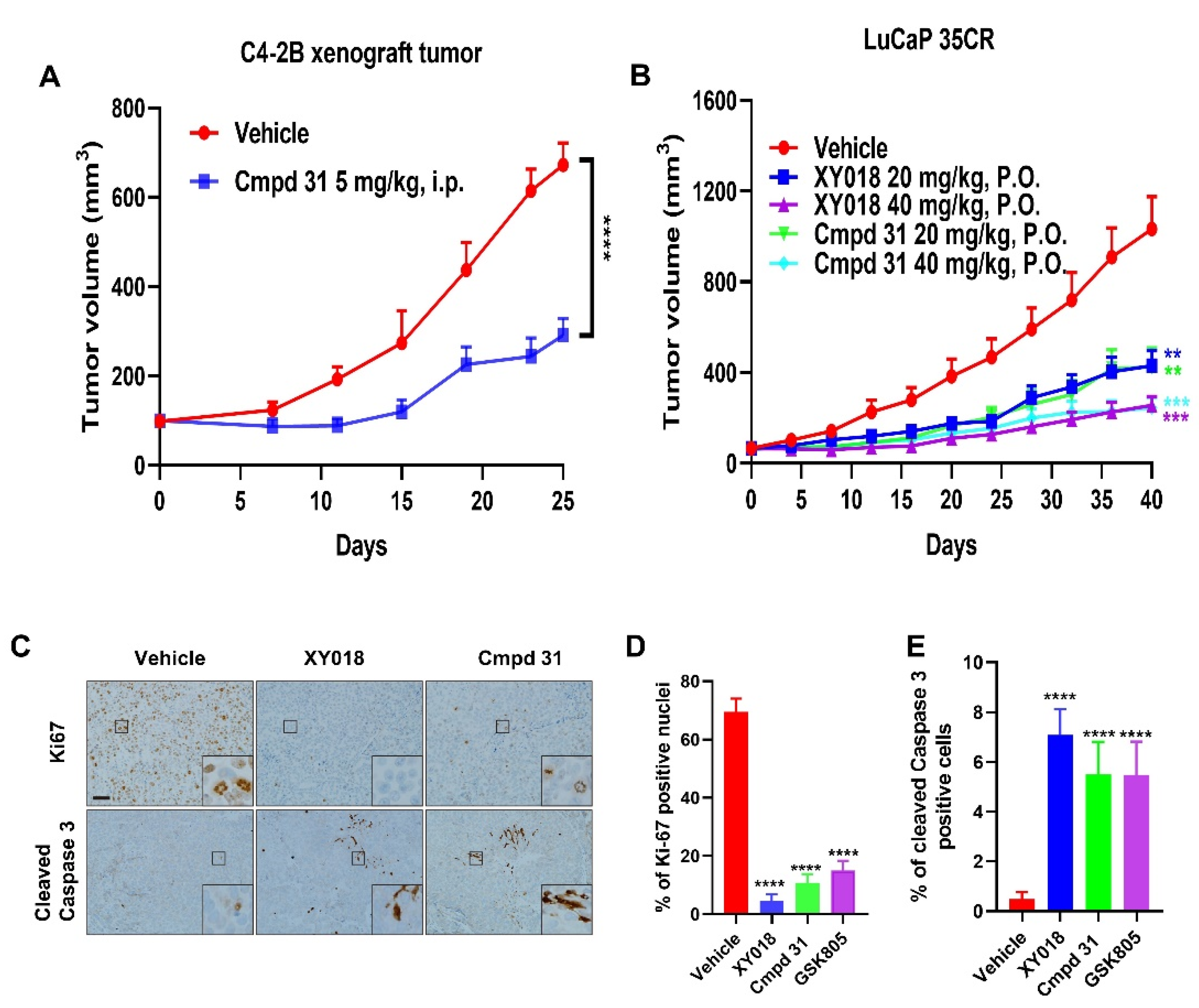
2.2. Orally Administered RORγ Antagonists/Inverse Agonists Potently Inhibit Growth of PDX Tumors
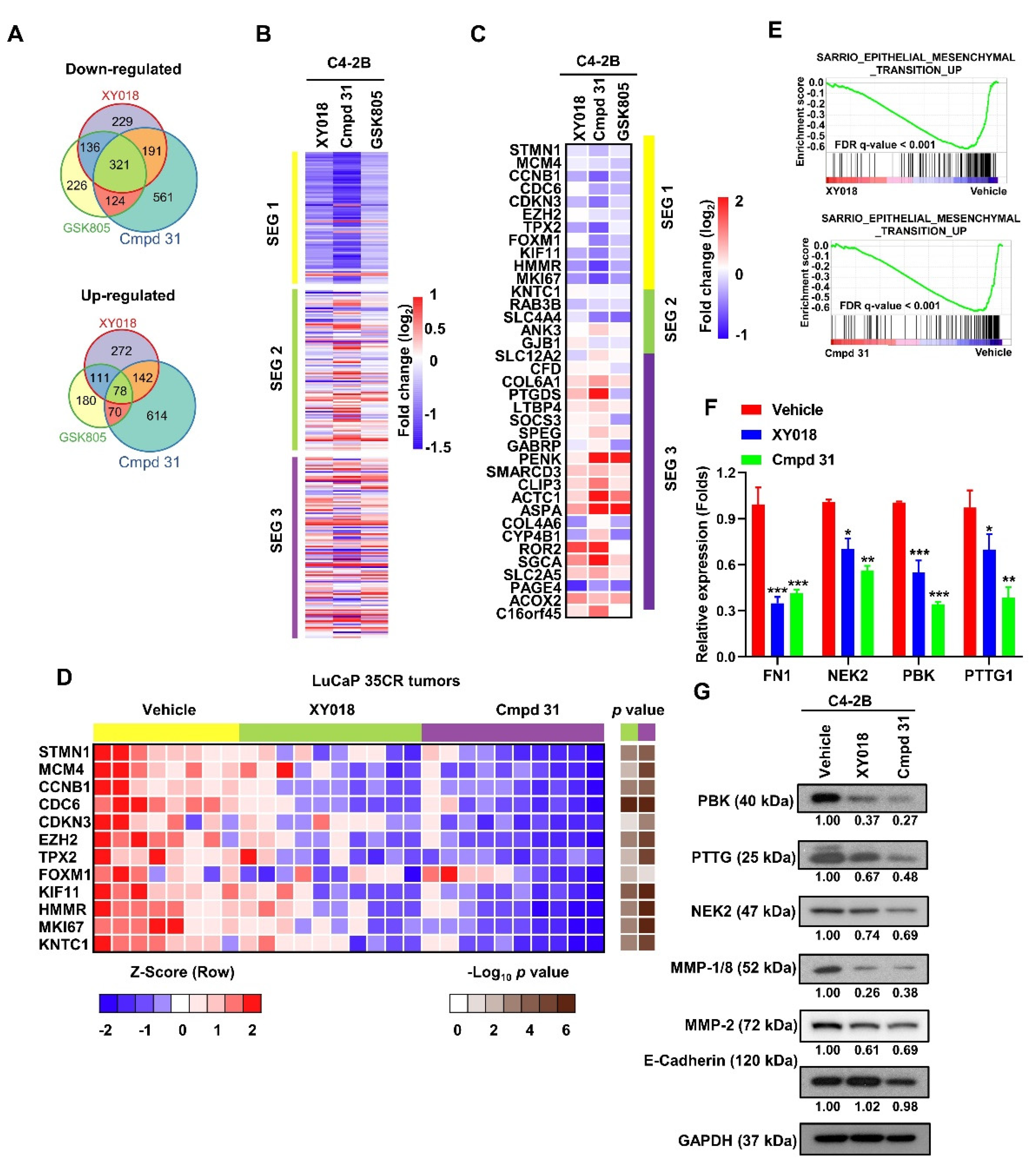
2.3. RORγ Antagonists/Inverse Agonists Suppress the Expression of Gene Programs Linked to Tumor Aggressiveness
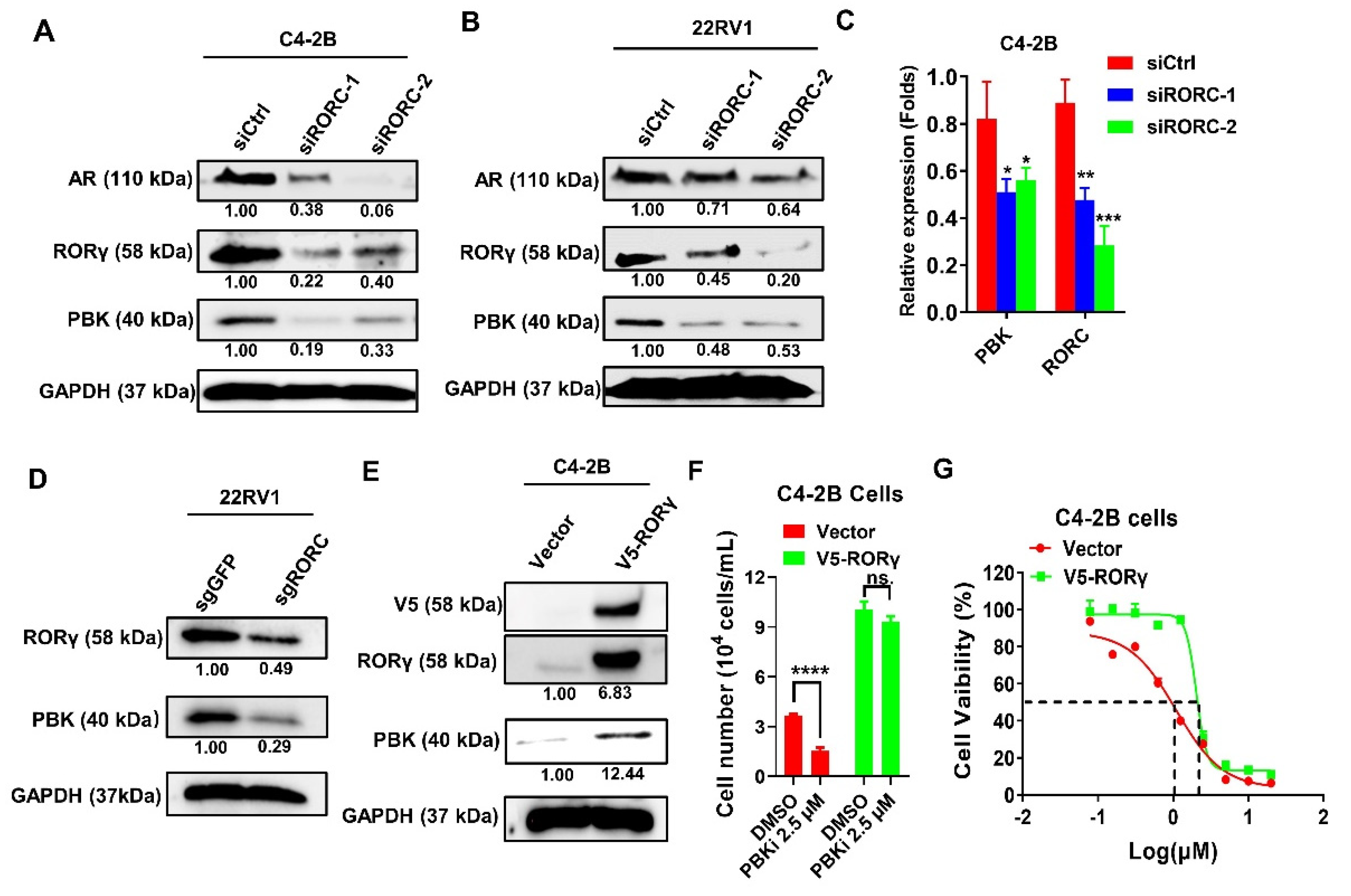
2.4. PBK Is a Downstream Target of RORγ
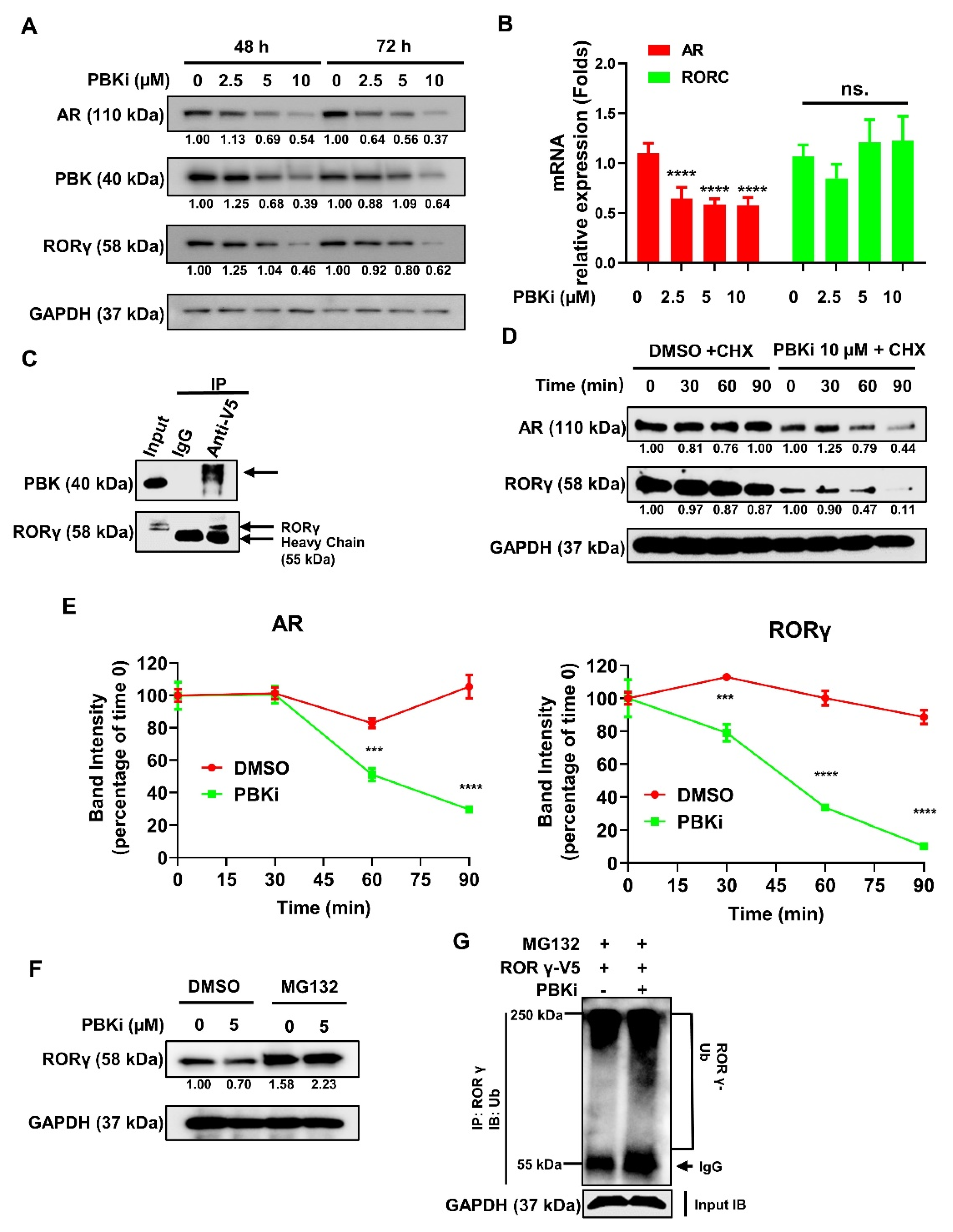
2.5. PBK Interacts With RORγ and Modulates RORγ Protein Stability
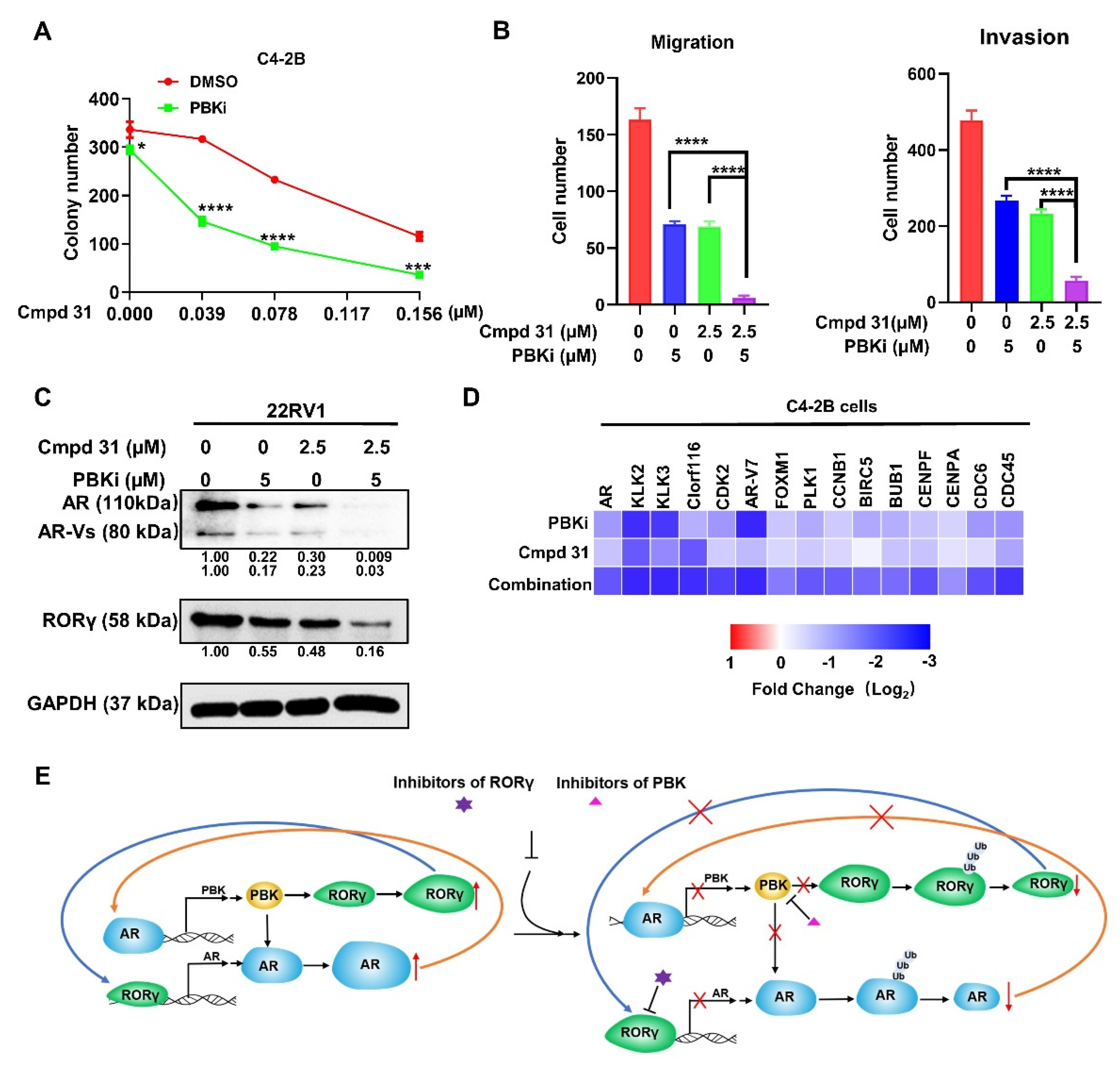
2.6. Dual Inhibition of PBK and RORγ Synergistically Inhibits CRPC Cell Survival and Invasion and AR Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture and siRNA Transfection
4.2. Chemicals and Reagents
4.3. Cell Viability and Growth Assay
4.4. Western Blotting and qRT-PCR
4.5. Immunohistochemistry (IHC)
4.6. Transwell Assay
4.7. Lentivirus Production, Cell Infection, co-IP, and Ubiquitin Pull Down
4.8. Xenograft Tumor Models and Treatments
4.9. RNA-Seq and Bioinformatics Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Einstein, D.J.; Arai, S.; Balk, S.P. Targeting the androgen receptor and overcoming resistance in prostate cancer. Curr. Opin. Oncol. 2019, 31, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Carceles-Cordon, M.; Kelly, W.K.; Gomella, L.; Knudsen, K.E.; Rodriguez-Bravo, V.; Domingo-Domenech, J. Cellular rewiring in lethal prostate cancer: The architect of drug resistance. Nat. Rev. Urol. 2020, 17, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Huang, H. Interplay among PI3K/AKT, PTEN/FOXO and AR Signaling in Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Dai, B.; Jiang, T.; Xu, K.; Xie, Y.; Kim, O.; Nesheiwat, I.; Kong, X.; Melamed, J.; Handratta, V.D.; et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006, 10, 309–319. [Google Scholar] [CrossRef]
- Asim, M.; A Siddiqui, I.; Hafeez, B.B.; Baniahmad, A.; Mukhtar, H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene 2008, 27, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Lamoureux, F.; Crafter, C.; Davies, B.R.; Beraldi, E.; Fazli, L.; Kim, S.; Thaper, D.; Gleave, M.E.; Zoubeidi, A. Synergistic Targeting of PI3K/AKT Pathway and Androgen Receptor Axis Significantly Delays Castration-Resistant Prostate Cancer Progression In Vivo. Mol. Cancer Ther. 2013, 12, 2342–2355. [Google Scholar] [CrossRef]
- Desai, S.J.; Ma, A.-H.; Tepper, C.G.; Chen, H.-W.; Kung, H.-J. Inappropriate Activation of the Androgen Receptor by Nonsteroids: Involvement of the Src Kinase Pathway and Its Therapeutic Implications. Cancer Res. 2006, 66, 10449–10459. [Google Scholar] [CrossRef]
- Warren, A.Y.; Massie, C.E.; Watt, K.; Luko, K.; Orafidiya, F.; Selth, L.A.; Mohammed, H.; Chohan, B.S.; Menon, S.; Baridi, A.; et al. A reciprocal feedback between the PDZ binding kinase and androgen receptor drives prostate cancer. Oncogene 2019, 38, 1136–1150. [Google Scholar] [CrossRef]
- Roggero, C.M.; Jin, L.; Cao, S.; Sonavane, R.; Kopplin, N.G.; Ta, H.Q.; Ekoue, D.N.; Witwer, M.; Ma, S.; Liu, H.; et al. A detailed characterization of stepwise activation of the androgen receptor variant 7 in prostate cancer cells. Oncogene 2021, 40, 1106–1117. [Google Scholar] [CrossRef]
- Maitland, N. Resistance to Antiandrogens in Prostate Cancer: Is It Inevitable, Intrinsic or Induced? Cancers 2021, 13, 327. [Google Scholar] [CrossRef]
- Paschalis, A.; Sharp, A.; Welti, J.C.; Neeb, A.; Raj, G.V.; Luo, J.; Plymate, S.R.; De Bono, J.S. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–675. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Atallah, N.M.; Pascuzzi, P.E.; Gupta, S.; Liu, X. Inhibition of the Wnt/β-Catenin Pathway Overcomes Resistance to Enzalutamide in Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar] [CrossRef]
- Adelaiye-Ogala, R.; Gryder, B.E.; Nguyen, Y.T.M.; Alilin, A.N.; Grayson, A.R.; Bajwa, W.; Jansson, K.H.; Beshiri, M.L.; Agarwal, S.; Rodriguez-Nieves, J.A.; et al. Targeting the PI3K/AKT Pathway Overcomes Enzalutamide Resistance by Inhibiting Induction of the Glucocorticoid Receptor. Mol. Cancer Ther. 2020, 19, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, D.; Johnson, J.K.; Pascal, L.E.; Takubo, K.; Avula, R.; Chakka, A.B.; Zhou, J.; Chen, W.; Zhong, M.; et al. A Novel Small Molecule Targets Androgen Receptor and Its Splice Variants in Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2019, 19, 75–88. [Google Scholar] [CrossRef]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.-K.; Watt, K.; Tam, T.; Yang, Y.C.; Bañuelos, C.A.; et al. Regression of Castrate-Recurrent Prostate Cancer by a Small-Molecule Inhibitor of the Amino-Terminus Domain of the Androgen Receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Fujimoto, N.; Kashiwagi, E.; Eto, M. The Role of Nuclear Receptors in Prostate Cancer. Cells 2019, 8, 602. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zou, J.X.; Xue, X.; Cai, D.; Zhang, Y.; Duan, Z.; Xiang, Q.; Yang, J.C.; Louie, M.C.; Borowsky, A.D.; et al. ROR-γ drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat. Med. 2016, 22, 488–496. [Google Scholar] [CrossRef]
- Jetten, A.M.; Cook, D.N. (Inverse) Agonists of Retinoic Acid–Related Orphan Receptor γ: Regulation of Immune Responses, Inflammation, and Autoimmune Disease. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, X.; Jin, X.; Song, Y.; Li, J.; Luo, X.; Song, M.; Yan, W.; Song, H.; Xu, Y. Discovery of 2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide derivatives as new RORγ inhibitors using virtual screening, synthesis and biological evaluation. Eur. J. Med. Chem. 2014, 78, 431–441. [Google Scholar] [CrossRef]
- Wu, X.-S.; Wang, R.; Xing, Y.-L.; Xue, X.-Q.; Zhang, Y.; Lu, Y.-Z.; Song, Y.; Luo, X.-Y.; Wu, C.; Zhou, Y.-L.; et al. Discovery and structural optimization of 4-(4-(benzyloxy)phenyl)-3,4-dihydropyrimidin-2(1H)-ones as RORc inverse agonists. Acta Pharmacol. Sin. 2016, 37, 1516–1524. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, X.; Xue, X.; Li, C.; Wang, J.; Wang, R.; Zhang, C.; Wang, C.; Shi, Y.; Zou, L.; et al. Discovery and Characterization of XY101, a Potent, Selective, and Orally Bioavailable RORγ Inverse Agonist for Treatment of Castration-Resistant Prostate Cancer. J. Med. Chem. 2019, 62, 4716–4730. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, J.; Wang, Q.; Zou, H.; Wang, H.; Zhang, Z.; Chen, J.; Wang, Q.; Wang, P.; Zhao, Y.; et al. Targeting castration-resistant prostate cancer with a novel RORγ antagonist elaiophylin. Acta Pharm. Sin. B 2020, 10, 2313–2322. [Google Scholar] [CrossRef]
- Cai, D.; Wang, J.; Gao, B.; Li, J.; Wu, F.; Zou, J.X.; Xu, J.; Jiang, Y.; Zou, H.; Huang, Z.; et al. RORγ is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Z.; Chen, C.Z.; Liu, C.; Evans, C.P.; Gao, A.C.; Zhou, F.; Chen, H.-W. Therapeutic Targeting of MDR1 Expression by RORγ Antagonists Resensitizes Cross-Resistant CRPC to Taxane via Coordinated Induction of Cell Death Programs. Mol. Cancer Ther. 2020, 19, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.-M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef]
- Lam, H.-M.; McMullin, R.; Nguyen, H.M.; Coleman, I.; Gormley, M.; Gulati, R.; Brown, L.G.; Holt, S.K.; Li, W.; Ricci, D.S.; et al. Characterization of an Abiraterone Ultraresponsive Phenotype in Castration-Resistant Prostate Cancer Patient-Derived Xenografts. Clin. Cancer Res. 2017, 23, 2301–2312. [Google Scholar] [CrossRef]
- You, S.; Knudsen, B.S.; Erho, N.; Alshalalfa, M.; Takhar, M.; Ashab, H.A.-D.; Davicioni, E.; Karnes, R.J.; Klein, E.A.; Den, R.B.; et al. Integrated Classification of Prostate Cancer Reveals a Novel Luminal Subtype with Poor Outcome. Cancer Res. 2016, 76, 4948–4958. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Attard, G.; Balk, S.P.; Bevan, C.; Burnstein, K.; Cato, L.; Cherkasov, A.; De Bono, J.S.; Dong, Y.; Gao, A.C.; et al. Role of Androgen Receptor Variants in Prostate Cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. Eur. Urol. 2018, 73, 715–723. [Google Scholar] [CrossRef]
- Wang, B.; Lo, U.-G.; Wu, K.; Kapur, P.; Liu, X.; Huang, J.; Chen, W.; Hernandez, E.; Santoyo, J.; Ma, S.-H.; et al. Developing new targeting strategy for androgen receptor variants in castration resistant prostate cancer. Int. J. Cancer 2017, 141, 2121–2130. [Google Scholar] [CrossRef]
- Yang, Q.-X.; Zhong, S.; He, L.; Jia, X.-J.; Tang, H.; Cheng, S.-T.; Ren, J.-H.; Yu, H.-B.; Zhou, L.; Zhou, H.-Z.; et al. PBK overexpression promotes metastasis of hepatocellular carcinoma via activating ETV4-uPAR signaling pathway. Cancer Lett. 2019, 452, 90–102. [Google Scholar] [CrossRef]
- Brown-Clay, J.D.; Shenoy, D.N.; Timofeeva, O.; Kallakury, B.V.; Nandi, A.K.; Banerjee, P.P. PBK/TOPK enhances aggressive phenotype in prostate cancer via β-catenin-TCF/LEF-mediated matrix metalloproteinases production and invasion. Oncotarget 2015, 6, 15594–15609. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Srivastava, A.K.; Dalela, D.; Rath, S.; Goel, M.; Bhatt, M. Expression of PDZ-binding kinase/T-LAK cell-originated protein kinase (PBK/TOPK) in human urinary bladder transitional cell carcinoma. Immunobiology 2014, 219, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Zhang, Y.; Yacob, B.W.; Saeed, J.; Tompkins, K.D.; Bagby, S.M.; Pitts, T.M.; Somerset, H.; Leong, S.; Wierman, M.E.; et al. Targeting PDZ-binding kinase is anti-tumorigenic in novel preclinical models of ACC. Endocr. Relat. Cancer 2019, 26, 765–778. [Google Scholar] [CrossRef]
- Rutz, S.; Eidenschenk, C.; Kiefer, J.R.; Ouyang, W. Post-translational regulation of RORγt—A therapeutic target for the modulation of interleukin-17-mediated responses in autoimmune diseases. Cytokine Growth Factor Rev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Kathania, M.; Khare, P.; Zeng, M.; Cantarel, B.; Zhang, H.; Ueno, H.; Venuprasad, K. Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-γt ubiquitination. Nat. Immunol. 2016, 17, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, P.; Han, L.; Guo, Z.; Wang, X.; Chen, Z.; Nie, J.; Yin, S.; Piccioni, M.; Tsun, A.; et al. Cutting Edge: Ubiquitin-Specific Protease 4 Promotes Th17 Cell Function under Inflammation by Deubiquitinating and Stabilizing RORγt. J. Immunol. 2015, 194, 4094–4097. [Google Scholar] [CrossRef]
- He, Z.; Ma, J.; Wang, R.; Zhang, J.; Huang, Z.; Wang, F.; Sen, S.; Rothenberg, E.V.; Sun, Z. A two-amino-acid substitution in the transcription factor RORγt disrupts its function in TH17 differentiation but not in thymocyte development. Nat. Immunol. 2017, 18, 1128–1138. [Google Scholar] [CrossRef]
- Shengwei, L.; Wang, F.; Zhang, J.; Sen, S.; Pang, Q.; Luo, S.; Gwack, Y.; Sun, Z. Regulation of Th17 Differentiation by IKKα-Dependent and -Independent Phosphorylation of RORγt. J. Immunol. 2017, 199, 955–964. [Google Scholar] [CrossRef]
- Rutz, S.; Kayagaki, N.; Phung, Q.T.; Eidenschenk, C.; Noubade, R.; Wang, X.; Lesch, J.; Lu, R.; Newton, K.; Huang, O.W.; et al. Deubiquitinase DUBA is a post-translational brake on interleukin-17 production in T cells. Nat. Cell Biol. 2015, 518, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Guo, L.; Duan, Z.J.; Tepper, C.G.; Xue, L.; Chen, X.; Kung, H.-J.; Gao, A.C.; Zou, J.X.; Chen, H.-W. Histone Methyltransferase NSD2/MMSET Mediates Constitutive NF- B Signaling for Cancer Cell Proliferation, Survival, and Tumor Growth via a Feed-Forward Loop. Mol. Cell. Biol. 2012, 32, 3121–3131. [Google Scholar] [CrossRef] [PubMed]
- Kalashnikova, E.V.; Revenko, A.S.; Gemo, A.T.; Andrews, N.P.; Tepper, C.G.; Zou, J.X.; Cardiff, R.D.; Borowsky, A.D.; Chen, H.-W. ANCCA/ATAD2 Overexpression Identifies Breast Cancer Patients with Poor Prognosis, Acting to Drive Proliferation and Survival of Triple-Negative Cells through Control of B-Myb and EZH2. Cancer Res. 2010, 70, 9402–9412. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; A Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-Mesenchymal Transition in Breast Cancer Relates to the Basal-like Phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Huang, Z.; Wang, J.; Ma, Z.; Yang, J.; Corey, E.; Evans, C.P.; Yu, A.-M.; Chen, H.-W. Targeting Feedforward Loops Formed by Nuclear Receptor RORγ and Kinase PBK in mCRPC with Hyperactive AR Signaling. Cancers 2021, 13, 1672. https://doi.org/10.3390/cancers13071672
Zhang X, Huang Z, Wang J, Ma Z, Yang J, Corey E, Evans CP, Yu A-M, Chen H-W. Targeting Feedforward Loops Formed by Nuclear Receptor RORγ and Kinase PBK in mCRPC with Hyperactive AR Signaling. Cancers. 2021; 13(7):1672. https://doi.org/10.3390/cancers13071672
Chicago/Turabian StyleZhang, Xiong, Zenghong Huang, Junjian Wang, Zhao Ma, Joy Yang, Eva Corey, Christopher P. Evans, Ai-Ming Yu, and Hong-Wu Chen. 2021. "Targeting Feedforward Loops Formed by Nuclear Receptor RORγ and Kinase PBK in mCRPC with Hyperactive AR Signaling" Cancers 13, no. 7: 1672. https://doi.org/10.3390/cancers13071672
APA StyleZhang, X., Huang, Z., Wang, J., Ma, Z., Yang, J., Corey, E., Evans, C. P., Yu, A.-M., & Chen, H.-W. (2021). Targeting Feedforward Loops Formed by Nuclear Receptor RORγ and Kinase PBK in mCRPC with Hyperactive AR Signaling. Cancers, 13(7), 1672. https://doi.org/10.3390/cancers13071672