
Introduction of a Variant Classification System for Analysis of Genotype-Phenotype Relationships in Heritable Retinoblastoma

 and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Data Acquisition
2.3. Data Analysis
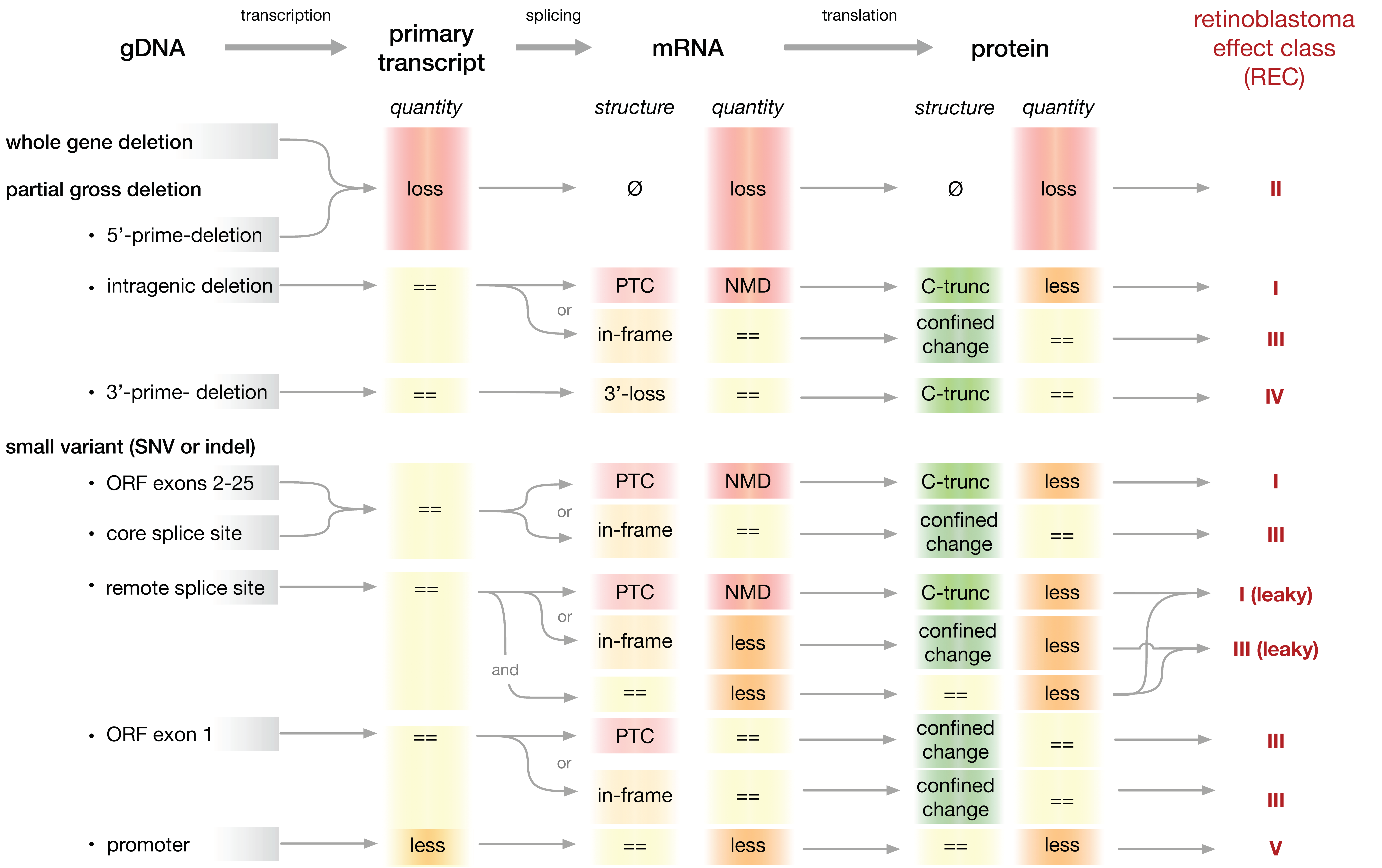
2.4. A System to Classify Pathogenic RB1 Gene Variants for Analysis of Genotype-Phenotype Relationships
3. Results
3.1. Clinical and Genetic Characteristics of the Study Cohort
3.2. Most Patients in the Study Cohort Are Heterozygous for Variants That Result in PTCs Expected to Elicit NMD (REC-I)
3.3. Phenotypic Expression in Patients Heterozygous for Variants Previously Associated with Incomplete Penetrance
3.4. Patients with REC-I Variants Show Earlier Age at Diagnosis and More Tumor Foci Compared to Patients with Variants of Other Effect Classes
3.5. Phenotype of Patients with REC-II Variants Depends on Involvement of MED4
3.6. Patients with REC-III Variants Show a Broad Variety of Phenotypic Expression
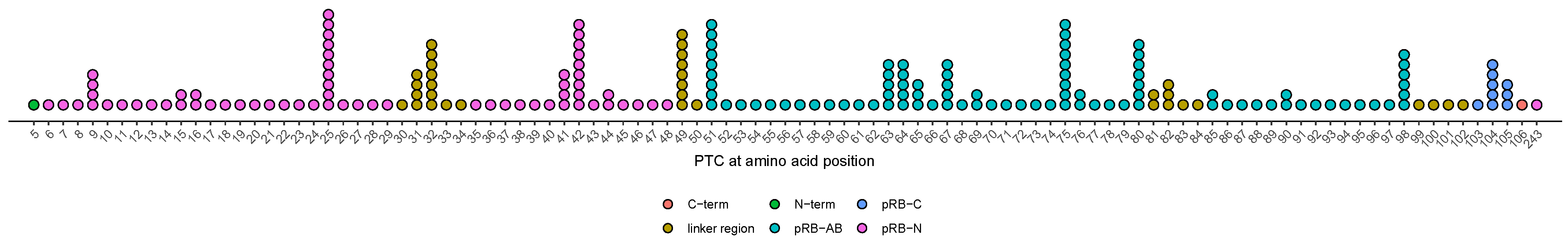
3.7. Age at Diagnosis of Patients with Variants Resulting in PTCs in Unstructured Linker Regions Appears to Be Earlier Compared to Patients with PTC in Other Regions
3.8. Effect of Alterations at Splice-Sites Depending on Their Relative Location
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant Description 1 | Type of RB1 Variant | Retinoblastoma Effect Class | Observations of IP |
|---|---|---|---|
| Contiguous gene deletions including MED4 | Whole RB1 deletion | REC-II | Multiple i.e., Mitter et.al. 2011 [19] (model proposed by Dehainault et al. [20]) |
| c.19dup | Frameshift PTC exon 1 | REC-III | 1 of 3 families with IP (2 unaffected) |
| c.54_79del | Frameshift PTC exon 1 | REC-III | 2 unaffected in one family |
| c.67_104del | Frameshift PTC exon 1 | REC-III | 2 unaffected in one family |
| c.1439_1441del | In-frame | REC-III | 2 families with IP published and unpublished Lohmann et al. 1994 [27] |
| c.1981C > T | Missense | REC-III | 18 families with IP |
| c.2116_2118dup | In-frame | REC-III | 3 relatives with IP in one family, unpublished |
| c.2211 + 1G > T | Splice-site | REC-III | 1 of 4 families with IP (1 unaffected) |
| c.2171T > G | Missense | REC-III | 2 relatives with IP in one family, unpublished |
| c.2717_2718dup | Frameshift PTC Exon 27 | REC-III | Mitter 2009 [59] |
| c.-198G > A | Promoter | REC-V | Taylor et al. 2007 [53] Two families, more than 2 ip |
| c.658C > G | Splice-site | REC I-leaky | Two families with 1 IP, one published in Zhang et al. 2008 [46] |
| c.1695 + 6T > C | Splice-site | REC I-leaky | One family with 6 IP |
| c.939G > A | Splice-site | REC III-leaky | One family with 1 IP |
References
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Otterson, G.A.; Chen, W.; Coxon, A.B.; Khleif, S.N.; Kaye, F.J. Incomplete penetrance of familial retinoblastoma linked to germ-line mutations that result in partial loss of RB function. Proc. Natl. Acad. Sci. USA 1997, 94, 12036–12040. [Google Scholar] [CrossRef]
- Salviat, F.; Gauthier-Villars, M.; Carton, M.; Cassoux, N.; Lumbroso-Le Rouic, L.; Dehainault, C.; Levy, C.; Golmard, L.; Aerts, I.; Doz, F.; et al. Association between Genotype and Phenotype in Consecutive Unrelated Individuals with Retinoblastoma. JAMA Ophthalmol. 2020, 138, 843–850. [Google Scholar] [CrossRef]
- Klutz, M.; Brockmann, D.; Lohmann, D.R. A parent-of-origin effect in two families with retinoblastoma is associated with a distinct splice mutation in the RB1 gene. Am. J. Hum. Genet. 2002, 71, 174–179. [Google Scholar] [CrossRef][Green Version]
- Eloy, P.; Dehainault, C.; Sefta, M.; Aerts, I.; Doz, F.; Cassoux, N.; Lumbroso le Rouic, L.; Stoppa-Lyonnet, D.; Radvanyi, F.; Millot, G.A.; et al. A Parent-of-Origin Effect Impacts the Phenotype in Low Penetrance Retinoblastoma Families Segregating the c.1981C>T/p.Arg661Trp Mutation of RB1. PLoS Genet. 2016, 12, e1005888. [Google Scholar] [CrossRef] [PubMed]
- Castera, L.; Sabbagh, A.; Dehainault, C.; Michaux, D.; Mansuet-Lupo, A.; Patillon, B.; Lamar, E.; Aerts, I.; Lumbroso-Le Rouic, L.; Couturier, J.; et al. MDM2 as a modifier gene in retinoblastoma. J. Natl. Cancer Inst. 2010, 102, 1805–1808. [Google Scholar] [CrossRef]
- Xu, X.L.; Singh, H.P.; Wang, L.; Qi, D.L.; Poulos, B.K.; Abramson, D.H.; Jhanwar, S.C.; Cobrinik, D. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature 2014, 514, 385–388. [Google Scholar] [CrossRef]
- Li, Z.; Qi, D.L.; Singh, H.P.; Zou, Y.; Shen, B.; Cobrinik, D. A novel thyroid hormone receptor isoform, TRbeta2-46, promotes SKP2 expression and retinoblastoma cell proliferation. J. Biol. Chem. 2019, 294, 2961–2969. [Google Scholar] [CrossRef]
- Qi, D.L.; Cobrinik, D. MDM2 but not MDM4 promotes retinoblastoma cell proliferation through p53-independent regulation of MYCN translation. Oncogene 2017, 36, 1760–1769. [Google Scholar] [CrossRef]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef]
- Lee, C.; Chang, J.H.; Lee, H.S.; Cho, Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002, 16, 3199–3212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Schweers, B.; Dyer, M.A. The first knockout mouse model of retinoblastoma. Cell Cycle 2004, 3, 952–959. [Google Scholar] [CrossRef]
- Rubin, S.M.; Gall, A.L.; Zheng, N.; Pavletich, N.P. Structure of the Rb C-terminal domain bound to E2F1-DP1: A mechanism for phosphorylation-induced E2F release. Cell 2005, 123, 1093–1106. [Google Scholar] [CrossRef]
- Burke, J.R.; Deshong, A.J.; Pelton, J.G.; Rubin, S.M. Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J. Biol. Chem. 2010, 285, 16286–16293. [Google Scholar] [CrossRef]
- Burke, J.R.; Hura, G.L.; Rubin, S.M. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 2012, 26, 1156–1166. [Google Scholar] [CrossRef]
- Rubin, S.M. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 2013, 38, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Lindeboom, R.G.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Mitter, D.; Ullmann, R.; Muradyan, A.; Klein-Hitpass, L.; Kanber, D.; Ounap, K.; Kaulisch, M.; Lohmann, D. Genotype-phenotype correlations in patients with retinoblastoma and interstitial 13q deletions. Eur. J. Hum. Genet. 2011, 19, 947–958. [Google Scholar] [CrossRef]
- Dehainault, C.; Garancher, A.; Castera, L.; Cassoux, N.; Aerts, I.; Doz, F.; Desjardins, L.; Lumbroso, L.; Montes de Oca, R.; Almouzni, G.; et al. The survival gene MED4 explains low penetrance retinoblastoma in patients with large RB1 deletion. Hum. Mol. Genet. 2014, 23, 5243–5250. [Google Scholar] [CrossRef][Green Version]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C., Jr.; Squire, J.A.; et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef]
- de Jong, M.C.; Kors, W.A.; Moll, A.C.; de Graaf, P.; Castelijns, J.A.; Jansen, R.W.; Gallie, B.; Soliman, S.E.; Shaikh, F.; Dimaras, H.; et al. Screening for Pineal Trilateral Retinoblastoma Revisited: A Meta-analysis. Ophthalmology 2020, 127, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Onadim, Z.; Woolford, A.J.; Kingston, J.E.; Hungerford, J.L. The RB1 gene mutation in a child with ectopic intracranial retinoblastoma. Br. J. Cancer 1997, 76, 1405–1409. [Google Scholar] [CrossRef][Green Version]
- Kleinerman, R.A.; Yu, C.L.; Little, M.P.; Li, Y.; Abramson, D.; Seddon, J.; Tucker, M.A. Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J. Clin. Oncol. 2012, 30, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Dommering, C.J.; Marees, T.; van der Hout, A.H.; Imhof, S.M.; Meijers-Heijboer, H.; Ringens, P.J.; van Leeuwen, F.E.; Moll, A.C. RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam. Cancer 2011. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, P.; Hulsenbeck, I.; Frank, M.; Schmidt, B.; Jockel, K.H.; Lohmann, D.R. The impact of RB1 genotype on incidence of second tumours in heritable retinoblastoma. Eur. J. Cancer 2020, 133, 47–55. [Google Scholar] [CrossRef]
- Lohmann, D.R.; Brandt, B.; Hopping, W.; Passarge, E.; Horsthemke, B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum. Genet. 1994, 94, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Onadim, Z.; Hogg, A.; Baird, P.N.; Cowell, J.K. Oncogenic point mutations in exon 20 of the RB1 gene in families showing incomplete penetrance and mild expression of the retinoblastoma phenotype. Proc. Natl. Acad. Sci. USA 1992, 89, 6177–6181. [Google Scholar] [CrossRef]
- Harbour, J.W. Molecular basis of low-penetrance retinoblastoma. Arch. Ophthalmol. 2001, 119, 1699–1704. [Google Scholar] [CrossRef]
- Imperatore, V.; Pinto, A.M.; Gelli, E.; Trevisson, E.; Morbidoni, V.; Frullanti, E.; Hadjistilianou, T.; De Francesco, S.; Toti, P.; Gusson, E.; et al. Parent-of-origin effect of hypomorphic pathogenic variants and somatic mosaicism impact on phenotypic expression of retinoblastoma. Eur. J. Hum. Genet. 2018, 26, 1026–1037. [Google Scholar] [CrossRef]
- Kanber, D.; Berulava, T.; Ammerpohl, O.; Mitter, D.; Richter, J.; Siebert, R.; Horsthemke, B.; Lohmann, D.; Buiting, K. The human retinoblastoma gene is imprinted. PLoS Genet. 2009, 5, e1000790. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Kanber, D.; Horsthemke, B.; Lohmann, D. Imprinting of RB1 (the new kid on the block). Brief. Funct. Genom. 2010, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Allderdice, P.W.; Davis, J.G.; Miller, O.J.; Klinger, H.P.; Warburton, D.; Miller, D.A.; Allen, F.H., Jr.; Abrams, C.A.; McGilvray, E. The 13q-deletion syndrome. Am. J. Hum. Genet. 1969, 21, 499–512. [Google Scholar] [PubMed]
- Lohmann, D.R.; Gerick, M.; Brandt, B.; Oelschlager, U.; Lorenz, B.; Passarge, E.; Horsthemke, B. Constitutional RB1-gene mutations in patients with isolated unilateral retinoblastoma. Am. J. Hum. Genet. 1997, 61, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Sippel, K.C.; Fraioli, R.E.; Smith, G.D.; Schalkoff, M.E.; Sutherland, J.; Gallie, B.L.; Dryja, T.P. Frequency of somatic and germ-line mosaicism in retinoblastoma: Implications for genetic counseling. Am. J. Hum. Genet. 1998, 62, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martin, C.; Robledo, C.; Gomez-Mariano, G.; Monzon, S.; Sastre, A.; Abelairas, J.; Sabado, C.; Martin-Begue, N.; Ferreres, J.C.; Fernandez-Teijeiro, A.; et al. Frequency of low-level and high-level mosaicism in sporadic retinoblastoma: Genotype-phenotype relationships. J. Hum. Genet. 2020, 65, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.G. Review and hypotheses: Somatic mosaicism: Observations related to clinical genetics. Am. J. Hum. Genet. 1988, 43, 355–363. [Google Scholar] [PubMed]
- Lohmann, D.R.; Brandt, B.; Hopping, W.; Passarge, E.; Horsthemke, B. The spectrum of RB1 germ-line mutations in hereditary retinoblastoma. Am. J. Hum. Genet. 1996, 58, 940–949. [Google Scholar]
- Jonsson, H.; Magnusdottir, E.; Eggertsson, H.P.; Stefansson, O.A.; Arnadottir, G.A.; Eiriksson, O.; Zink, F.; Helgason, E.A.; Jonsdottir, I.; Gylfason, A.; et al. Differences between germline genomes of monozygotic twins. Nat. Genet. 2021, 53, 27–34. [Google Scholar] [CrossRef]
- Lohmann, D.R.; Gallie, B.L. Retinoblastoma: Revisiting the model prototype of inherited cancer. Am. J. Med. Genet. Part C Semin. Med. Genet. 2004, 129C, 23–28. [Google Scholar] [CrossRef]
- Pearl, J. Causal inference in statistics: An overview. Stat. Surv. 2009, 3, 96–146. [Google Scholar] [CrossRef]
- Zielenski, J.; Tsui, L.C. Cystic fibrosis: Genotypic and phenotypic variations. Annu. Rev. Genet. 1995, 29, 777–807. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Cuppens, H.; Macek, M., Jr.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef]
- Albrecht, P.; Ansperger-Rescher, B.; Schuler, A.; Zeschnigk, M.; Gallie, B.; Lohmann, D.R. Spectrum of gross deletions and insertions in the RB1 gene in patients with retinoblastoma and association with phenotypic expression. Hum. Mutat. 2005, 26, 437–445. [Google Scholar] [CrossRef]
- Zeschnigk, M.; Bohringer, S.; Price, E.A.; Onadim, Z.; Masshofer, L.; Lohmann, D.R. A novel real-time PCR assay for quantitative analysis of methylated alleles (QAMA): Analysis of the retinoblastoma locus. Nucleic Acids. Res. 2004, 32, e125. [Google Scholar] [CrossRef]
- Zhang, K.; Nowak, I.; Rushlow, D.; Gallie, B.L.; Lohmann, D.R. Patterns of missplicing caused by RB1 gene mutations in patients with retinoblastoma and association with phenotypic expression. Hum. Mutat. 2008, 29, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sanchez, F.; Ramirez-Castillejo, C.; Weekes, D.B.; Beneyto, M.; Prieto, F.; Najera, C.; Mittnacht, S. Attenuation of disease phenotype through alternative translation initiation in low-penetrance retinoblastoma. Hum. Mutat. 2007, 28, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Dommering, C.J.; Mol, B.M.; Moll, A.C.; Burton, M.; Cloos, J.; Dorsman, J.C.; Meijers-Heijboer, H.; van der Hout, A.H. RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J. Med. Genet. 2014, 51, 366–374. [Google Scholar] [CrossRef]
- Price, E.A.; Price, K.; Kolkiewicz, K.; Hack, S.; Reddy, M.A.; Hungerford, J.L.; Kingston, J.E.; Onadim, Z. Spectrum of RB1 mutations identified in 403 retinoblastoma patients. J. Med. Genet. 2014, 51, 208–214. [Google Scholar] [CrossRef]
- Valverde, J.R.; Alonso, J.; Palacios, I.; Pestana, A. RB1 gene mutation up-date, a meta-analysis based on 932 reported mutations available in a searchable database. BMC Genet. 2005, 6, 53. [Google Scholar] [CrossRef]
- Bremner, R.; Du, D.C.; Connolly-Wilson, M.J.; Bridge, P.; Ahmad, K.F.; Mostachfi, H.; Rushlow, D.; Dunn, J.M.; Gallie, B.L. Deletion of RB exons 24 and 25 causes low-penetrance retinoblastoma. Am. J. Hum. Genet. 1997, 61, 556–570. [Google Scholar] [CrossRef][Green Version]
- Taylor, M.; Dehainault, C.; Desjardins, L.; Doz, F.; Levy, C.; Sastre, X.; Couturier, J.; Stoppa-Lyonnet, D.; Houdayer, C.; Gauthier-Villars, M. Genotype-phenotype correlations in hereditary familial retinoblastoma. Hum. Mutat. 2007, 28, 284–293. [Google Scholar] [CrossRef]
- Lohmann, D.R.; Gallie, B.L. Retinoblastoma. 18 July 2000 [Updated 21 November 2018]. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Eds.; University of Washington: Seattle, WA, USA, 1993–2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1452/ (accessed on 1 September 2020).
- Zeggini, E.; Ioannidis, J.P. Meta-analysis in genome-wide association studies. Pharmacogenomics 2009, 10, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Abramson, D.H.; Frank, C.M. Second nonocular tumors in survivors of bilateral retinoblastoma: A possible age effect on radiation-related risk. Ophthalmology 1998, 105, 573–579. [Google Scholar] [CrossRef]
- Kanber, D.; Buiting, K.; Roos, C.; Gromoll, J.; Kaya, S.; Horsthemke, B.; Lohmann, D. The origin of the RB1 imprint. PLoS ONE 2013, 8, e81502. [Google Scholar] [CrossRef]
- Tomar, A.S.; Finger, P.T.; Gallie, B.; Kivela, T.T.; Mallipatna, A.; Zhang, C.; Zhao, J.; Wilson, M.W.; Brenna, R.C.; Burges, M.; et al. Global Retinoblastoma Treatment Outcomes: Association with National Income Level. Ophthalmology 2020. [Google Scholar] [CrossRef] [PubMed]
- Mitter, D.; Rushlow, D.; Nowak, I.; Ansperger-Rescher, B.; Gallie, B.L.; Lohmann, D.R. Identification of a mutation in exon 27 of the RB1 gene associated with incomplete penetrance retinoblastoma. Fam. Cancer 2009, 8, 55–58. [Google Scholar] [CrossRef] [PubMed]






| Effect on pRB | Type of gDNA Alteration | Examples | ||
|---|---|---|---|---|
| Structure | Quantity | |||
| REC-I | C-truncated | Less | Intragenic deletions Small variants (SNV or indels) ORF exons 2–25 Splice-site | Nonsense or Frameshift variant |
| REC-I (leaky) | C-truncated and no change | Less | Small variants (SNV or indels) Remote splice-site | Splice-site variant |
| REC-II | NA | Complete loss | Whole gene deletions Partial gross deletions including 5′ end | Whole RB1 gene deletions |
| REC-III | Confined change | No change | Intragenic deletions Small variants (SNV or indels) ORF exons 1–25 Splice-site | Missense or in-frame SNV |
| REC-III (leaky) | Confined change and no change | No change | Small variants (SNV or indels) Remote splice-site | Splice-site variant |
| REC-IV | C-truncated | No change | Partial gross deletions including 3′-end Small variants ORF exons 25–27 (no NMD) | SNV or indels 3′ end |
| REC-V | No change | Less | Small variants regulatory promoter motifs | SNV in promotor region |
| Clinical Characteristics | Study Cohort |
|---|---|
| All | 287 |
| Sex | |
| Female | 137 |
| Male | 149 |
| Unknown | 1 |
| Laterality | |
| Unilateral | 32 |
| Bilateral | 255 |
| ICRB Group of 415 of 542 affected eyes | |
| ICRB A | 48 |
| ICRB B | 147 |
| ICRB C | 27 |
| ICRB D | 46 |
| ICRB E | 147 |
| Missing information | 127 |
| Family history | |
| Isolated | 245 |
| Familial | 39 |
| Unknown | 3 |
| REC Class | n (%) | Laterality n (% Within Laterality) | Age at Diagnosis (Months) | Number of Tumors in the Less-Affected Eye at Diagnosis of Each Patient | |||
|---|---|---|---|---|---|---|---|
| Unilateral | Bilateral | Median | Range | Median | Range | ||
| REC-I | 199 (69.3) | 13 (6.5) | 186 (93.5) | 7.3 | 0.2–48.0 | 2 | 0–8 |
| REC-I-non-leaky | 190 (66.2) | 11 (5.8) | 179 (94.2) | 7.2 | 0.2–48.0 | 2 | 0–8 |
| REC-I-leaky | 9 (2.8) | 2 (22.2) | 7 (77.8) | 12.0 | 2.5–19.3 | 1 | 0–2 |
| REC-II | 39 (13.6) | 9 (23.1) | 30 (76.9) | 10.3 | 0.4–40.9 | 1 | 0–3 |
| REC-II− | 6 (2.1) | / | 6 (100) | 11.0 | 2.6–27.8 | 1 | 1–2 |
| REC-II+ | 27 (9.4) | 8 (29.6) | 19 (70.4) | 8.4 | 0.4–40.9 | 1 | 0–2 |
| REC-III | 45 (15.7) | 9 (20.0) | 36 (80.0) | 11.6 | 0.9–45.1 | 1 | 0–4 |
| REC-III-non-leaky | 39 (13.6) | 9 (23.1) | 30 (76.9) | 12.3 | 0.9–45.1 | 1 | 0–4 |
| REC-III-leaky | 6 (2.1) | / | 6 (100) | 8.2 | 4.1–14.6 | 1 | 1–4 |
| REC-IV | 1 (0.3) | 0 | 1 (100) | 13.1 | / | 2 | / |
| REC-V | 3 (1.0) | 1 (33.3) | 2 (66.7) | 18.4 | 18.0–34.8 | 1 | 0–2 |
| Total | 287 (100.0) | 255 (88.9) | 32 (11.1) | 8.3 | 0.2–48.0 | 1 | 0–8 |
| Feature of Phenotypic Expression | Regular Penetrance (n = 240) | Incomplete Penetrance (n = 47) |
|---|---|---|
| Age of diagnosis | ||
| Median | 8.0 months | 12.4 months |
| Range | 0.2–48.0 months | 0.4–40.9 months |
| Number of tumors in the less-affected eye at diagnosis per patient | ||
| Mean | 2 | 1 |
| Range | 0–8 | 0–4 |
| Laterality | ||
| Unilateral (in % per column) | 15 (6.3%) | 17 (36.2%) |
| Bilateral (in % per column) | 225 (93.8%) | 30 (63.8%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hülsenbeck, I.; Frank, M.; Biewald, E.; Kanber, D.; Lohmann, D.R.; Ketteler, P. Introduction of a Variant Classification System for Analysis of Genotype-Phenotype Relationships in Heritable Retinoblastoma. Cancers 2021, 13, 1605. https://doi.org/10.3390/cancers13071605
Hülsenbeck I, Frank M, Biewald E, Kanber D, Lohmann DR, Ketteler P. Introduction of a Variant Classification System for Analysis of Genotype-Phenotype Relationships in Heritable Retinoblastoma. Cancers. 2021; 13(7):1605. https://doi.org/10.3390/cancers13071605
Chicago/Turabian StyleHülsenbeck, Isabel, Mirjam Frank, Eva Biewald, Deniz Kanber, Dietmar R. Lohmann, and Petra Ketteler. 2021. "Introduction of a Variant Classification System for Analysis of Genotype-Phenotype Relationships in Heritable Retinoblastoma" Cancers 13, no. 7: 1605. https://doi.org/10.3390/cancers13071605
APA StyleHülsenbeck, I., Frank, M., Biewald, E., Kanber, D., Lohmann, D. R., & Ketteler, P. (2021). Introduction of a Variant Classification System for Analysis of Genotype-Phenotype Relationships in Heritable Retinoblastoma. Cancers, 13(7), 1605. https://doi.org/10.3390/cancers13071605