
Musashi1 Contribution to Glioblastoma Development via Regulation of a Network of DNA Replication, Cell Cycle and Division Genes

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
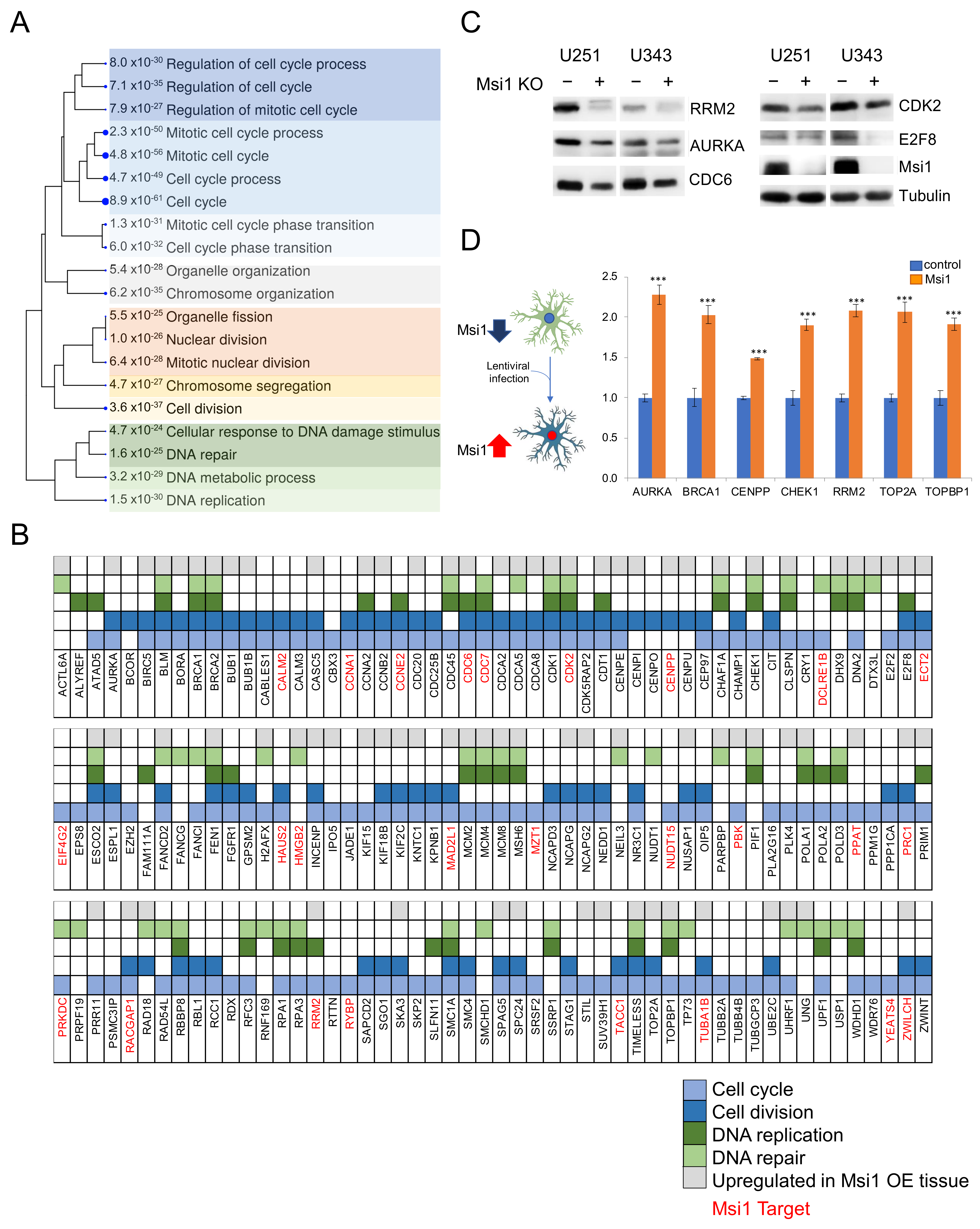
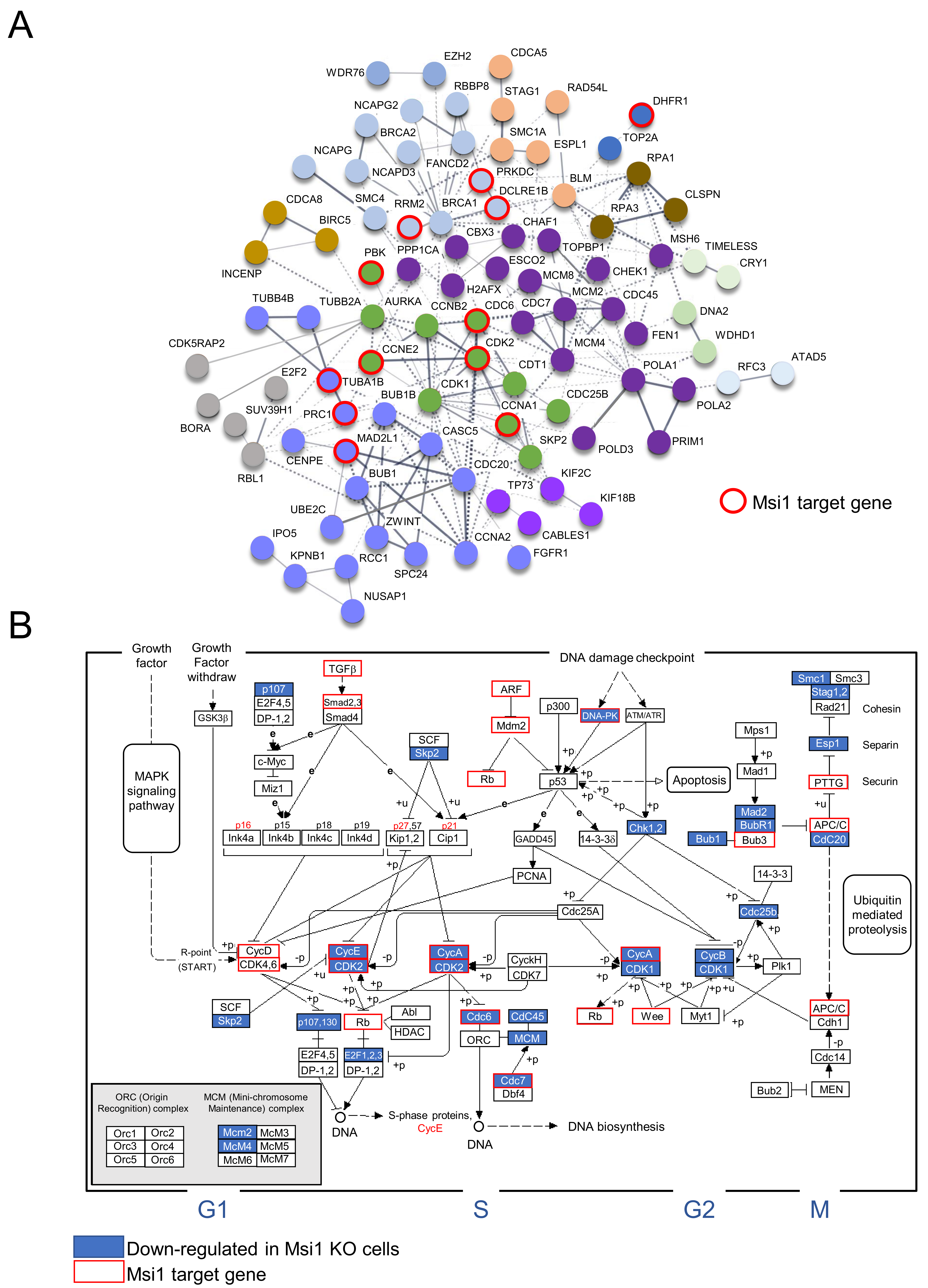
2.1. Genomic Analyses Define Regulation of DNA Replication and Cell Cycle/Division as Musashi1 Core Functions
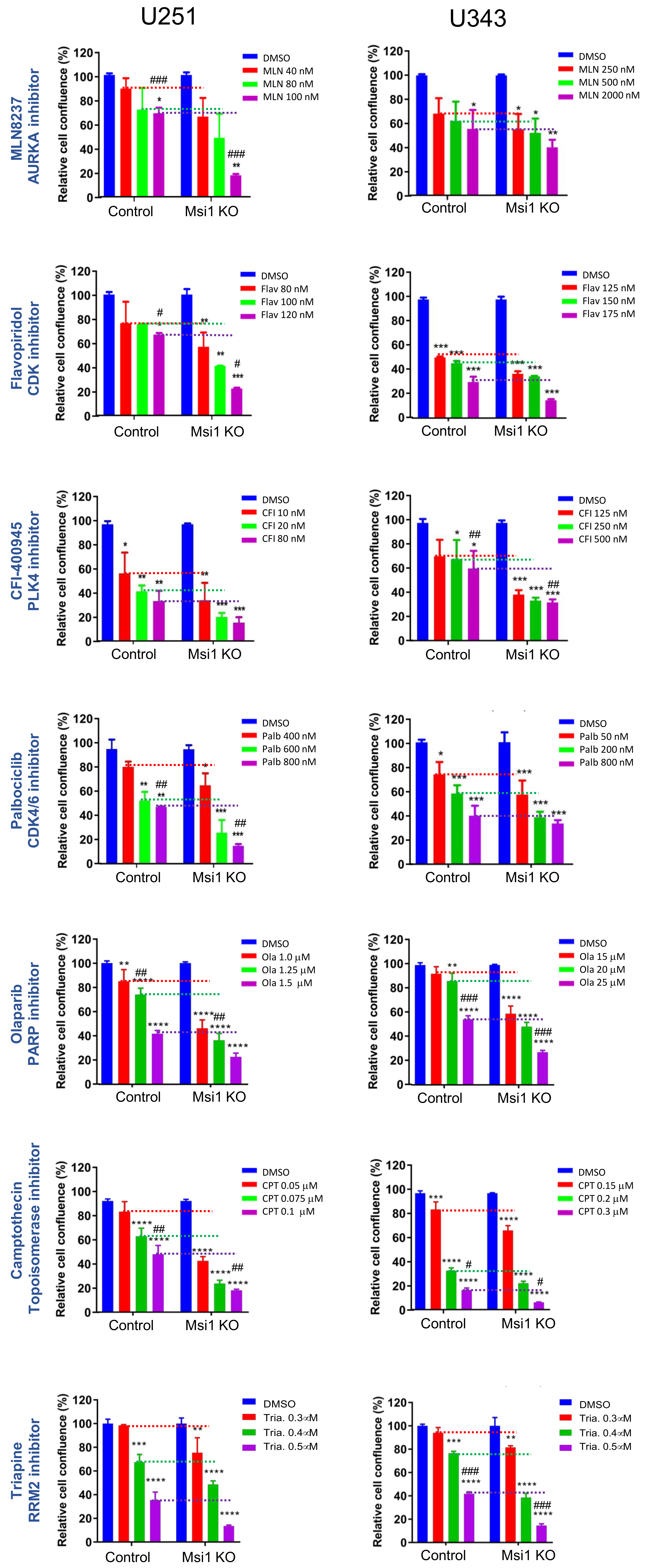
2.2. Glioblastoma Msi1 KO Cells Are More Sensitive to DNA Replication and Cell Cycle Inhibitors
2.3. E2F2 and E2F8 are Mediators of Msi1 Impact on DNA Replication and Cell Cycle/Division
3. Discussion
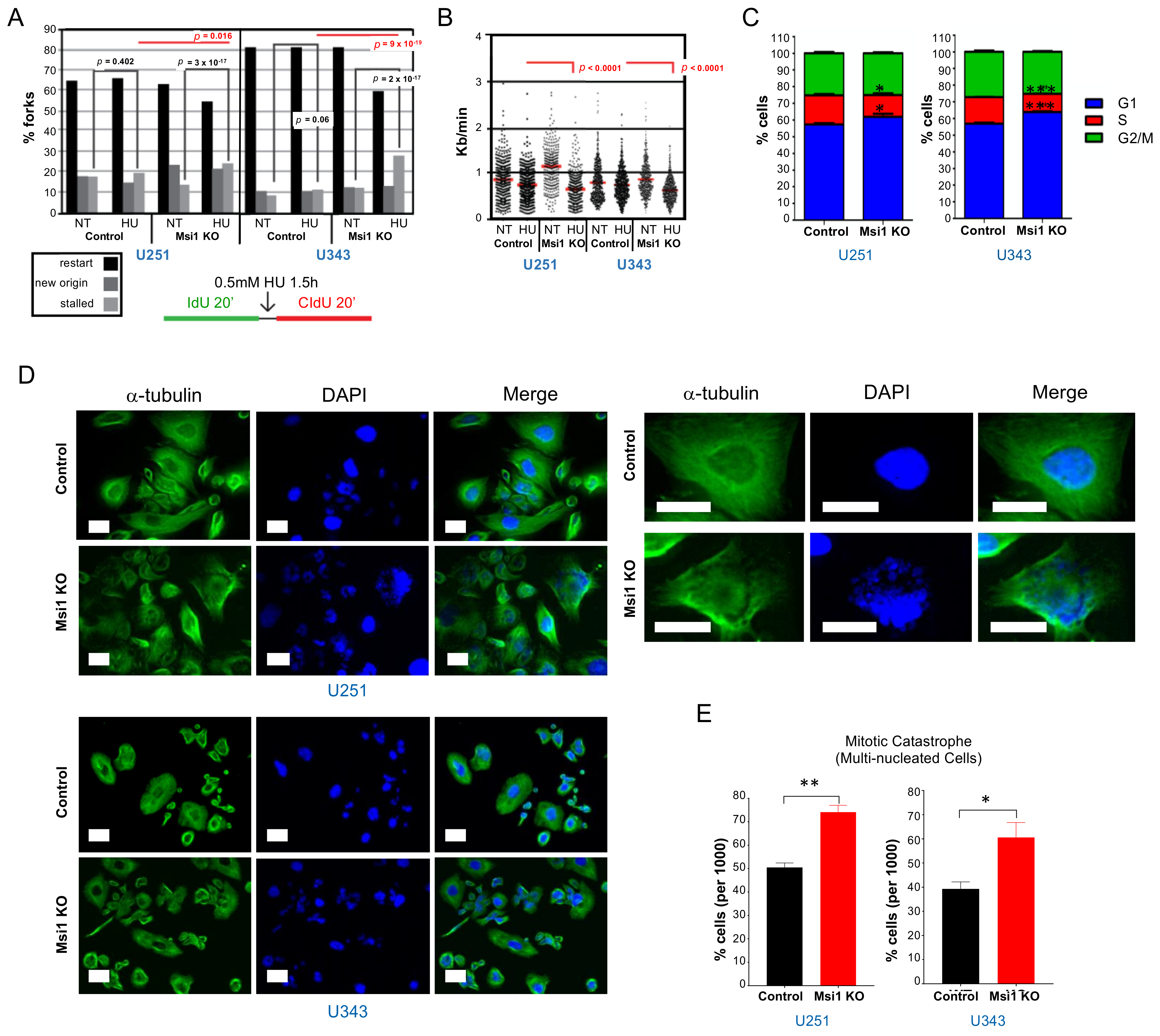
3.1. Musashi1 Modulates Cell Cycle Progression and DNA Replication in GBM Cells
3.2. Musashi1 Regulates the Expression of Members of the Centromeric Complex
3.3. E2F2 and E2F8 Are Critical Mediators of Msi1 Impact on Cell Cycle and DNA Replication
3.4. Targeting RNA-Binding Proteins in Cancer Therapy
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Transfection and siRNA Knockdown
4.3. Astrocyte Cell Culture and Lentivirus Infection
4.4. RNA Extraction and qRT-PCR Analysis
4.5. RNA-Seq Analysis
4.6. Gene Ontology and Network Analyses
4.7. Expression Correlation and Expression Analyses
4.8. Western Blot
4.9. Cell Cycle Analysis
4.10. Cell Division Analysis
4.11. Cell Proliferation Assay
4.12. DNA Replication
4.13. Impact of Cell Cycle and DNA Replication Inhibitors on Msi1 KO Cells
4.14. Analysis of Drug Synergism
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Vo, D.T.; Abdelmohsen, K.; Martindale, J.L.; Qiao, M.; Tominaga, K.; Burton, T.L.; Gelfond, J.A.; Brenner, A.J.; Patel, V.; Trageser, D.; et al. The oncogenic RNA-binding protein Musashi1 is regulated by HuR via mRNA translation and stability in glioblastoma cells. Mol. Cancer Res. 2012, 10, 143–155. [Google Scholar] [CrossRef]
- Uren, P.J.; Vo, D.T.; de Araujo, P.R.; Pötschke, R.; Burns, S.C.; Bahrami-Samani, E.; Qiao, M.; de Sousa Abreu, R.; Nakaya, H.I.; Correa, B.R.; et al. RNA-Binding Protein Musashi1 Is a Central Regulator of Adhesion Pathways in Glioblastoma. Mol. Cell. Biol. 2015, 35, 2965–2978. [Google Scholar] [CrossRef] [PubMed]
- Velasco, M.X.; Kosti, A.; Guardia, G.D.A.; Santos, M.C.; Tegge, A.; Qiao, M.; Correa, B.R.S.; Hernández, G.; Kokovay, E.; Galante, P.A.F.; et al. Antagonism between the RNA-binding protein Musashi1 and miR-137 and its potential impact on neurogenesis and glioblastoma development. RNA 2019, 25, 768–782. [Google Scholar] [CrossRef]
- Kudinov, A.E.; Karanicolas, J.; Golemis, E.A.; Boumber, Y. Musashi RNA-Binding Proteins as Cancer Drivers and Novel Therapeutic Targets. Clin. Cancer Res. 2017, 23, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Tokunaga, A.; Yoshida, T.; Hashimoto, M.; Mikoshiba, K.; Weinmaster, G.; Nakafuku, M.; Okano, H. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol. Cell. Biol. 2001, 21, 3888–3900. [Google Scholar] [CrossRef]
- Yi, C.; Li, G.; Ivanov, D.N.; Wang, Z.; Velasco, M.X.; Hernández, G.; Kaundal, S.; Villarreal, J.; Gupta, Y.K.; Qiao, M.; et al. Luteolin inhibits Musashi1 binding to RNA and disrupts cancer phenotypes in glioblastoma cells. RNA Biol. 2018, 15, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, P.R.; Gorthi, A.; da Silva, A.E.; Tonapi, S.S.; Vo, D.T.; Burns, S.C.; Qiao, M.; Uren, P.J.; Yuan, Z.M.; Bishop, A.J.; et al. Musashi1 Impacts Radio-Resistance in Glioblastoma by Controlling DNA-Protein Kinase Catalytic Subunit. Am. J. Pathol. 2016, 186, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Vo, D.T.; Subramaniam, D.; Remke, M.; Burton, T.L.; Uren, P.J.; Gelfond, J.A.; de Sousa Abreu, R.; Burns, S.C.; Qiao, M.; Suresh, U.; et al. The RNA-binding protein Musashi1 affects medulloblastoma growth via a network of cancer-related genes and is an indicator of poor prognosis. Am. J. Pathol. 2012, 181, 1762–1772. [Google Scholar] [CrossRef]
- Vo, D.T.; Qiao, M.; Smith, A.D.; Burns, S.C.; Brenner, A.J.; Penalva, L.O. The oncogenic RNA-binding protein Musashi1 is regulated by tumor suppressor miRNAs. RNA Biol. 2011, 8, 817–828. [Google Scholar] [CrossRef]
- Glazer, R.I.; Vo, D.T.; Penalva, L.O. Musashi1: An RBP with versatile functions in normal and cancer stem cells. Front. Biosci. 2012, 17, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Lang, Y.; Kong, X.; He, C.; Wang, F.; Liu, B.; Zhang, S.; Ning, J.; Zhu, K.; Xu, S. Musashi1 Promotes Non-Small Cell Lung Carcinoma Malignancy and Chemoresistance via Activating the Akt Signaling Pathway. Cell. Physiol. Biochem. 2017, 44, 455–466. [Google Scholar] [CrossRef]
- Chiou, G.Y.; Yang, T.W.; Huang, C.C.; Tang, C.Y.; Yen, J.Y.; Tsai, M.C.; Chen, H.Y.; Fadhilah, N.; Lin, C.C.; Jong, Y.J. Musashi-1 promotes a cancer stem cell lineage and chemoresistance in colorectal cancer cells. Sci. Rep. 2017, 7, 2172. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, J.; Wang, H.; Cheng, Q.; Zhou, C.; Chen, X.; Ye, F. Inhibition of RNA-Binding Protein Musashi-1 Suppresses Malignant Properties and Reverses Paclitaxel Resistance in Ovarian Carcinoma. J. Cancer 2019, 10, 1580–1592. [Google Scholar] [CrossRef] [PubMed]
- Pötschke, R.; Gielen, G.; Pietsch, T.; Kramm, C.; Klusmann, J.H.; Hüttelmaier, S.; Kühnöl, C.D. Musashi1 enhances chemotherapy resistance of pediatric glioblastoma cells in vitro. Pediatr. Res. 2020, 87, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Lin, L.T.; Wang, M.L.; Tsai, K.L.; Huang, P.I.; Yang, Y.P.; Lee, Y.Y.; Chen, Y.W.; Lo, W.L.; Lan, Y.T.; et al. Musashi-1 promotes chemoresistant granule formation by PKR/eIF2α signalling cascade in refractory glioblastoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Diaz, P.C.; Burton, T.L.; Burns, S.C.; Hung, J.Y.; Penalva, L.O. Musashi1 modulates cell proliferation genes in the medulloblastoma cell line Daoy. BMC Cancer 2008, 8, 280. [Google Scholar] [CrossRef]
- Chen, H.Y.; Lin, L.T.; Wang, M.L.; Lee, S.H.; Tsai, M.L.; Tsai, C.C.; Liu, W.H.; Chen, T.C.; Yang, Y.P.; Lee, Y.Y.; et al. Musashi-1 regulates AKT-derived IL-6 autocrinal/paracrinal malignancy and chemoresistance in glioblastoma. Oncotarget 2016, 7, 42485–42501. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef]
- Wang, X.Y.; Penalva, L.O.; Yuan, H.; Linnoila, R.I.; Lu, J.; Okano, H.; Glazer, R.I. Musashi1 regulates breast tumor cell proliferation and is a prognostic indicator of poor survival. Mol. Cancer 2010, 9, 221. [Google Scholar] [CrossRef]
- Cambuli, F.M.; Correa, B.R.; Rezza, A.; Burns, S.C.; Qiao, M.; Uren, P.J.; Kress, E.; Boussouar, A.; Galante, P.A.; Penalva, L.O.; et al. A Mouse Model of Targeted Musashi1 Expression in Whole Intestinal Epithelium Suggests Regulatory Roles in Cell Cycle and Stemness. Stem Cells 2015, 33, 3621–3634. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yu, H.; Linnoila, R.I.; Li, L.; Li, D.; Mo, B.; Okano, H.; Penalva, L.O.; Glazer, R.I. Musashi1 as a potential therapeutic target and diagnostic marker for lung cancer. Oncotarget 2013, 4, 739–750. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Abreu, R.; Sanchez-Diaz, P.C.; Vogel, C.; Burns, S.C.; Ko, D.; Burton, T.L.; Vo, D.T.; Chennasamudaram, S.; Le, S.Y.; Shapiro, B.A.; et al. Genomic analyses of musashi1 downstream targets show a strong association with cancer-related processes. J. Biol. Chem. 2009, 284, 12125–12135. [Google Scholar] [CrossRef]
- Battelli, C.; Nikopoulos, G.N.; Mitchell, J.G.; Verdi, J.M. The RNA-binding protein Musashi-1 regulates neural development through the translational repression of p21WAF-1. Mol. Cell. Neurosci. 2006, 31, 85–96. [Google Scholar] [CrossRef]
- Niu, J.; Zhao, X.; Liu, Q.; Yang, J. Knockdown of MSI1 inhibited the cell proliferation of human osteosarcoma cells by targeting p21 and p27. Oncol. Lett. 2017, 14, 5271–5278. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; George, R.J.; Dieckgraefe, B.K.; McLeod, H.L.; Ramalingam, S.; Bishnupuri, K.S.; Natarajan, G.; Anant, S.; Houchen, C.W. Knockdown of RNA binding protein musashi-1 leads to tumor regression in vivo. Gastroenterology 2008, 134, 1448–1458. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Niedzwiedz, W. Visualization of DNA replication in the vertebrate model system DT40 using the DNA fiber technique. J. Vis. Exp. 2011, e3255. [Google Scholar] [CrossRef]
- Szekeres, T.; Fritzer-Szekeres, M.; Elford, H.L. The enzyme ribonucleotide reductase: Target for antitumor and anti-HIV therapy. Crit. Rev. Clin. Lab. Sci. 1997, 34, 503–528. [Google Scholar] [CrossRef]
- Kim, T.M.; Son, M.Y.; Dodds, S.; Hu, L.; Hasty, P. Deletion of BRCA2 exon 27 causes defects in response to both stalled and collapsed replication forks. Mutat. Res. 2014, 766–767, 66–72. [Google Scholar] [CrossRef]
- Hu, L.; Kim, T.M.; Son, M.Y.; Kim, S.A.; Holland, C.L.; Tateishi, S.; Kim, D.H.; Yew, P.R.; Montagna, C.; Dumitrache, L.C.; et al. Two replication fork maintenance pathways fuse inverted repeats to rearrange chromosomes. Nature 2013, 501, 569–572. [Google Scholar] [CrossRef]
- Forouzanfar, M.; Lachinani, L.; Dormiani, K.; Nasr-Esfahani, M.H.; Gure, A.O.; Ghaedi, K. Intracellular functions of RNA-binding protein, Musashi1, in stem and cancer cells. Stem Cell Res. Ther. 2020, 11, 193. [Google Scholar] [CrossRef]
- MacNicol, A.M.; Hardy, L.L.; Spencer, H.J.; MacNicol, M.C. Neural stem and progenitor cell fate transition requires regulation of Musashi1 function. BMC Dev. Biol. 2015, 15, 15. [Google Scholar] [CrossRef]
- Okano, H.; Kawahara, H.; Toriya, M.; Nakao, K.; Shibata, S.; Imai, T. Function of RNA-binding protein Musashi-1 in stem cells. Exp. Cell Res. 2005, 306, 349–356. [Google Scholar] [CrossRef]
- Lan, L.; Appelman, C.; Smith, A.R.; Yu, J.; Larsen, S.; Marquez, R.T.; Liu, H.; Wu, X.; Gao, P.; Roy, A.; et al. Natural product (-)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol. Oncol. 2015, 9, 1406–1420. [Google Scholar] [CrossRef]
- Minuesa, G.; Albanese, S.K.; Xie, W.; Kazansky, Y.; Worroll, D.; Chow, A.; Schurer, A.; Park, S.M.; Rotsides, C.Z.; Taggart, J.; et al. Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia. Nat. Commun. 2019, 10, 2691. [Google Scholar] [CrossRef] [PubMed]
- Minuesa, G.; Antczak, C.; Shum, D.; Radu, C.; Bhinder, B.; Li, Y.; Djaballah, H.; Kharas, M.G. A 1536-well fluorescence polarization assay to screen for modulators of the MUSASHI family of RNA-binding proteins. Comb. Chem. High Throughput Screen. 2014, 17, 596–609. [Google Scholar] [CrossRef]
- Clingman, C.C.; Deveau, L.M.; Hay, S.A.; Genga, R.M.; Shandilya, S.M.; Massi, F.; Ryder, S.P. Allosteric inhibition of a stem cell RNA-binding protein by an intermediary metabolite. Elife 2014, 3. [Google Scholar] [CrossRef]
- Slinker, B.K. The statistics of synergism. J. Mol. Cell. Cardiol. 1998, 30, 723–731. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- McGrath, E.L.; Rossi, S.L.; Gao, J.; Widen, S.G.; Grant, A.C.; Dunn, T.J.; Azar, S.R.; Roundy, C.M.; Xiong, Y.; Prusak, D.J.; et al. Differential Responses of Human Fetal Brain Neural Stem Cells to Zika Virus Infection. Stem Cell Rep. 2017, 8, 715–727. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, D.; Wei, W.; Cao, W.; Zhang, R.; Dong, Q.; Zhang, J.; Wang, Y.; Liu, N. MiR-218 Inhibited Growth and Metabolism of Human Glioblastoma Cells by Directly Targeting E2F2. Cell. Mol. Neurobiol. 2015, 35, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Xiao, L.; Zhao, X.; Zhao, J.; Wang, J.; Wang, F.; Cao, S.; Lin, X. miR-125b regulates the proliferation of glioblastoma stem cells by targeting E2F2. FEBS Lett. 2012, 586, 3831–3839. [Google Scholar] [CrossRef]
- Okamoto, O.K.; Oba-Shinjo, S.M.; Lopes, L.; Nagahashi Marie, S.K. Expression of HOXC9 and E2F2 are up-regulated in CD133(+) cells isolated from human astrocytomas and associate with transformation of human astrocytes. Biochim. Biophys. Acta 2007, 1769, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, F.; Cui, D.; Jiang, R.; Chen, J.; Huang, Q.; Shi, J. HOXD-AS1/miR-130a sponge regulates glioma development by targeting E2F8. Int. J. Cancer 2018, 142, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986. [Google Scholar] [CrossRef]
- Pang, B.; Xu, J.; Hu, J.; Guo, F.; Wan, L.; Cheng, M.; Pang, L. Single-cell RNA-seq reveals the invasive trajectory and molecular cascades underlying glioblastoma progression. Mol. Oncol. 2019, 13, 2588–2603. [Google Scholar] [CrossRef]
- Nikpour, P.; Baygi, M.E.; Steinhoff, C.; Hader, C.; Luca, A.C.; Mowla, S.J.; Schulz, W.A. The RNA binding protein Musashi1 regulates apoptosis, gene expression and stress granule formation in urothelial carcinoma cells. J. Cell. Mol. Med. 2011, 15, 1210–1224. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Møllgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398. [Google Scholar] [CrossRef] [PubMed]
- Troschel, F.M.; Minte, A.; Ismail, Y.M.; Kamal, A.; Abdullah, M.S.; Ahmed, S.H.; Deffner, M.; Kemper, B.; Kiesel, L.; Eich, H.T.; et al. Knockdown of Musashi RNA Binding Proteins Decreases Radioresistance but Enhances Cell Motility and Invasion in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 2169. [Google Scholar] [CrossRef]
- Li, J.; Yan, K.; Yang, Y.; Li, H.; Wang, Z.; Xu, X. Musashi-1 positively regulates growth and proliferation of hepatoma cells. Nan Fang Yi Ke Da Xue Xue Bao 2019, 39, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Greve, B.; Kelsch, R.; Müller-Uthoff, H.; Weiss, K.; Kharabi Masouleh, B.; Sibrowski, W.; Kiesel, L.; Buchweitz, O. The adult stem cell marker Musashi-1 modulates endometrial carcinoma cell cycle progression and apoptosis via Notch-1 and p21WAF1/CIP1. Int. J. Cancer 2011, 129, 2042–2049. [Google Scholar] [CrossRef]
- Wang, C.F.; Zhang, H.C.; Feng, X.M.; Song, X.M.; Wu, Y.N. Knockdown of MSI1 inhibits the proliferation of human oral squamous cell carcinoma by inactivating STAT3 signaling. Int. J. Mol. Med. 2019, 44, 115–124. [Google Scholar] [CrossRef]
- Gao, C.; Han, C.; Yu, Q.; Guan, Y.; Li, N.; Zhou, J.; Tian, Y.; Zhang, Y. Downregulation of Msi1 suppresses the growth of human colon cancer by targeting p21cip1. Int. J. Oncol. 2015, 46, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, W.T.; Zheng, P.S. Msi1 promotes tumor growth and cell proliferation by targeting cell cycle checkpoint proteins p21, p27 and p53 in cervical carcinomas. Oncotarget 2014, 5, 10870–10885. [Google Scholar] [CrossRef]
- Kaldis, P.; Aleem, E. Cell cycle sibling rivalry: Cdc2 vs. Cdk2. Cell Cycle 2005, 4, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, T.; Xu, G.; Liu, H.; Ren, C.; Xie, W.; Wang, M. Cyclin-Dependent Kinase 2 Promotes Tumor Proliferation and Induces Radio Resistance in Glioblastoma. Transl. Oncol. 2016, 9, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Gudas, J.M.; Payton, M.; Thukral, S.; Chen, E.; Bass, M.; Robinson, M.O.; Coats, S. Cyclin E2, a novel G1 cyclin that binds Cdk2 and is aberrantly expressed in human cancers. Mol. Cell. Biol. 1999, 19, 612–622. [Google Scholar] [CrossRef]
- Ferreira, W.A.; Araújo, M.D.; Anselmo, N.P.; de Oliveira, E.H.; Brito, J.R.; Burbano, R.R.; Harada, M.L.; Borges, B.O.N. Expression Analysis of Genes Involved in the RB/E2F Pathway in Astrocytic Tumors. PLoS ONE 2015, 10, e0137259. [Google Scholar] [CrossRef]
- Stangeland, B.; Mughal, A.A.; Grieg, Z.; Sandberg, C.J.; Joel, M.; Nygård, S.; Meling, T.; Murrell, W.; Vik Mo, E.O.; Langmoen, I.A. Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget 2015, 6, 26192–26215. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Masai, H.; Bartek, J. Regulation and roles of Cdc7 kinase under replication stress. Cell Cycle 2014, 13, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xie, W.; Wang, N.; Li, C.; Wang, M. CDC7-dependent transcriptional regulation of RAD54L is essential for tumorigenicity and radio-resistance of glioblastoma. Transl. Oncol. 2018, 11, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Erbayraktar, Z.; Alural, B.; Erbayraktar, R.S.; Erkan, E.P. Cell division cycle 7-kinase inhibitor PHA-767491 hydrochloride suppresses glioblastoma growth and invasiveness. Cancer Cell Int. 2016, 16, 88. [Google Scholar] [CrossRef][Green Version]
- Aye, Y.; Li, M.; Long, M.J.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Kohnken, R.; Kodigepalli, K.M.; Wu, L. Regulation of deoxynucleotide metabolism in cancer: Novel mechanisms and therapeutic implications. Mol. Cancer 2015, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zheng, J.; Chen, S.; Huang, B.; Li, G.; Feng, Z.; Wang, J.; Xu, S. RRM2 promotes the progression of human glioblastoma. J. Cell. Physiol. 2018, 233, 6759–6767. [Google Scholar] [CrossRef]
- Bonner, M.K.; Haase, J.; Swinderman, J.; Halas, H.; Miller Jenkins, L.M.; Kelly, A.E. Enrichment of Aurora B kinase at the inner kinetochore controls outer kinetochore assembly. J. Cell Biol. 2019, 218, 3237–3257. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, S.; Ibrahim, A.N.; Deng, Z.; Wang, M. Serine/threonine kinase BUB1 promotes proliferation and radio-resistance in glioblastoma. Pathol. Res. Pract. 2019, 215, 152508. [Google Scholar] [CrossRef] [PubMed]
- Bie, L.; Zhao, G.; Cheng, P.; Rondeau, G.; Porwollik, S.; Ju, Y.; Xia, X.Q.; McClelland, M. The accuracy of survival time prediction for patients with glioma is improved by measuring mitotic spindle checkpoint gene expression. PLoS ONE 2011, 6, e25631. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Hubert, C.G.; Herman, J.; Corrin, P.; Toledo, C.M.; Skutt-Kakaria, K.; Vazquez, J.; Basom, R.; Zhang, B.; Risler, J.K.; et al. Cancer-Specific requirement for BUB1B/BUBR1 in human brain tumor isolates and genetically transformed cells. Cancer Discov. 2013, 3, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.G.; Pezuk, J.A.; Brassesco, M.S.; de Oliveira, J.C.; de Paula Queiroz, R.G.; Machado, H.R.; Carlotti, C.G.; Neder, L.; de Oliveira, H.F.; Scrideli, C.A.; et al. BUB1 and BUBR1 inhibition decreases proliferation and colony formation, and enhances radiation sensitivity in pediatric glioblastoma cells. Childs Nerv. Syst. 2013, 29, 2241–2248. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.; Davenport, J.; Neale, G.; Goorha, R. The mitotic checkpoint gene BubR1 has two distinct functions in mitosis. Exp. Cell Res. 2005, 308, 85–100. [Google Scholar] [CrossRef]
- Logarinho, E.; Resende, T.; Torres, C.; Bousbaa, H. The human spindle assembly checkpoint protein Bub3 is required for the establishment of efficient kinetochore-microtubule attachments. Mol. Biol. Cell 2008, 19, 1798–1813. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Listovsky, T.; Sale, J.E. Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. J. Cell Biol. 2013, 203, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Kapanidou, M.; Curtis, N.L.; Bolanos-Garcia, V.M. Cdc20: At the Crossroads between Chromosome Segregation and Mitotic Exit. Trends Biochem. Sci. 2017, 42, 193–205. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef]
- Marucci, G.; Morandi, L.; Magrini, E.; Farnedi, A.; Franceschi, E.; Miglio, R.; Calò, D.; Pession, A.; Foschini, M.P.; Eusebi, V. Gene expression profiling in glioblastoma and immunohistochemical evaluation of IGFBP-2 and CDC20. Virchows Arch. 2008, 453, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.D.; Gujar, A.D.; Mahlokozera, T.; Chen, I.; Pan, Y.; Luo, J.; Brost, T.; Thompson, E.A.; Turski, A.; Leuthardt, E.C.; et al. A CDC20-APC/SOX2 Signaling Axis Regulates Human Glioblastoma Stem-like Cells. Cell Rep. 2015, 11, 1809–1821. [Google Scholar] [CrossRef]
- Ding, Y.; Yu, S.; Bao, Z.; Liu, Y.; Liang, T. CDC20 with malignant progression and poor prognosis of astrocytoma revealed by analysis on gene expression. J. Neurooncol. 2017, 133, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wu, Q.; Mack, S.C.; Yang, K.; Kim, L.; Hubert, C.G.; Flavahan, W.A.; Chu, C.; Bao, S.; Rich, J.N. CDC20 maintains tumor initiating cells. Oncotarget 2015, 6, 13241–13254. [Google Scholar] [CrossRef] [PubMed]
- De, K.; Grubb, T.M.; Zalenski, A.A.; Pfaff, K.E.; Pal, D.; Majumder, S.; Summers, M.K.; Venere, M. Hyperphosphorylation of CDH1 in Glioblastoma Cancer Stem Cells Attenuates APC/C. Mol. Cancer Res. 2019, 17, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Donaires, F.S.; Godoy, P.R.; Leandro, G.S.; Puthier, D.; Sakamoto-Hojo, E.T. E2F transcription factors associated with up-regulated genes in glioblastoma. Cancer Biomark. 2017, 18, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Burns, S.C.; Mirsafian, H.; Li, W.; Vo, D.T.; Qiao, M.; Lei, X.; Smith, A.D.; Penalva, L.O. Genomic analyses of early responses to radiation inglioblastoma reveal new alterations at transcription, splicing, and translation levels. Sci. Rep. 2020, 10, 8979. [Google Scholar] [CrossRef]
- Yu, H.; Li, Z.; Wang, M. Expression and prognostic role of E2F transcription factors in high-grade glioma. CNS Neurosci. Ther. 2020. [Google Scholar] [CrossRef]
- Gujar, A.D.; Le, S.; Mao, D.D.; Dadey, D.Y.; Turski, A.; Sasaki, Y.; Aum, D.; Luo, J.; Dahiya, S.; Yuan, L.; et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc. Natl. Acad. Sci. USA 2016, 113, E8247–E8256. [Google Scholar] [CrossRef]
- Sharma, N.; Timmers, C.; Trikha, P.; Saavedra, H.I.; Obery, A.; Leone, G. Control of the p53-p21CIP1 Axis by E2f1, E2f2, and E2f3 is essential for G1/S progression and cellular transformation. J. Biol. Chem. 2006, 281, 36124–36131. [Google Scholar] [CrossRef]
- Chen, C.; Wells, A.D. Comparative analysis of E2F family member oncogenic activity. PLoS ONE 2007, 2, e912. [Google Scholar] [CrossRef]
- Kim, L.K.; Park, S.A.; Eoh, K.J.; Heo, T.H.; Kim, Y.T.; Kim, H.J. E2F8 regulates the proliferation and invasion through epithelial-mesenchymal transition in cervical cancer. Int. J. Biol. Sci. 2020, 16, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Iino, K.; Mitobe, Y.; Ikeda, K.; Takayama, K.I.; Suzuki, T.; Kawabata, H.; Suzuki, Y.; Horie-Inoue, K.; Inoue, S. RNA-binding protein NONO promotes breast cancer proliferation by post-transcriptional regulation of SKP2 and E2F8. Cancer Sci. 2020, 111, 148–159. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Tang, L.; Luo, J.; Ji, H.; Zhang, W.; Zhou, J.; Li, Q.; Miao, L. Long noncoding RNA SNHG6 promotes proliferation and angiogenesis of cholangiocarcinoma cells through sponging miR-101-3p and activation of E2F8. J. Cancer 2020, 11, 3002–3012. [Google Scholar] [CrossRef] [PubMed]
- Lü, Y.; Zhang, J.; Li, L.; Li, S.; Yang, Z. Carcinogenesis effects of E2F transcription factor 8 (E2F8) in hepatocellular carcinoma outcomes: An integrated bioinformatic report. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Yan, P.Y.; Zhang, X.A. Knockdown of E2F8 Suppresses Cell Proliferation in Colon Cancer Cells by Modulating the NF-κB Pathway. Ann. Clin. Lab. Sci. 2019, 49, 474–480. [Google Scholar]
- Dou, L.; Han, K.; Xiao, M.; Lv, F. miR-223-5p Suppresses Tumor Growth and Metastasis in Non-Small Cell Lung Cancer by Targeting E2F8. Oncol. Res. 2019, 27, 261–268. [Google Scholar] [CrossRef]
- Lv, Y.; Xiao, J.; Liu, J.; Xing, F. E2F8 is a Potential Therapeutic Target for Hepatocellular Carcinoma. J. Cancer 2017, 8, 1205–1213. [Google Scholar] [CrossRef]
- Lee, S.; Park, Y.R.; Kim, S.H.; Park, E.J.; Kang, M.J.; So, I.; Chun, J.N.; Jeon, J.H. Geraniol suppresses prostate cancer growth through down-regulation of E2F8. Cancer Med. 2016, 5, 2899–2908. [Google Scholar] [CrossRef]
- Deng, Q.; Wang, Q.; Zong, W.Y.; Zheng, D.L.; Wen, Y.X.; Wang, K.S.; Teng, X.M.; Zhang, X.; Huang, J.; Han, Z.G. E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res. 2010, 70, 782–791. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, D.; Li, Z.; Wang, M. E2F transcription factor 8 promotes proliferation and radioresistance in glioblastoma. Pathol. Res. Pract. Pathol. Res. Pract. 2020, 216, 10. [Google Scholar] [CrossRef]
- Xu, L.; Chen, Y.; Mayakonda, A.; Koh, L.; Chong, Y.K.; Buckley, D.L.; Sandanaraj, E.; Lim, S.W.; Lin, R.Y.; Ke, X.Y.; et al. Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc. Natl. Acad. Sci. USA 2018, 115, E5086–E5095. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Berenguer-Daizé, C.; Astorgues-Xerri, L.; Odore, E.; Cayol, M.; Cvitkovic, E.; Noel, K.; Bekradda, M.; MacKenzie, S.; Rezai, K.; Lokiec, F.; et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 2016, 139, 2047–2055. [Google Scholar] [CrossRef]
- Moros, A.; Rodríguez, V.; Saborit-Villarroya, I.; Montraveta, A.; Balsas, P.; Sandy, P.; Martínez, A.; Wiestner, A.; Normant, E.; Campo, E.; et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 2014, 28, 2049–2059. [Google Scholar] [CrossRef]
- Hong, S. RNA Binding Protein as an Emerging Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 203–210. [Google Scholar] [CrossRef]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef]
- Diouf, B.; Lin, W.; Goktug, A.; Grace, C.R.R.; Waddell, M.B.; Bao, J.; Shao, Y.; Heath, R.J.; Zheng, J.J.; Shelat, A.A.; et al. Alteration of RNA Splicing by Small-Molecule Inhibitors of the Interaction between NHP2L1 and U4. SLAS Discov. 2018, 23, 164–173. [Google Scholar] [CrossRef]
- Morita, Y.; Leslie, M.; Kameyama, H.; Volk, D.E.; Tanaka, T. Aptamer Therapeutics in Cancer: Current and Future. Cancers 2018, 10, 80. [Google Scholar] [CrossRef]
- Thumma, S.C.; Jacobson, B.A.; Patel, M.R.; Konicek, B.W.; Franklin, M.J.; Jay-Dixon, J.; Sadiq, A.; De, A.; Graff, J.R.; Kratzke, R.A. Antisense oligonucleotide targeting eukaryotic translation initiation factor 4E reduces growth and enhances chemosensitivity of non-small-cell lung cancer cells. Cancer Gene Ther. 2015, 22, 396–401. [Google Scholar] [CrossRef]
- Lan, L.; Liu, J.; Xing, M.; Smith, A.R.; Wang, J.; Wu, X.; Appelman, C.; Li, K.; Roy, A.; Gowthaman, R.; et al. Identification and Validation of an Aspergillus nidulans Secondary Metabolite Derivative as an Inhibitor of the Musashi-RNA Interaction. Cancers 2020, 12, 2221. [Google Scholar] [CrossRef]
- Wang, M.; Sun, X.Y.; Zhou, Y.C.; Zhang, K.J.; Lu, Y.Z.; Liu, J.; Huang, Y.; Wang, G.; Jiang, S.; Zhou, G. Suppression of Musashi-2 by the small compound largazole exerts inhibitory effects on malignant cells. Int. J. Oncol. 2020, 56, 1274–1283. [Google Scholar] [CrossRef]
- Lan, L.; Liu, H.; Smith, A.R.; Appelman, C.; Yu, J.; Larsen, S.; Marquez, R.T.; Wu, X.; Liu, F.Y.; Gao, P.; et al. Natural product derivative Gossypolone inhibits Musashi family of RNA-binding proteins. BMC Cancer 2018, 18, 809. [Google Scholar] [CrossRef]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Son, M.Y.; Deng, C.X.; Hoeijmarkers, J.H.; Rebel, V.I.; Hasty, P. A mechanism for 1,4-Benzoquinone-induced genotoxicity. Oncotarget 2016, 7, 46433–46447. [Google Scholar] [CrossRef]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baroni, M.; Yi, C.; Choudhary, S.; Lei, X.; Kosti, A.; Grieshober, D.; Velasco, M.; Qiao, M.; Burns, S.S.; Araujo, P.R.; et al. Musashi1 Contribution to Glioblastoma Development via Regulation of a Network of DNA Replication, Cell Cycle and Division Genes. Cancers 2021, 13, 1494. https://doi.org/10.3390/cancers13071494
Baroni M, Yi C, Choudhary S, Lei X, Kosti A, Grieshober D, Velasco M, Qiao M, Burns SS, Araujo PR, et al. Musashi1 Contribution to Glioblastoma Development via Regulation of a Network of DNA Replication, Cell Cycle and Division Genes. Cancers. 2021; 13(7):1494. https://doi.org/10.3390/cancers13071494
Chicago/Turabian StyleBaroni, Mirella, Caihong Yi, Saket Choudhary, Xiufen Lei, Adam Kosti, Denise Grieshober, Mitzli Velasco, Mei Qiao, Suzanne S. Burns, Patricia R. Araujo, and et al. 2021. "Musashi1 Contribution to Glioblastoma Development via Regulation of a Network of DNA Replication, Cell Cycle and Division Genes" Cancers 13, no. 7: 1494. https://doi.org/10.3390/cancers13071494
APA StyleBaroni, M., Yi, C., Choudhary, S., Lei, X., Kosti, A., Grieshober, D., Velasco, M., Qiao, M., Burns, S. S., Araujo, P. R., DeLambre, T., Son, M. Y., Plateroti, M., Ferreira, M. A. R., Hasty, P., & Penalva, L. O. F. (2021). Musashi1 Contribution to Glioblastoma Development via Regulation of a Network of DNA Replication, Cell Cycle and Division Genes. Cancers, 13(7), 1494. https://doi.org/10.3390/cancers13071494