
Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
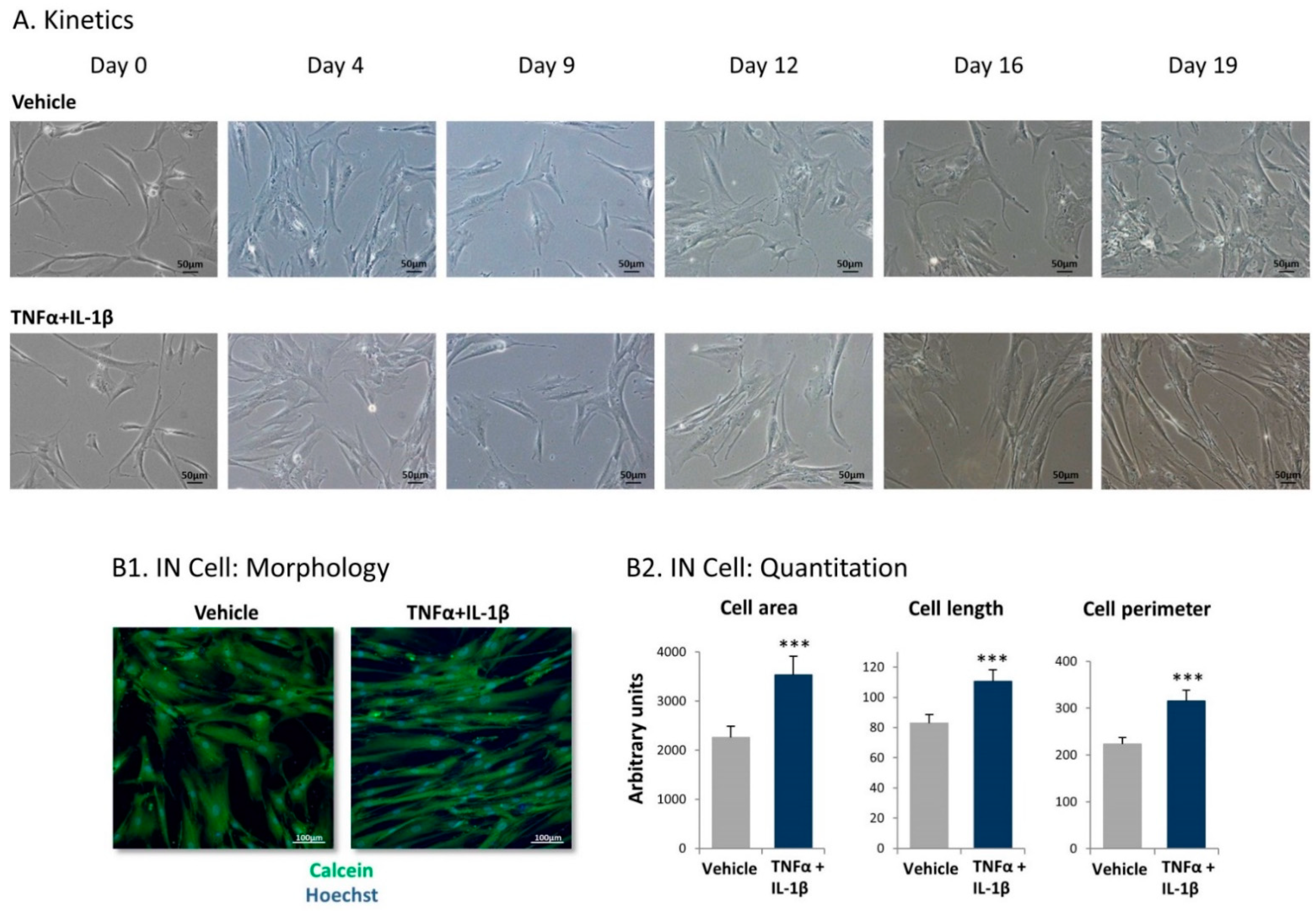
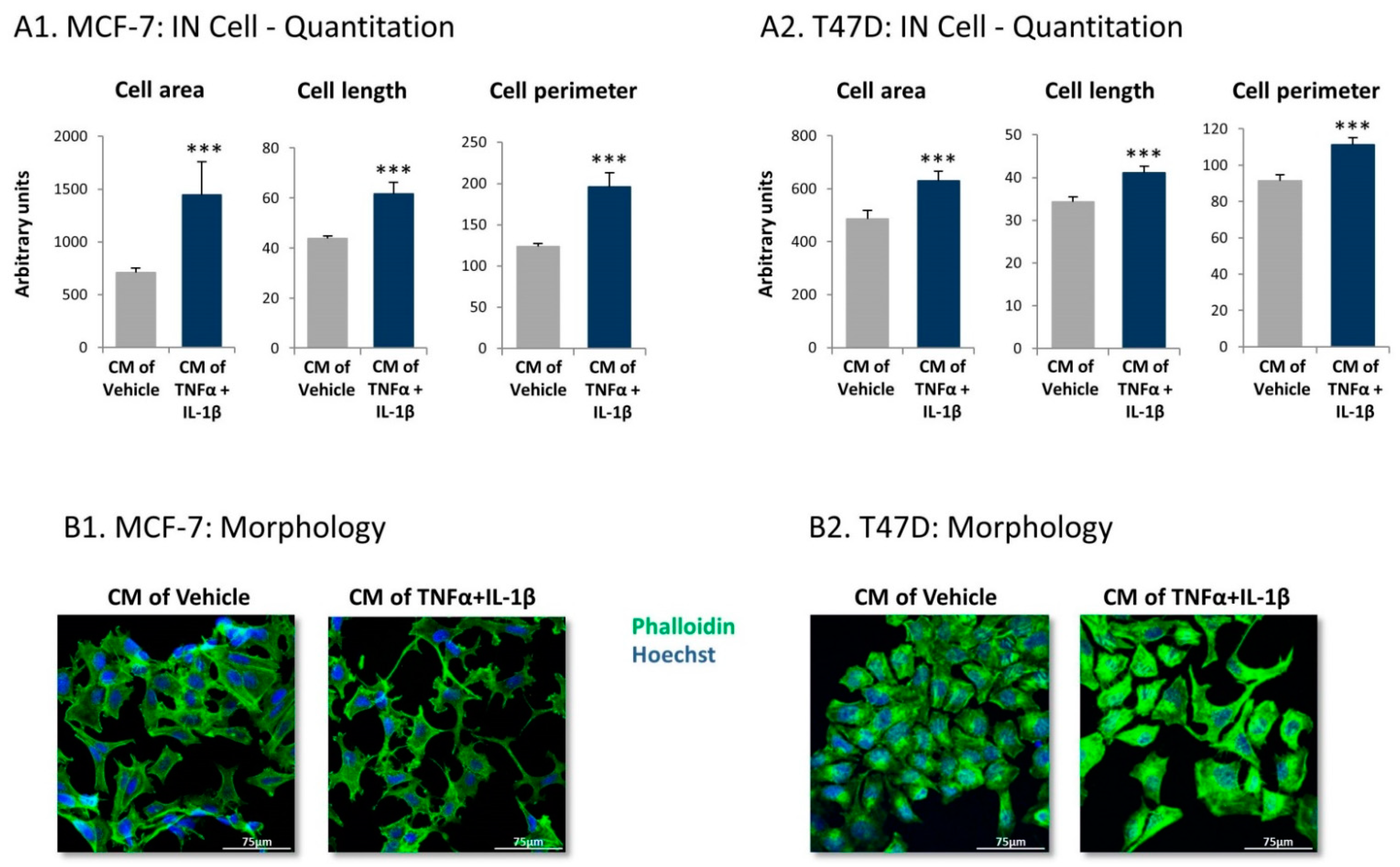
2.1. Following Persistent Stimulation by TNFα + IL-1β, MSCs Acquire a CAF-Like Morphology Accompanied by Modifications in Fibroblast-Related Transcriptional Programs
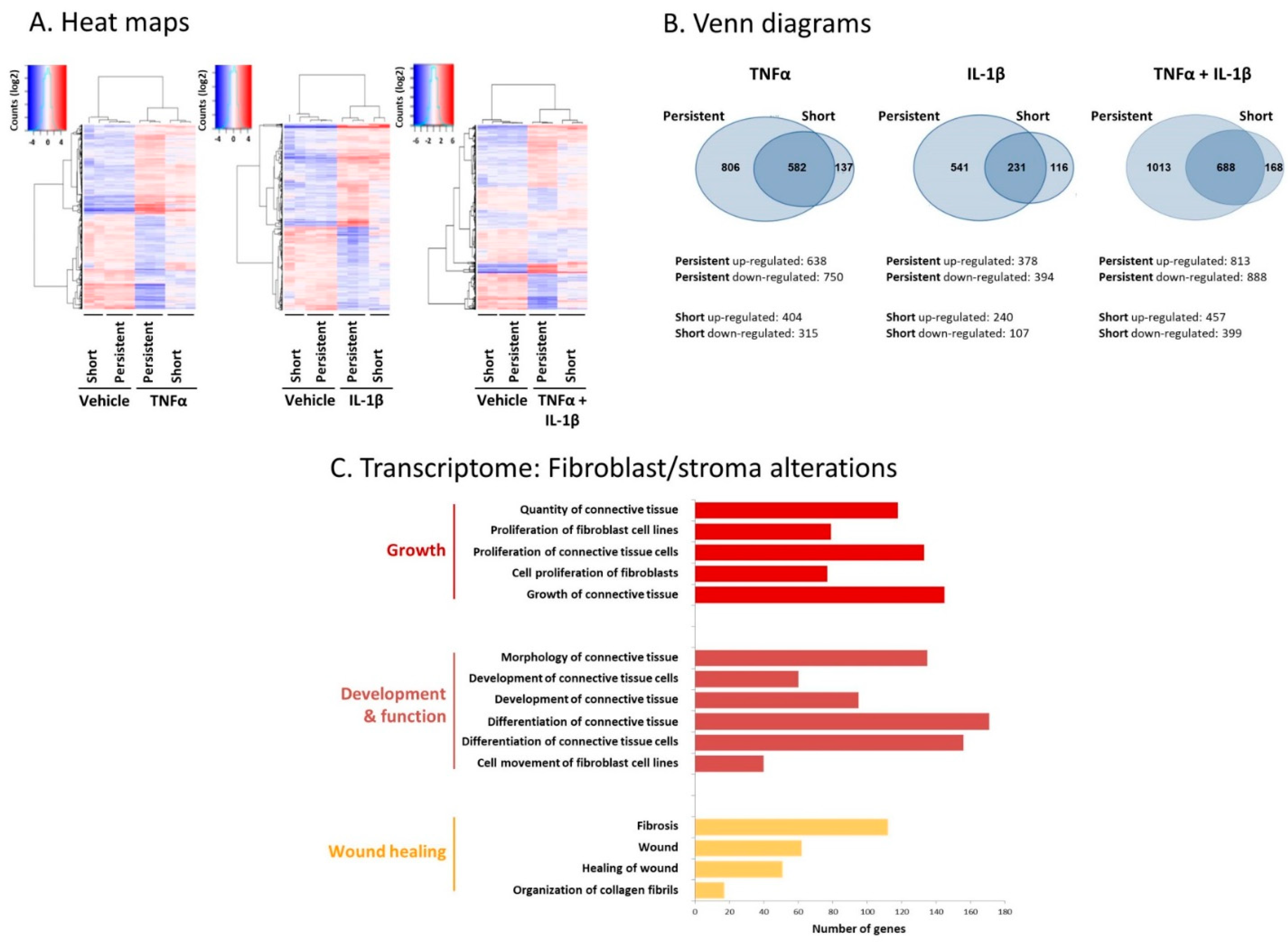
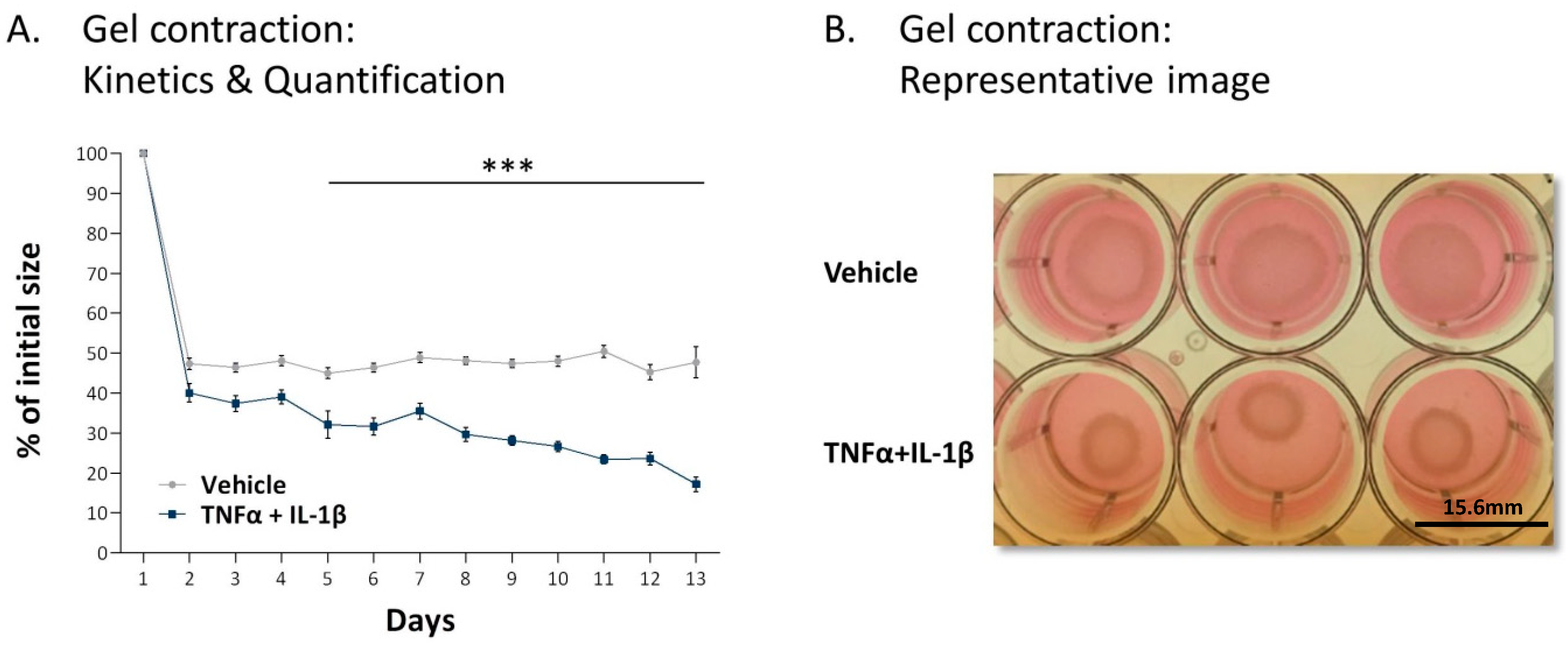
2.2. Following Persistent Stimulation by TNFα + IL-1β, MSCs Undergo Conversion to Inflammatory CAFs
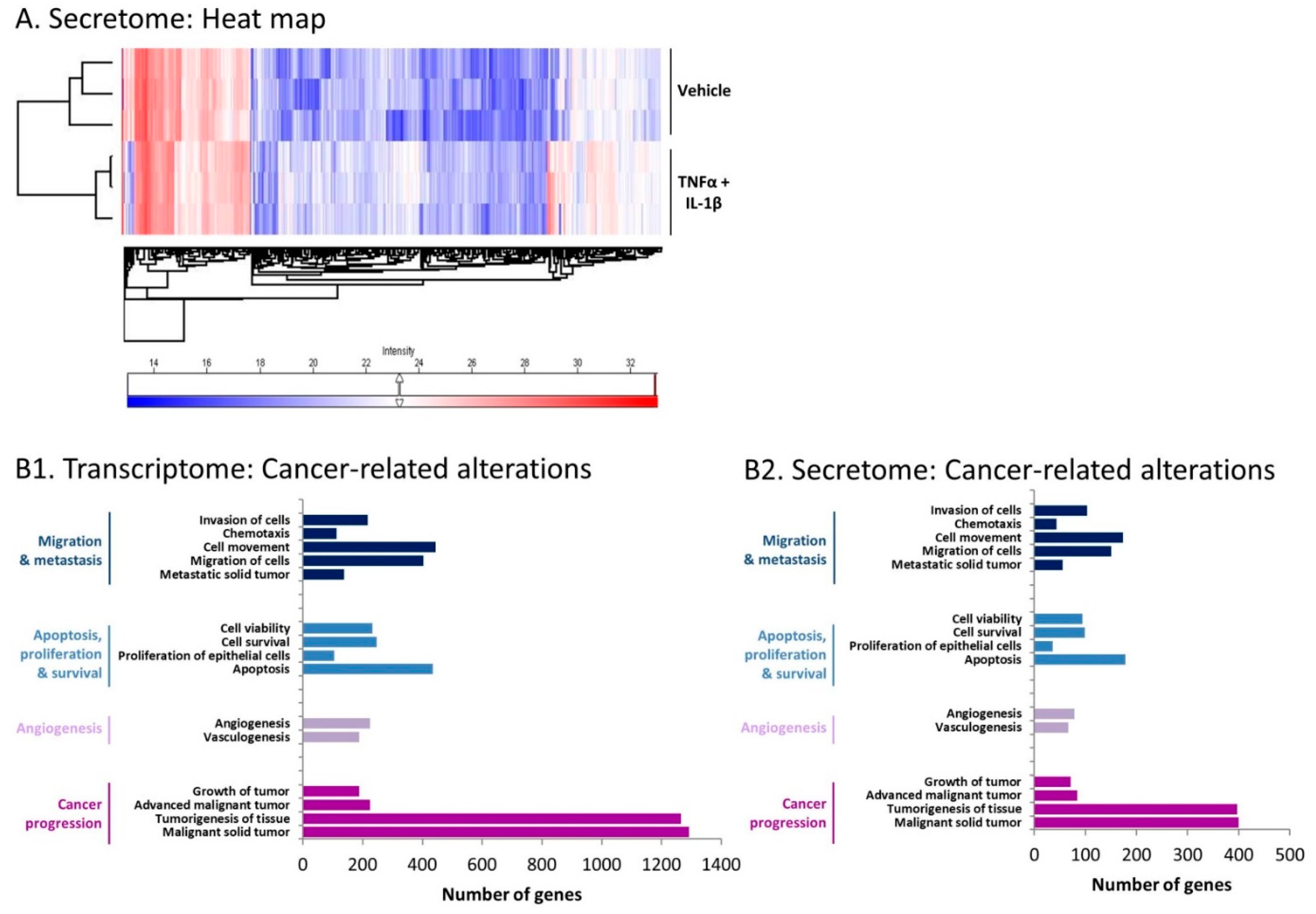
2.3. Inflammation-Driven CAFs Express Prominent Alterations in the Expression of Cancer-Related Genes and Proteins
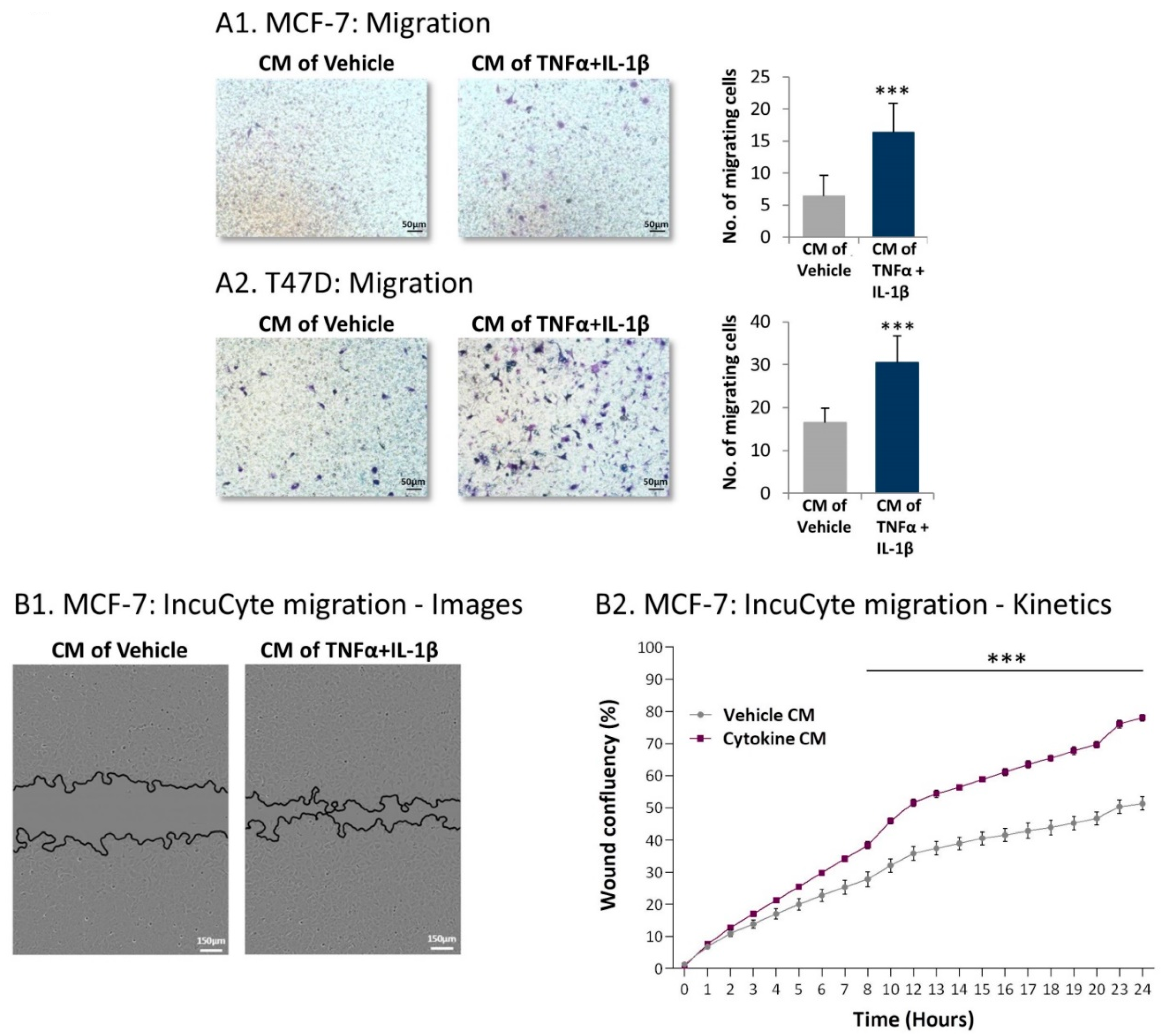
2.4. Inflammation-Driven CAFs Secrete Factors That Promote Pro-Metastatic Activities in Luminal-A BC Cells
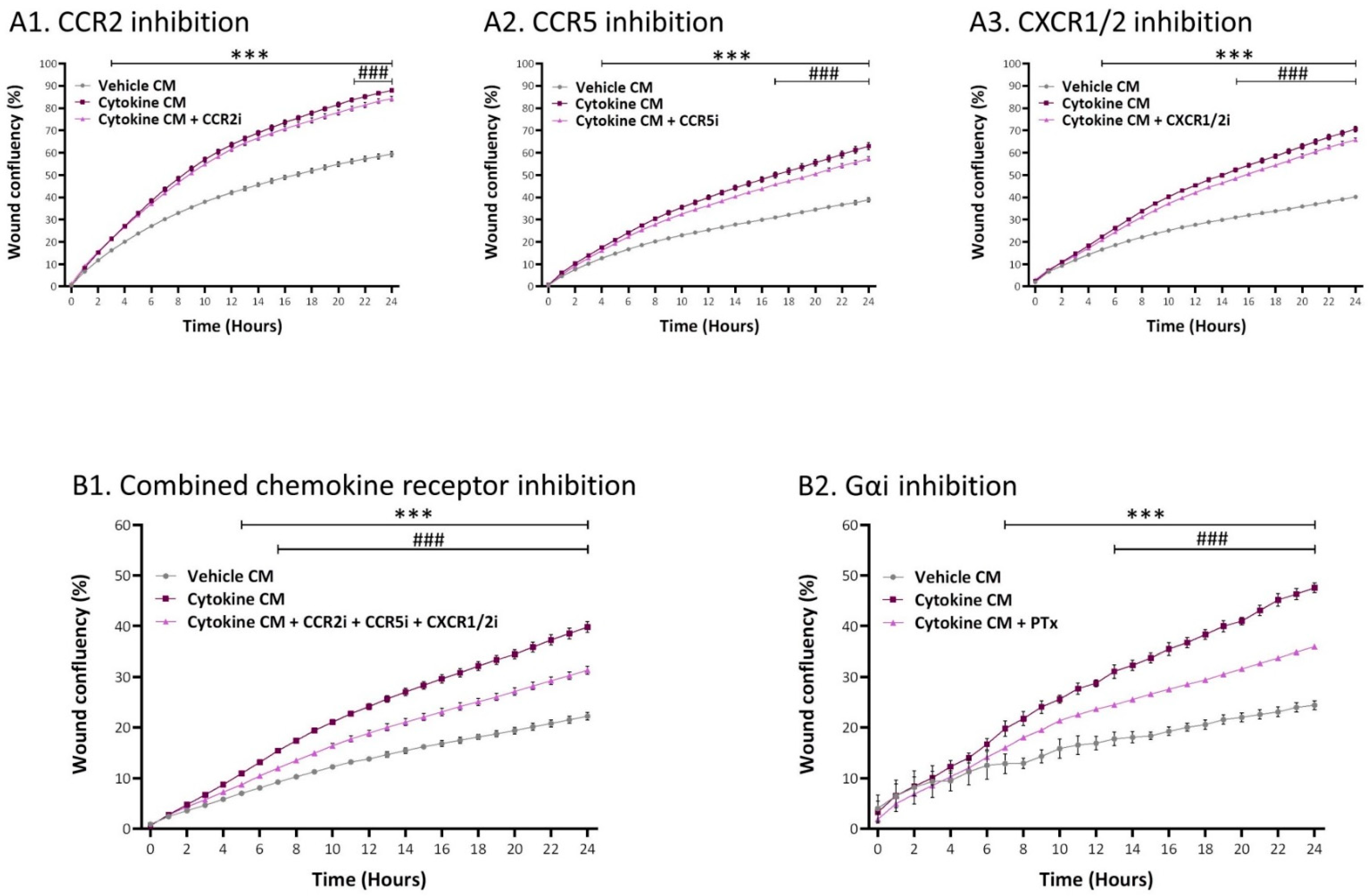
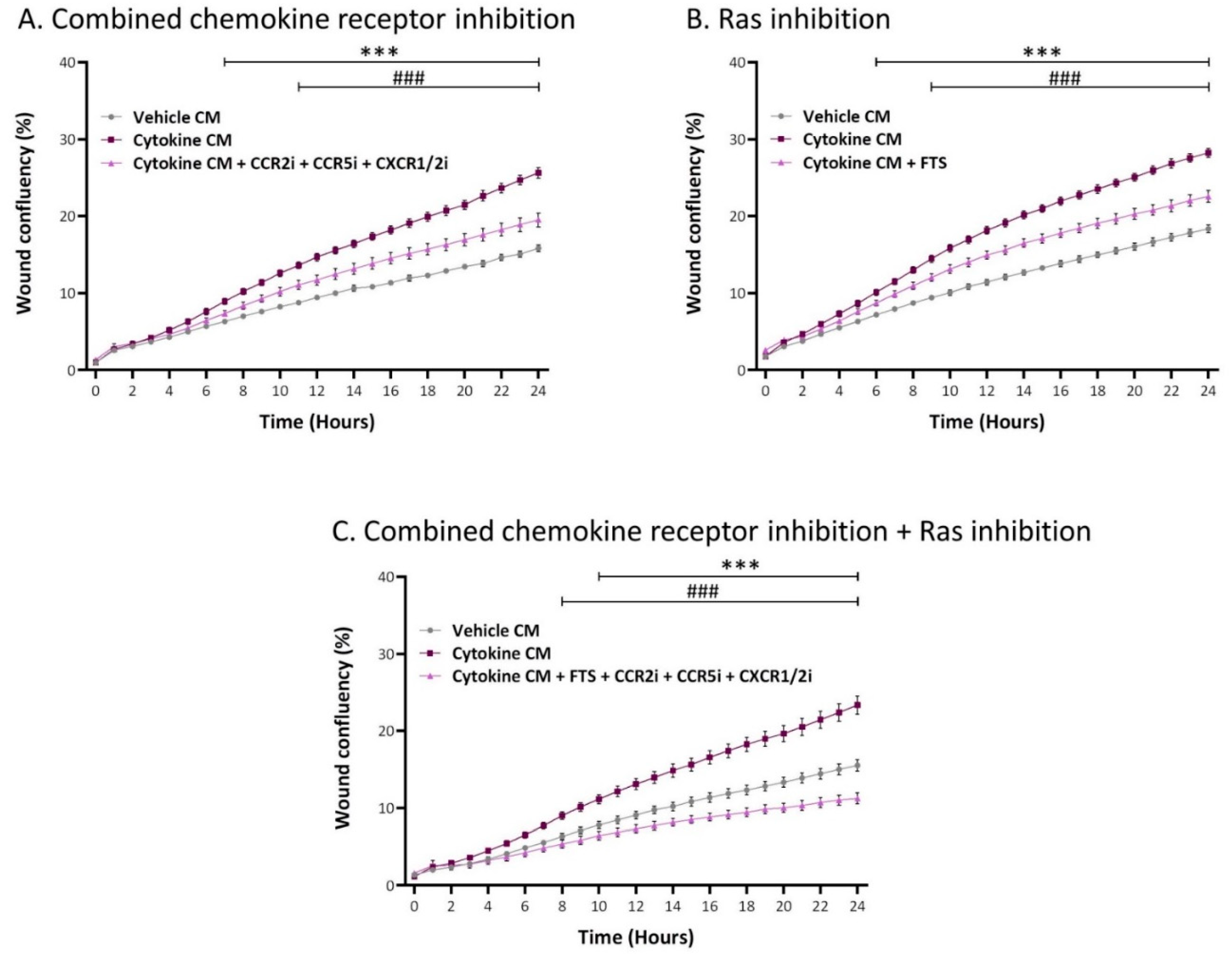
2.5. Inflammation-Driven CAFs Promote Tumor Cell Migration via Ligands of Ras-Activating Receptors and via Inflammatory Chemokines
3. Discussion
4. Materials and Methods
4.1. Cell Growth and Stimulation by Cytokines
4.2. Morphological Assessment and IN Cell Analyses
4.3. Transcriptome Analyses
4.4. Confocal Analyses
4.5. Secretome Analyses
4.6. Collagen Contraction Assays
4.7. Spheroid Formation Assays
4.8. Flow Cytometry Analyses
4.9. Transwell and IncuCyte Migration Assays
4.10. Determination of Cell Number and Viability
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molnár, I.A.; Molnár, B.Á.; Vízkeleti, L.; Fekete, K.; Tamás, J.; Deák, P.; Szundi, C.; Székely, B.; Moldvay, J.; Vári-Kakas, S.; et al. Breast carcinoma subtypes show different patterns of metastatic behavior. Virchows Archiv für Patholog. Anatom. Physiol. und für Klin. Med. 2017, 470, 275–283. [Google Scholar] [CrossRef]
- Abdelhakiem, M.K.; Johnstone, C.; Bergom, C.; Currey, A.; Robbins, J.R. The influence of breast cancer subtype on survival after palliative radiation for osseous metastases. Cancer Med. 2020, 9, 8979–8988. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Maman, S.; Witz, I.P. A history of exploring cancer in context. Nat. Rev. Cancer 2018, 18, 359–376. [Google Scholar] [CrossRef]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Kalluri, R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a003244. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Pas de deux: Control of anti-tumor immunity by cancer-associated inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef]
- Jiang, X.; Shapiro, D.J. The immune system and inflammation in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 673–682. [Google Scholar] [CrossRef]
- Baumgarten, S.C.; Frasor, J. Minireview: Inflammation: An instigator of more aggressive estrogen receptor (ER) Positive breast cancers. Mol. Endocrinol. 2012, 26, 360–371. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Cuiffo, B.G.; Karnoub, A.E. Mesenchymal stem cells in tumor development: Emerging roles and concepts. Cell Adh. Migr. 2012, 6, 220–230. [Google Scholar] [CrossRef]
- Lazennec, G.; Lam, P.Y. Recent discoveries concerning the tumor-mesenchymal stem cell interactions. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1866, 290–299. [Google Scholar] [CrossRef]
- Hill, B.S.; Sarnella, A.; D’Avino, G.; Zannetti, A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin. Cancer Biol. 2020, 60, 202–213. [Google Scholar] [CrossRef]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast cancer tumor stroma: Cellular components, phenotypic heterogeneity, intercellular communication, prognostic implications and therapeutic opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef] [PubMed]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef]
- Martínez-Reza, I.; Díaz, L.; García-Becerra, R. Preclinical and clinical aspects of TNF-α and its receptors TNFR1 and TNFR2 in breast cancer. J. Biomed. Sci. 2017, 24, 90. [Google Scholar] [CrossRef]
- Al-Hatamleh, M.A.I.; Ahmad, S.; Boer, J.C.; Lim, J.; Chen, X.; Plebanski, M.; Mohamud, R. A perspective review on the role of nanomedicine in the modulation of TNF-TNFR2 axis in breast cancer immunotherapy. J. Oncol. 2019, 2019, 1–13. [Google Scholar] [CrossRef]
- Salamanna, F.; Borsari, V.; Contartese, D.; Costa, V.; Giavaresi, G.; Fini, M. What is the role of interleukins in breast cancer bone metastases? A systematic review of preclinical and clinical evidence. Cancers 2019, 11, 2018. [Google Scholar] [CrossRef]
- Tulotta, C.; Ottewell, P.D. The role of IL-1B in breast cancer bone metastasis. Endocr. Relat. Cancer 2018, 25, R421–R434. [Google Scholar] [CrossRef]
- Apte, R.N.; Voronov, E. Immunotherapeutic approaches of IL-1 neutralization in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 293–306. [Google Scholar] [CrossRef]
- Zhou, X.L.; Fan, W.; Yang, G.; Yu, M.X. The clinical significance of PR, ER, NF- kappa B, and TNF- alpha in breast cancer. Dis. Mark. 2014, 2014, 494581. [Google Scholar]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130. [Google Scholar] [CrossRef]
- Jin, L.; Yuan, R.Q.; Fuchs, A.; Yao, Y.; Joseph, A.; Schwall, R.; Schnitt, S.J.; Guida, A.; Hastings, H.M.; Andres, J.; et al. Expression of interleukin-1beta in human breast carcinoma. Cancer 1997, 80, 421–434. [Google Scholar] [CrossRef]
- Semesiuk, N.I.; Zhylchuk, A.; Bezdenezhnykh, N.; Lykhova, A.; Vorontsova, A.L.; Zhylchuk, V.E.; Kudryavets, Y.I. Disseminated tumor cells and enhanced level of some cytokines in bone marrow and peripheral blood of breast cancer patients as predictive factors of tumor progression. Exp. Oncol. 2013, 35, 295–302. [Google Scholar]
- Abrahamsson, A.; Morad, V.; Saarinen, N.M.; Dabrosin, C. Estradiol, tamoxifen, and flaxseed alter IL-1beta and IL-1Ra levels in normal human breast tissue in vivo. J. Clin. Endocrinol. Metab. 2012, 97, E2044–E2054. [Google Scholar] [CrossRef]
- Perrier, S.; Caldefie-Chézet, F.; Vasson, M.-P. IL-1 family in breast cancer: Potential interplay with leptin and other adipocytokines. FEBS Lett. 2008, 583, 259–265. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; Martin, E.E.; Ge, C.; Franceschi, R.T.; Kleer, C.G.; Anwar, T.; Arellano-Garcia, C.; Medhora, N.; Lama, A.; Chen, Y.-C.; et al. Mesenchymal stem cell-induced DDR2 mediates stromal-breast cancer interactions and metastasis growth. Cell Rep. 2017, 18, 1215–1228. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Friedman, R.; Varro, A.; Tycko, B.; Wang, T.C.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef]
- Kidd, S.; Spaeth, E.; Watson, K.; Burks, J.; Lu, H.; Klopp, A.; Andreeff, M.; Marini, F.C. Origins of the tumor microenvironment: Quantitative assessment of adipose-derived and bone marrow–derived stroma. PLoS ONE 2012, 7, e30563. [Google Scholar] [CrossRef] [PubMed]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Develop. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, R.J.; Oh, S.Y. Breast cancer-associated fibroblasts: Where we are and where we need to go. Cancers 2016, 8, 19. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Kanzaki, R.; Pietras, K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci. 2020, 111, 2708–2717. [Google Scholar] [CrossRef]
- Louault, K.; Li, R.R.; DeClerck, Y.A. Cancer-associated fibroblasts: Understanding their heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463.e10–479.e10. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFalpha. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef]
- Han, Z.; Tian, Z.; Lv, G.; Zhang, L.; Jiang, G.; Sun, K.; Wang, C.; Bu, X.; Li, R.; Shi, Y.; et al. Immunosuppressive effect of bone marrow-derived mesenchymal stem cells in inflammatory microenvironment favours the growth of B16 melanoma cells. J. Cell. Mol. Med. 2011, 15, 2343–2352. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Han, Z.; Cai, X.; Wu, M.; Wei, L.; Liu, Y.; Sun, K.; Zhang, S.; Jiang, G.; Li, R.; et al. Mesenchymal stem cells in inflammation microenvironment accelerates hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition. PLoS ONE 2012, 7, e43272. [Google Scholar] [CrossRef]
- Lerrer, S.; Liubomirski, Y.; Bott, A.; Abnaof, K.; Oren, N.; Yousaf, A.; Korner, C.; Meshel, T.; Wiemann, S.; Ben-Baruch, A. Co-inflammatory roles of TGFbeta1 in the presence of tnfalpha drive a pro-inflammatory fate in mesenchymal stem cells. Front. Immunol. 2017, 8, 479. [Google Scholar] [CrossRef] [PubMed]
- Katanov, C.; Lerrer, S.; Liubomirski, Y.; Leider-Trejo, L.; Meshel, T.; Bar, J.; Feniger-Barish, R.; Kamer, I.; Soria-Artzi, G.; Kahani, H.; et al. Regulation of the inflammatory profile of stromal cells in human breast cancer: Prominent roles for TNF-alpha and the NF-kappaB pathway. Stem Cell Res. Ther. 2015, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Körner, C.; Ben-Baruch, A. Tumor-stroma-inflammation networks promote pro-metastatic chemokines and aggressiveness characteristics in triple-negative breast cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef]
- Escobar, P.; Bouclier, C.; Serret, J.; Bieche, I.; Brigitte, M.; Caicedo, A.; Sanchez, E.; Vacher, S.; Vignais, M.L.; Bourin, P.; et al. IL-1beta produced by aggressive breast cancer cells is one of the factors that dictate their interactions with mesenchymal stem cells through chemokine production. Oncotarget 2015, 6, 29034–29047. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Reinhardt, F.; Herschman, H.R.; Weinberg, R.A. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012, 2, 840–855. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow–derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Daubriac, J.; Han, S.; Grahovac, J.; Smith, E.; Hosein, A.; Buchanan, M.; Basik, M.; Boucher, Y. The crosstalk between breast carcinoma-associated fibroblasts and cancer cells promotes RhoA-dependent invasion via IGF-1 and PAI-1. Oncotarget 2018, 9, 10375–10387. [Google Scholar] [CrossRef]
- Saxena, K.; Jolly, M.K.; Balamurugan, K. Hypoxia, partial EMT and collective migration: Emerging culprits in metastasis. Transl. Oncol. 2020, 13, 100845. [Google Scholar] [CrossRef] [PubMed]
- Khaled, N.; Bidet, Y. New insights into the implication of epigenetic alterations in the emt of triple negative breast cancer. Cancers 2019, 11, 559. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Smaill, J.B.; Liu, T.; Ding, K.; Lu, X. Small-molecule inhibitors directly targeting KRAS as anticancer therapeutics. J. Med. Chem. 2020, 63, 14404–14424. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G protein–coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef]
- Arang, N.; Gutkind, J.S. G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett. 2020, 594, 4201–4232. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Bachelerie, F.; Ben-Baruch, A.; Mantovani, A.; Matsushima, K.; Murphy, P.M.; Nibbs, R.J.B.; Nomiyama, H.; Power, C.A.; Proudfoot, A.E.I.; Rosenkilde, M.M.; et al. International union of basic and clinical pharmacology. [corrected] LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [PubMed]
- Ksiazkiewicz, M.; Gottfried, E.; Kreutz, M.; Mack, M.; Hofstaedter, F.; Kunz-Schughart, L.A. Importance of CCL2-CCR2A/2B signaling for monocyte migration into spheroids of breast cancer-derived fibroblasts. Immunobiology 2010, 215, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M. Chemokine receptor CCR5: Insights into structure, function, and regulation. Cell. Signal. 2004, 16, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.-H. Adipose tissue derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor derived factors. Cell. Oncol. 2011, 34, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.; Franzen, C.A.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1-TGF-beta1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015, 34, 4821–4833. [Google Scholar] [CrossRef]
- Shangguan, L.; Ti, X.; Krause, U.; Hai, B.; Zhao, Y.; Yang, Z.; Liu, F. Inhibition of TGF-beta/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells 2012, 30, 2810–2819. [Google Scholar] [CrossRef]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Morein, D.; Rubinstein-Achiasaf, L.; Sprinzak, D.; Wiemann, S.; Körner, C.; Ehrlich, M.; Ben-Baruch, A. Notch-mediated tumor-stroma-inflammation networks promote invasive properties and CXCL8 expression in triple-negative breast cancer. Front. Immunol. 2019, 10, 804. [Google Scholar] [CrossRef]
- Schauer, I.G.; Zhang, J.; Xing, Z.; Guo, X.; Mercado-Uribe, I.; Sood, A.K.; Huang, P.; Liu, J. Interleukin-1beta promotes ovarian tumorigenesis through a p53/NF-kappaB-mediated inflammatory response in stromal fibroblasts. Neoplasia 2013, 15, 409–420. [Google Scholar] [CrossRef]
- Shin, S.Y.; Nam, J.S.; Lim, Y.; Lee, Y.H. TNFalpha-exposed bone marrow-derived mesenchymal stem cells promote locomotion of MDA-MB-231 breast cancer cells through transcriptional activation of CXCR3 ligand chemokines. J. Biol. Chem. 2010, 285, 30731–30740. [Google Scholar] [CrossRef]
- Dudas, J.; Fullar, A.; Bitsche, M.; Schartinger, V.; Kovalszky, I.; Sprinzl, G.M.; Riechelmann, H. Tumor-produced, active interleukin-1beta regulates gene expression in carcinoma-associated fibroblasts. Exp. Cell Res. 2011, 317, 2222–2229. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Heo, S.C.; Jeong, G.O.; Yoon, J.W.; Mo, W.M.; Lee, M.J.; Jang, I.-H.; Kwon, S.M.; Lee, J.S.; Kim, J.H. Tumor necrosis factor-α-activated mesenchymal stem cells promote endothelial progenitor cell homing and angiogenesis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 2136–2144. [Google Scholar] [CrossRef]
- Ziaei, R.; Ayatollahi, M.; Yaghobi, R.; Sahraeian, Z.; Zarghami, N. Involvement of TNF-α in differential gene expression pattern of CXCR4 on human marrow-derived mesenchymal stem cells. Mol. Biol. Rep. 2014, 41, 1059–1066. [Google Scholar] [CrossRef]
- Uchibori, R.; Tsukahara, T.; Mizuguchi, H.; Saga, Y.; Urabe, M.; Mizukami, H.; Kume, A.; Ozawa, K. NF-kappaB activity regulates mesenchymal stem cell accumulation at tumor sites. Cancer Res. 2013, 73, 364–372. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Shi, Y.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Crisostomo, P.R.; Wang, Y.; Markel, T.A.; Wang, M.; Lahm, T.; Meldrum, D.R. Human mesenchymal stem cells stimulated by TNF-alpha, LPS, or hypoxia produce growth factors by an NF kappa B- but not JNK-dependent mechanism. Am. J. Physiol. Cell Physiol. 2008, 294, C675–C682. [Google Scholar] [CrossRef] [PubMed]
- Romieu-Mourez, R.; Francois, M.; Abate, A.; Boivin, M.N.; Birman, E.; Bailey, D.; Bramson, J.L.; Forner, K.; Young, Y.K.; Medin, J.A.; et al. Mesenchymal stromal cells expressing ErbB-2/neu elicit protective antibreast tumor immunity in vivo, which is paradoxically suppressed by IFN-gamma and tumor necrosis factor-alpha priming. Cancer Res. 2010, 70, 7742–7747. [Google Scholar] [CrossRef]
- Yu, P.F.; Huang, Y.; Han, Y.Y.; Lin, L.Y.; Sun, W.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. TNFalpha-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2(+) neutrophils. Oncogene 2017, 36, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.-P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 2017, 15, 1–18. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, A.; Dittmer, J. Long-term exposure to carcinoma-associated fibroblasts makes breast cancer cells addictive to integrin beta1. Oncotarget 2018, 9, 22079–22094. [Google Scholar] [CrossRef]
- Morein, D.; Erlichman, N.; Ben-Baruch, A. Beyond cell motility: The expanding roles of chemokines and their receptors in malignancy. Front. Immunol. 2020, 11, 952. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their receptors: Multifaceted roles in cancer progression and potential value as cancer prognostic markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L.; Wolf, M.J.; Roblek, M.; Lorentzen, A.; Heikenwalder, M. Inflammatory chemokines and metastasis—Tracing the accessory. Oncogene 2013, 33, 3217–3224. [Google Scholar] [CrossRef]
- Mollica Poeta, V.; Massara, M.; Capucett, A.; Bonecch, R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef]
- Alfaro, C.; Sanmamed, M.F.; Rodríguez-Ruiz, M.E.; Teijeira, Á.; Oñate, C.; González, Á.; Ponz, M.; Schalper, K.A.; Pérez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Schioppa, T.; Tiberio, L.; Stabile, H.; Sozzani, S. Leukocyte trafficking in tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 40–47. [Google Scholar] [CrossRef]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.-K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef]
- Liubomirski, Y.; Ben-Baruch, A. Notch-inflammation networks in regulation of breast cancer progression. Cells 2020, 9, 1576. [Google Scholar] [CrossRef]
- Gaziano, J.M. Aspirin for primary prevention. Clinical considerations in 2019. JAMA 2019, 321, 253–255. [Google Scholar] [CrossRef]
- Kehm, R.D.; Hopper, J.L.; John, E.M.; Phillips, K.-A.; MacInnis, R.J.; Dite, G.S.; Milne, R.L.; Liao, Y.; Zeinomar, N.; Terry, M.B.; et al. Regular use of aspirin and other non-steroidal anti-inflammatory drugs and breast cancer risk for women at familial or genetic risk: A cohort study. Breast Cancer Res. 2019, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef]
- Li, P.; Zheng, Y.; Chen, X. Drugs for autoimmune inflammatory diseases: From small molecule compounds to anti-TNF biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Bettiol, A.; Lopalco, G.; Venerito, V.; Fornaro, M.; Vannacci, A.; Rigante, D.; Cimaz, R.; Iannone, F.; Emmi, G.; Cantarini, L.; et al. Unveiling the efficacy, safety, and tolerability of anti-interleukin-1 treatment in monogenic and multifactorial autoinflammatory diseases. Int. J. Mol. Sci. 2019, 20, 1898. [Google Scholar] [CrossRef] [PubMed]
- Keydar, I.; Chen, L.; Karby, S.; Weiss, F.; Delarea, J.; Radu, M.; Chaitcik, S.; Brenner, H. Establishment and characterization of a cell line of human breast carcinoma origin. Eur. J. Cancer 1979, 15, 659–670. [Google Scholar] [CrossRef]
- Kim, T.J.; Kang, Y.J.; Lim, Y.; Lee, H.W.; Bae, K.; Lee, Y.S.; Yoo, J.M.; Yoo, H.S.; Yun, Y.P. Ceramide 1-phosphate induces neointimal formation via cell proliferation and cell cycle progression upstream of ERK1/2 in vascular smooth muscle cells. Exp. Cell Res. 2011, 317, 2041–2051. [Google Scholar] [CrossRef]
- Kaplan, O.; Ruíz-Cabello, J.; Cohen, J.S. In vitro cytotoxic effects of tumor necrosis factor-α in human breast cancer cells may be associated with increased glucose consumption. FEBS Lett. 1997, 406, 175–178. [Google Scholar] [CrossRef][Green Version]
- El Mahdani, N.E.; Ameyar, M.; Cai, Z.; Colard, O.; Masliah, J.; Chouaib, S. Resistance to TNF-induced cytotoxicity correlates with an abnormal cleavage of cytosolic phospholipase A2. J. Immunol. 2000, 165, 6756–6761. [Google Scholar] [CrossRef]
- Rossi, M.; Sharkey, A.M.; Vigano, P.; Fiore, G.; Furlong, R.; Florio, P.; Ambrosini, G.; Smith, S.K.; Petraglia, F. Identification of genes regulated by interleukin-1beta in human endometrial stromal cells. Reproduction 2005, 130, 721–729. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhat, V.; Berdnikov, A.; Liu, J.; Zhang, G.; Buchel, E.; Safneck, J.; Marshall, A.J.; Murphy, L.C.; Postovit, L.M.; et al. Paracrine crosstalk between fibroblasts and ER(+) Breast cancer cells creates an il1beta-enriched niche that promotes tumor growth. iScience 2019, 19, 388–401. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubinstein-Achiasaf, L.; Morein, D.; Ben-Yaakov, H.; Liubomirski, Y.; Meshel, T.; Elbaz, E.; Dorot, O.; Pichinuk, E.; Gershovits, M.; Weil, M.; et al. Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers 2021, 13, 1472. https://doi.org/10.3390/cancers13061472
Rubinstein-Achiasaf L, Morein D, Ben-Yaakov H, Liubomirski Y, Meshel T, Elbaz E, Dorot O, Pichinuk E, Gershovits M, Weil M, et al. Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers. 2021; 13(6):1472. https://doi.org/10.3390/cancers13061472
Chicago/Turabian StyleRubinstein-Achiasaf, Linor, Dina Morein, Hagar Ben-Yaakov, Yulia Liubomirski, Tsipi Meshel, Eti Elbaz, Orly Dorot, Edward Pichinuk, Michael Gershovits, Miguel Weil, and et al. 2021. "Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells" Cancers 13, no. 6: 1472. https://doi.org/10.3390/cancers13061472
APA StyleRubinstein-Achiasaf, L., Morein, D., Ben-Yaakov, H., Liubomirski, Y., Meshel, T., Elbaz, E., Dorot, O., Pichinuk, E., Gershovits, M., Weil, M., & Ben-Baruch, A. (2021). Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers, 13(6), 1472. https://doi.org/10.3390/cancers13061472