
A MAPK/miR-29 Axis Suppresses Melanoma by Targeting MAFG and MYBL2

 , , , , , and
, , , , , and {kind=link}
{kind=link}
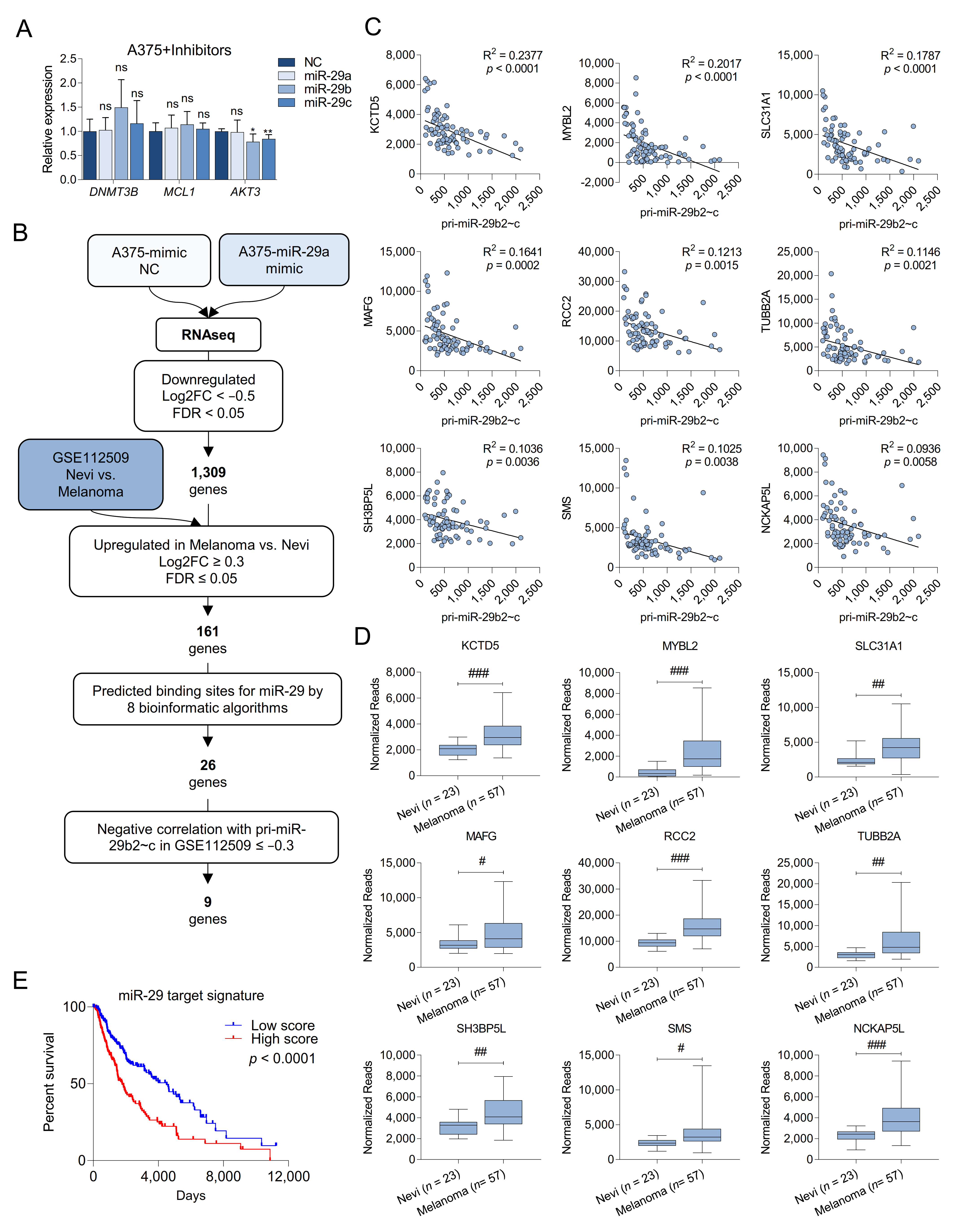{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Oncogenic BRAF Promotes miR-29 Expression in MEFs
2.2. The MAPK Pathway Regulates miR-29 Expression in Melanocytes and Melanoma Cells
2.3. miR-29 Inactivation Promotes Melanoma Formation
2.4. Melanoma Cells Are Addicted to High Levels of miR-29 Target Genes
2.5. MAFG and MYBL2 Are Putative miR-29 Targets with Roles in Melanoma
2.6. MAFG Is a Bona Fide Target of miR-29 in Melanocytes and Melanoma
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. RNA Isolation and Quantitative RT-PCR
4.3. RNA-Sequencing
4.4. Plasmids, Cell Transfection, and Lentiviral Transduction
4.5. Proliferation and Colony Formation Assays
4.6. Immunoblotting
4.7. ES Cell Targeting, Mouse Generation, and ESC-GEMM Experiments
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Romano, G.; Kwong, L.N. miRNAs, Melanoma and Microenvironment: An Intricate Network. Int. J. Mol. Sci. 2017, 18, 2354. [Google Scholar] [CrossRef]
- Wozniak, M.; Mielczarek, A.; Czyz, M. miRNAs in Melanoma: Tumor Suppressors and Oncogenes with Prognostic Potential. Curr. Med. Chem. 2016, 23, 3136–3153. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, L.; Qiu, Y.; Liu, L. MicroRNA-29a Inhibits Growth, Migration and Invasion of Melanoma A375 Cells in Vitro by Directly Targeting BMI1. Cell. Physiol. Biochem. 2018, 50, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.; Safarzadeh, A.; Beyranvand, F.; Ahmadpour, F.; Hajiasgharzadeh, K.; Baghbanzadeh, A.; Baradaran, B. The potential role of miR-29 in health and cancer diagnosis, prognosis, and therapy. J. Cell. Physiol. 2019, 234, 19280–19297. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, A.J.; Liu, Y.; Fang, Y.; Ding, X.; Liang, M. The miR-29 family: Genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol. Genom. 2012, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; He, H.-B.; Zhang, W.-Y.; Zhang, H.-X.; Bai, J.-B.; Liu, H.-Z.; Cao, J.-H.; Chang, K.-C.; Li, X.-Y.; Zhao, S.-H. miR-29 targets Akt3 to reduce proliferation and facilitate differentiation of myoblasts in skeletal muscle development. Cell Death Dis. 2013, 4, e668. [Google Scholar] [CrossRef]
- Ugalde, A.P.; Ramsay, A.J.; De La Rosa, J.; Varela, I.; Mariño, G.; Cadiñanos, J.; Lu, J.; Freije, J.M.P.; López-Otín, C. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 2011, 30, 2219–2232. [Google Scholar] [CrossRef]
- Nguyen, T.; Kuo, C.; Nicholl, M.B.; Sim, M.-S.; Turner, R.R.; Morton, D.L.; Hoon, D.S. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics 2011, 6, 388–394. [Google Scholar] [CrossRef]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar] [CrossRef]
- Zhao, J.-J.; Lin, J.; Lwin, T.; Yang, H.; Guo, J.; Kong, W.; Dessureault, S.; Moscinski, L.C.; Rezania, D.; Dalton, W.S.; et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood 2010, 115, 2630–2639. [Google Scholar] [CrossRef] [PubMed]
- Havelange, V.; Ms, N.S.; Ms, C.C.E.H.; Volinia, S.; Andreeff, M.; Marcucci, G.; Croce, C.M.; Garzon, R. Functional implications of microRNAs in acute myeloid leukemia by integrating microRNA and messenger RNA expression profiling. Cancer 2011, 117, 4696–4706. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Zhang, Y.; Qu, K.; Yang, X.; Fu, J.; Chen, W.; Li, X. MicroRNA-29B (mir-29b) regulates the Warburg effect in ovarian cancer by targeting AKT2 and AKT3. Oncotarget 2015, 6, 40799–40814. [Google Scholar] [CrossRef]
- Mott, J.L.; Kurita, S.; Cazanave, S.C.; Bronk, S.F.; Werneburg, N.W.; Fernandez-Zapico, M.E. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J. Cell. Biochem. 2010, 110, 1155–1164. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated Silencing of MYC-Mediated miR-29 by HDAC3 and EZH2 as a Therapeutic Target of Histone Modification in Aggressive B-Cell Lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar] [CrossRef]
- Kurinna, S.; Schafer, M.; Ostano, P.; Karouzakis, E.; Chiorino, G.; Bloch, W.; Bachmann, A.; Gay, S.; Garrod, D.; Lefort, K.; et al. A novel Nrf2-miR-29-desmocollin-2 axis regulates desmosome function in keratinocytes. Nat. Commun. 2014, 5, 5099. [Google Scholar] [CrossRef]
- Shah, N.M.; Zaitseva, L.; Bowles, K.M.; MacEwan, D.J.; A Rushworth, S. NRF2-driven miR-125B1 and miR-29B1 transcriptional regulation controls a novel anti-apoptotic miRNA regulatory network for AML survival. Cell Death Differ. 2014, 22, 654–664. [Google Scholar] [CrossRef]
- Perna, D.; Karreth, F.A.; Rust, A.G.; Perez-Mancera, P.A.; Rashid, M.; Iorio, F.; Alifrangis, C.; Arends, M.J.; Bosenberg, M.W.; Bollag, G.; et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc. Natl. Acad. Sci. USA 2015, 112, E536–E545. [Google Scholar] [CrossRef]
- Schönwasser, D.C.; Marais, R.M.; Marshall, C.J.; Parker, P.J. Activation of the Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Pathway by Conventional, Novel, and Atypical Protein Kinase C Isotypes. Mol. Cell. Biol. 1998, 18, 790–798. [Google Scholar] [CrossRef]
- Kunz, M.; Löffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef]
- Bok, I.; Vera, O.; Xu, X.; Jasani, N.; Nakamura, K.; Reff, J.; Nenci, A.; Gonzalez, J.G.; Karreth, F.A. A Versatile ES Cell–Based Melanoma Mouse Modeling Platform. Cancer Res. 2019, 80, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Cazalla, D.; Almstead, L.L.; Steitz, J.A.; DiMaio, D. miR-29 and miR-30 regulate B-Myb expression during cellular senescence. Proc. Natl. Acad. Sci. USA 2010, 108, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Klein, J.D.; Mitch, W.E.; Zhang, L.; Martinez, I.; Wang, X.H. MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging 2014, 6, 160–175. [Google Scholar] [CrossRef]
- Fang, M.; Hutchinson, L.; Deng, A.; Green, M.R. Common BRAF(V600E)-directed pathway mediates widespread epigenetic silencing in colorectal cancer and melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1250–1255. [Google Scholar] [CrossRef]
- Chen, B.; Wang, J.; Wang, J.; Wang, H.; Gu, X.; Tang, L.; Feng, X. A regulatory circuitry comprising TP53, miR-29 family, and SETDB1 in non-small cell lung cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Wu, G.S. The functional Interactions Between the MAPK and p53 Signaling Pathways. Cancer Biol. Ther. 2004, 3, 156–161. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Yu, H.; McDaid, R.; Lee, J.; Possik, P.; Li, L.; Kumar, S.M.; Elder, D.E.; Van Belle, P.; Gimotty, P.; Guerra, M.; et al. The Role of BRAF Mutation and p53 Inactivation during Transformation of a Subpopulation of Primary Human Melanocytes. Am. J. Pathol. 2009, 174, 2367–2377. [Google Scholar] [CrossRef]
- Ko, T.; Sharma, R.; Li, S. Genome-wide screening identifies novel genes implicated in cellular sensitivity to BRAFV600E expression. Oncogene 2019, 39, 723–738. [Google Scholar] [CrossRef]
- Lloyd, A.C.; Obermuller, F.; Staddon, S.; Barth, C.F.; McMahon, M.; Land, H. Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes Dev. 1997, 11, 663–677. [Google Scholar] [CrossRef]
- Sheu, J.J.-C.; Guan, B.; Tsai, F.-J.; Hsiao, E.Y.-T.; Chen, C.-M.; Seruca, R.; Wang, T.-L.; Shih, I.-M. Mutant BRAF Induces DNA Strand Breaks, Activates DNA Damage Response Pathway, and Up-Regulates Glucose Transporter-1 in Nontransformed Epithelial Cells. Am. J. Pathol. 2012, 180, 1179–1188. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF–MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.C.; Dias, S.D.R.; Marais, R. The role of B-RAF in melanoma. Cancer Metastasis Rev. 2005, 24, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Satyamoorthy, K.; Li, G.; Gerrero, M.R.; Brose, M.S.; Volpe, P.; Weber, B.L.; Van Belle, P.; Elder, D.E.; Herlyn, M. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003, 63, 756–759. [Google Scholar] [PubMed]
- Wu, J.; Rosenbaum, E.; Begum, S.; Westra, W.H. Distribution of BRAF T1799A(V600E) Mutations Across Various Types of Benign Nevi: Implications for Melanocytic Tumorigenesis. Am. J. Dermatopathol. 2007, 29, 534–537. [Google Scholar] [CrossRef]
- Yeh, I.; Von Deimling, A.; Bastian, B.C. Clonal BRAF Mutations in Melanocytic Nevi and Initiating Role of BRAF in Melanocytic Neoplasia. J. Natl. Cancer Inst. 2013, 105, 917–919. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nat. Cell Biol. 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Terzian, T.; Torchia, E.C.; Dai, D.; Robinson, S.E.; Murao, K.; Stiegmann, R.A.; Gonzalez, V.; Boyle, G.M.; Powell, M.B.; Pollock, P.M.; et al. p53 prevents progression of nevi to melanoma predominantly through cell cycle regulation. Pigment. Cell Melanoma Res. 2010, 23, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: A role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef]
- Viros, A.; Sanchez-Laorde, B.L.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nat. Cell Biol. 2014, 511, 478–482. [Google Scholar] [CrossRef]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment. Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Muthusamy, V.; Hobbs, C.; Nogueira, C.; Cordon-Cardo, C.; McKee, P.H.; Chin, L.; Bosenberg, M.W. Amplification ofCDK4 andMDM2 in malignant melanoma. Genes Chromosom. Cancer 2006, 45, 447–454. [Google Scholar] [CrossRef]
- Kogure, T.; Costinean, S.; Yan, I.; Braconi, C.; Croce, C.; Patel, T. Hepatic miR-29ab1 expression modulates chronic hepatic injury. J. Cell. Mol. Med. 2012, 16, 2647–2654. [Google Scholar] [CrossRef]
- Hwang, H.-W.; Wentzel, E.A.; Mendell, J.T. A Hexanucleotide Element Directs MicroRNA Nuclear Import. Science 2007, 315, 97–100. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, Z.; Qie, J.; Wang, Y.; Qian, J.; Wang, J.; Wu, J.; Li, Q.; Bai, C.; Han, B.; et al. Genetic polymorphism of SLC31A1 is associated with clinical outcomes of platinum-based chemotherapy in non-small-cell lung cancer patients through modulating microRNA-mediated regulation. Oncotarget 2018, 9, 23860–23877. [Google Scholar] [CrossRef]
- Matsuo, M.; Nakada, C.; Tsukamoto, Y.; Noguchi, T.; Uchida, T.; Hijiya, N.; Matsuura, K.; Moriyama, M. MiR-29c is downregulated in gastric carcinomas and regulates cell proliferation by targeting RCC2. Mol. Cancer 2013, 12, 15. [Google Scholar] [CrossRef]
- Vera, O.; Jimenez, J.; Pernia, O.; Rodríguez-Antolín, C.; Rodriguez, C.; Cabo, F.S.; Soto, J.; Rosas, R.; Lopez-Magallon, S.; Rodriguez, I.E.; et al. DNA Methylation of miR-7 is a Mechanism Involved in Platinum Response through MAFG Overexpression in Cancer Cells. Theranostics 2017, 7, 4118–4134. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yang, H.; Fan, W.; Tu, J.; Li, T.W.; Wang, J.; Shen, H.; Yang, J.; Xiong, T.; Steggerda, J.; et al. Mechanisms of MAFG Dysregulation in Cholestatic Liver Injury and Development of Liver Cancer. Gastroenterology 2018, 155, 557–571.e14. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Ou, J.; Hutchinson, L.; Green, M.R. The BRAF Oncoprotein Functions through the Transcriptional Repressor MAFG to Mediate the CpG Island Methylator Phenotype. Mol. Cell 2014, 55, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Thorner, A.R.; Hoadley, K.A.; Parker, J.S.; Winkel, S.; Millikan, R.C.; Perou, C.M. In vitro and in vivo analysis of B-Myb in basal-like breast cancer. Oncogene 2008, 28, 742–751. [Google Scholar] [CrossRef]
- Chen, J.; Chen, X. MYBL2 Is Targeted by miR-143-3p and Regulates Breast Cancer Cell Proliferation and Apoptosis. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 913–922. [Google Scholar] [CrossRef]
- Jin, Y.; Zhu, H.; Cai, W.; Fan, X.; Wang, Y.; Niu, Y.; Song, F.; Bu, Y. B-Myb Is Up-Regulated and Promotes Cell Growth and Motility in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 860. [Google Scholar] [CrossRef]
- Ren, F.; Wang, L.; Shen, X.; Xiao, X.; Liu, Z.; Wei, P.; Wang, Y.; Qi, P.; Shen, C.; Sheng, W.; et al. MYBL2 is an independent prognostic marker that has tumor-promoting functions in colorectal cancer. Am. J. Cancer Res. 2015, 5, 1542–1552. [Google Scholar] [PubMed]
- Cicirò, Y.; Sala, A. MYB oncoproteins: Emerging players and potential therapeutic targets in human cancer. Oncogene 2021, 10, 1–15. [Google Scholar] [CrossRef]
- Yuspa, S.; Harris, C. Altered differentiation of mouse epidermal cells treated with retinyl acetate in vitro. Exp. Cell Res. 1974, 86, 95–105. [Google Scholar] [CrossRef]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. J. Investig. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef]
- Karreth, F.A.; DeNicola, G.M.; Winter, S.P.; Tuveson, D.A. C-Raf Inhibits MAPK Activation and Transformation by B-RafV600E. Mol. Cell 2009, 36, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vera, O.; Bok, I.; Jasani, N.; Nakamura, K.; Xu, X.; Mecozzi, N.; Angarita, A.; Wang, K.; Tsai, K.Y.; Karreth, F.A. A MAPK/miR-29 Axis Suppresses Melanoma by Targeting MAFG and MYBL2. Cancers 2021, 13, 1408. https://doi.org/10.3390/cancers13061408
Vera O, Bok I, Jasani N, Nakamura K, Xu X, Mecozzi N, Angarita A, Wang K, Tsai KY, Karreth FA. A MAPK/miR-29 Axis Suppresses Melanoma by Targeting MAFG and MYBL2. Cancers. 2021; 13(6):1408. https://doi.org/10.3390/cancers13061408
Chicago/Turabian StyleVera, Olga, Ilah Bok, Neel Jasani, Koji Nakamura, Xiaonan Xu, Nicol Mecozzi, Ariana Angarita, Kaizhen Wang, Kenneth Y. Tsai, and Florian A. Karreth. 2021. "A MAPK/miR-29 Axis Suppresses Melanoma by Targeting MAFG and MYBL2" Cancers 13, no. 6: 1408. https://doi.org/10.3390/cancers13061408
APA StyleVera, O., Bok, I., Jasani, N., Nakamura, K., Xu, X., Mecozzi, N., Angarita, A., Wang, K., Tsai, K. Y., & Karreth, F. A. (2021). A MAPK/miR-29 Axis Suppresses Melanoma by Targeting MAFG and MYBL2. Cancers, 13(6), 1408. https://doi.org/10.3390/cancers13061408