
Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel

 ,
,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Role of Genomic Instability in Tumorigenesis
1.2. DNA Repair Deregulation Triggers Genomic Instability
2. Alt-NHEJ and De-Regulation in Cancer
2.1. Background on Alt-NHEJ Machinery Repair
2.2. Transcriptional and Post-Transcriptional Alt-NHEJ Regulation
3. Alt-NHEJ Involvement in Tumors: An Overview
3.1. Ovarian Cancer
3.2. Breast Cancer
3.3. Neuroblastoma
3.4. Acute Leukemias
3.5. Chronic Myeloid Leukemia
3.6. Multiple Myeloma
4. Targeting Alt-NHEJ Repair
4.1. Drugging Alt-NHEJ Major Proteins
4.2. Alt-NHEJ Could Stimulate Immune Recognition of Tumor Cells
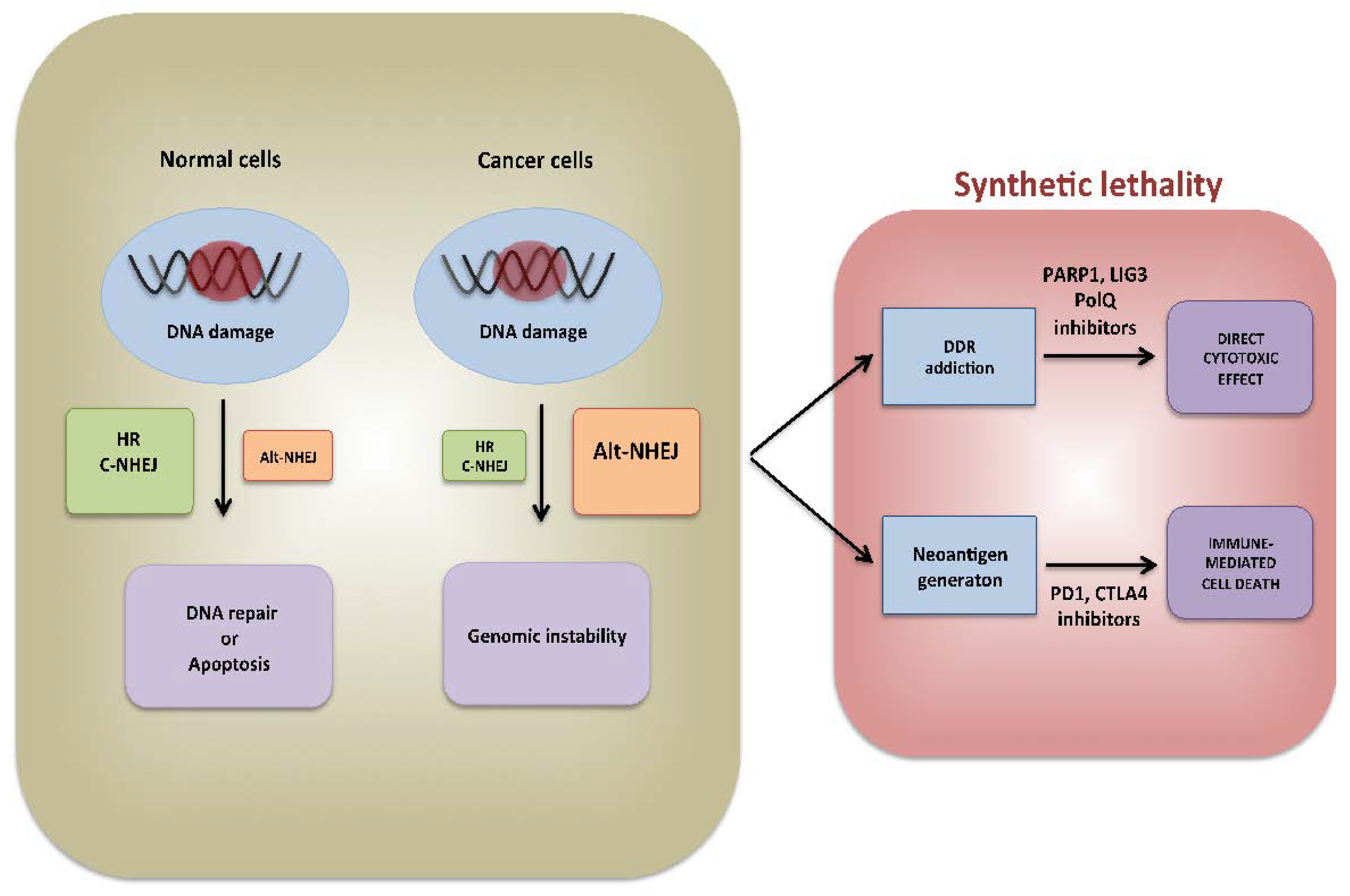
5. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. Chromosomal biomarkers of genomic instability relevant to cancer. Drug Discov. Today 2002, 7, 1128–1137. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst.) 2006, 5, 1042–1048. [Google Scholar] [CrossRef]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Rodriguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2015, 793, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Du, Z.; Wang, Y.; Feng, Z.; Fan, P.; Yan, C.; Willers, H.; Zhang, J. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res. 2015, 43, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, C.; Chen, S.H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef]
- Okano, S.; Lan, L.; Tomkinson, A.E.; Yasui, A. Translocation of XRCC1 and DNA ligase IIIalpha from centrosomes to chromosomes in response to DNA damage in mitotic human cells. Nucleic Acids Res. 2005, 33, 422–429. [Google Scholar] [CrossRef][Green Version]
- Della-Maria, J.; Zhou, Y.; Tsai, M.S.; Kuhnlein, J.; Carney, J.P.; Paull, T.T.; Tomkinson, A.E. Human Mre11/human Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J. Biol. Chem. 2011, 286, 33845–33853. [Google Scholar] [CrossRef]
- Lu, G.; Duan, J.; Shu, S.; Wang, X.; Gao, L.; Guo, J.; Zhang, Y. Ligase I and ligase III mediate the DNA double-strand break ligation in alternative end-joining. Proc. Natl. Acad. Sci. USA 2016, 113, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Paddock, M.N.; Bauman, A.T.; Higdon, R.; Kolker, E.; Takeda, S.; Scharenberg, A.M. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair (Amst.) 2011, 10, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Kent, T.; Deng, S.K.; McDevitt, S.; Kashkina, E.; Hoang, T.M.; Pomerantz, R.T.; Sfeir, A. The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol. 2017, 24, 1116–1123. [Google Scholar] [CrossRef]
- Ahrabi, S.; Sarkar, S.; Pfister, S.X.; Pirovano, G.; Higgins, G.S.; Porter, A.C.; Humphrey, T.C. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. 2016, 44, 5743–5757. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Ruan, C.; Huen, M.S.Y.; Wang, J.; Xie, A.; Fu, C.; Liu, T.; Huang, J. BRCA2 antagonizes classical and alternative nonhomologous end-joining to prevent gross genomic instability. Nat. Commun. 2017, 8, 1470. [Google Scholar] [CrossRef]
- Kee, Y.; D’Andrea, A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010, 24, 1680–1694. [Google Scholar] [CrossRef]
- Dutta, A.; Eckelmann, B.; Adhikari, S.; Ahmed, K.M.; Sengupta, S.; Pandey, A.; Hegde, P.M.; Tsai, M.S.; Tainer, J.A.; Weinfeld, M.; et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2017, 45, 2585–2599. [Google Scholar] [CrossRef]
- Simsek, D.; Furda, A.; Gao, Y.; Artus, J.; Brunet, E.; Hadjantonakis, A.K.; Van Houten, B.; Shuman, S.; McKinnon, P.J.; Jasin, M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011, 471, 245–248. [Google Scholar] [CrossRef]
- Arana, M.E.; Seki, M.; Wood, R.D.; Rogozin, I.B.; Kunkel, T.A. Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res. 2008, 36, 3847–3856. [Google Scholar] [CrossRef]
- Wood, R.D.; Doublie, S. DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair (Amst.) 2016, 44, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Jiang, G.; Willers, H.; Xia, F. Exonuclease function of human Mre11 promotes deletional nonhomologous end joining. J. Biol. Chem. 2009, 284, 30565–30573. [Google Scholar] [CrossRef]
- Taylor, R.M.; Whitehouse, C.J.; Caldecott, K.W. The DNA ligase III zinc finger stimulates binding to DNA secondary structure and promotes end joining. Nucleic Acids Res. 2000, 28, 3558–3563. [Google Scholar] [CrossRef][Green Version]
- Muvarak, N.; Kelley, S.; Robert, C.; Baer, M.R.; Perrotti, D.; Gambacorti-Passerini, C.; Civin, C.; Scheibner, K.; Rassool, F.V. c-MYC Generates Repair Errors via Increased Transcription of Alternative-NHEJ Factors, LIG3 and PARP1, in Tyrosine Kinase-Activated Leukemias. Mol. Cancer Res. 2015, 13, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Lu, Y.; Lv, J.; Wu, H.; Lu, J.; Zhang, C.; Zhang, S.; Bao, Q.; Zhang, X.; Xie, C.; et al. The long noncoding RNA lncPARP1 contributes to progression of hepatocellular carcinoma through up-regulation of PARP1. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Stamato, M.A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M.E.G.; Romeo, E.; et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 2018, 32, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Raimondi, L.; Juli, G.; Stamato, M.A.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. MALAT1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018, 11, 63. [Google Scholar] [CrossRef]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef]
- Neff, R.T.; Senter, L.; Salani, R. BRCA mutation in ovarian cancer: Testing, implications and treatment considerations. Ther. Adv. Med. Oncol. 2017, 9, 519–531. [Google Scholar] [CrossRef]
- Tagliaferri, P.; Ventura, M.; Baudi, F.; Cucinotto, I.; Arbitrio, M.; Di Martino, M.T.; Tassone, P. BRCA1/2 genetic background-based therapeutic tailoring of human ovarian cancer: Hope or reality? J. Ovarian Res. 2009, 2, 14. [Google Scholar] [CrossRef]
- Staropoli, N.; Ciliberto, D.; Del Giudice, T.; Iuliano, E.; Cuce, M.; Grillone, F.; Salvino, A.; Barbieri, V.; Russo, A.; Tassone, P.; et al. The Era of PARP inhibitors in ovarian cancer: “Class Action” or not? A systematic review and meta-analysis. Crit. Rev. Oncol./Hematol. 2018, 131, 83–89. [Google Scholar] [CrossRef]
- Santana Dos Santos, E.; Lallemand, F.; Petitalot, A.; Caputo, S.M.; Rouleau, E. HRness in Breast and Ovarian Cancers. Int. J. Mol. Sci. 2020, 21, 3850. [Google Scholar] [CrossRef]
- De Talhouet, S.; Peron, J.; Vuilleumier, A.; Friedlaender, A.; Viassolo, V.; Ayme, A.; Bodmer, A.; Treilleux, I.; Lang, N.; Tille, J.C.; et al. Clinical outcome of breast cancer in carriers of BRCA1 and BRCA2 mutations according to molecular subtypes. Sci. Rep. 2020, 10, 7073. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Tassone, P.; Tagliaferri, P.; Perricelli, A.; Blotta, S.; Quaresima, B.; Martelli, M.L.; Goel, A.; Barbieri, V.; Costanzo, F.; Boland, C.R.; et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer 2003, 88, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Tassone, P.; Blotta, S.; Palmieri, C.; Masciari, S.; Quaresima, B.; Montagna, M.; D’Andrea, E.; Eramo, O.P.; Migale, L.; Costanzo, F.; et al. Differential sensitivity of BRCA1-mutated HCC1937 human breast cancer cells to microtubule-interfering agents. Int. J. Oncol. 2005, 26, 1257–1263. [Google Scholar] [CrossRef]
- Tassone, P.; Di Martino, M.T.; Ventura, M.; Pietragalla, A.; Cucinotto, I.; Calimeri, T.; Bulotta, A.; Neri, P.; Caraglia, M.; Tagliaferri, P. Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol. Ther. 2009, 8, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Tobin, L.A.; Robert, C.; Nagaria, P.; Chumsri, S.; Twaddell, W.; Ioffe, O.B.; Greco, G.E.; Brodie, A.H.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA repair in therapy-resistant breast cancers. Mol. Cancer Res. 2012, 10, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Caren, H.; Kryh, H.; Nethander, M.; Sjoberg, R.M.; Trager, C.; Nilsson, S.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 2010, 107, 4323–4328. [Google Scholar] [CrossRef]
- Newman, E.A.; Lu, F.; Bashllari, D.; Wang, L.; Opipari, A.W.; Castle, V.P. Alternative NHEJ Pathway Components Are Therapeutic Targets in High-Risk Neuroblastoma. Mol. Cancer Res. 2015, 13, 470–482. [Google Scholar] [CrossRef]
- Newman, E.A.; Chukkapalli, S.; Bashllari, D.; Thomas, T.T.; Van Noord, R.A.; Lawlor, E.R.; Hoenerhoff, M.J.; Opipari, A.W.; Opipari, V.P. Alternative NHEJ pathway proteins as components of MYCN oncogenic activity in human neural crest stem cell differentiation: Implications for neuroblastoma initiation. Cell Death Dis. 2017, 8, 3208. [Google Scholar] [CrossRef]
- Pashaiefar, H.; Yaghmaie, M.; Tavakkoly-Bazzaz, J.; Hamidollah Ghaffari, S.; Alimoghaddam, K.; Izadi, P.; Ghavamzadeh, A. The Association between PARP1 and LIG3 Expression Levels and Chromosomal Translocations in Acute Myeloid Leukemia Patients. Cell J. 2018, 20, 204–210. [Google Scholar] [CrossRef]
- Fan, J.; Li, L.; Small, D.; Rassool, F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: Implications for genomic instability and therapy. Blood 2010, 116, 5298–5305. [Google Scholar] [CrossRef]
- Hahnel, P.S.; Enders, B.; Sasca, D.; Roos, W.P.; Kaina, B.; Bullinger, L.; Theobald, M.; Kindler, T. Targeting components of the alternative NHEJ pathway sensitizes KRAS mutant leukemic cells to chemotherapy. Blood 2014, 123, 2355–2366. [Google Scholar] [CrossRef]
- Sallmyr, A.; Tomkinson, A.E.; Rassool, F.V. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: Consequences for the repair of DNA double-strand breaks. Blood 2008, 112, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Di Martino, M.T.; Amodio, N.; Morelli, E.; Montesano, M.; Botta, C.; Scionti, F.; Talarico, D.; Altomare, E.; Gallo Cantafio, M.E.; et al. miR-22 suppresses DNA ligase III addiction in multiple myeloma. Leukemia 2019, 33, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Montesano, M.; Altomare, E.; Scionti, F.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. The potential role of miRNAs in multiple myeloma therapy. Expert Rev. Hematol. 2018, 11, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Gulla, A.; Di Martino, M.T.; Gallo Cantafio, M.E.; Morelli, E.; Amodio, N.; Botta, C.; Pitari, M.R.; Lio, S.G.; Britti, D.; Stamato, M.A.; et al. A 13 mer LNA-i-miR-221 Inhibitor Restores Drug Sensitivity in Melphalan-Refractory Multiple Myeloma Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1222–1233. [Google Scholar] [CrossRef]
- Lionetti, M.; Musto, P.; Di Martino, M.T.; Fabris, S.; Agnelli, L.; Todoerti, K.; Tuana, G.; Mosca, L.; Gallo Cantafio, M.E.; Grieco, V.; et al. Biological and clinical relevance of miRNA expression signatures in primary plasma cell leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3130–3142. [Google Scholar] [CrossRef]
- Amodio, N.; Di Martino, M.T.; Neri, A.; Tagliaferri, P.; Tassone, P. Non-coding RNA: A novel opportunity for the personalized treatment of multiple myeloma. Expert Opin. Biol. Ther. 2013, 13 (Suppl. 1), S125–S137. [Google Scholar] [CrossRef]
- Amodio, N.; Gallo Cantafio, M.E.; Botta, C.; Agosti, V.; Federico, C.; Caracciolo, D.; Ronchetti, D.; Rossi, M.; Driessen, C.; Neri, A.; et al. Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma. Cancers 2019, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica 2020, 106, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [PubMed]
- Burdak-Rothkamm, S.; Rothkamm, K. DNA Damage Repair Deficiency and Synthetic Lethality for Cancer Treatment. Trends Mol. Med. 2021, 27, 91–92. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- O’Sullivan, C.C.; Moon, D.H.; Kohn, E.C.; Lee, J.M. Beyond Breast and Ovarian Cancers: PARP Inhibitors for BRCA Mutation-Associated and BRCA-Like Solid Tumors. Front. Oncol. 2014, 4, 42. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef]
- Garcia-Diaz, M.; Bebenek, K. Multiple functions of DNA polymerases. Crit. Rev. Plant Sci. 2007, 26, 105–122. [Google Scholar] [CrossRef]
- Kawamura, K.; Bahar, R.; Seimiya, M.; Chiyo, M.; Wada, A.; Okada, S.; Hatano, M.; Tokuhisa, T.; Kimura, H.; Watanabe, S.; et al. DNA polymerase theta is preferentially expressed in lymphoid tissues and upregulated in human cancers. Int. J. Cancer 2004, 109, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.S.; Harris, A.L.; Prevo, R.; Helleday, T.; McKenna, W.G.; Buffa, F.M. Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget 2010, 1, 175–184. [Google Scholar] [CrossRef]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem. 2008, 77, 313–338. [Google Scholar] [CrossRef]
- Howes, T.R.; Tomkinson, A.E. DNA ligase I, the replicative DNA ligase. Sub-Cell. Biochem. 2012, 62, 327–341. [Google Scholar] [CrossRef]
- Grawunder, U.; Zimmer, D.; Fugmann, S.; Schwarz, K.; Lieber, M.R. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell 1998, 2, 477–484. [Google Scholar] [CrossRef]
- Wang, H.; Rosidi, B.; Perrault, R.; Wang, M.; Zhang, L.; Windhofer, F.; Iliakis, G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005, 65, 4020–4030. [Google Scholar] [CrossRef]
- Simsek, D.; Jasin, M. DNA ligase III: A spotty presence in eukaryotes, but an essential function where tested. Cell Cycle 2011, 10, 3636–3644. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, S.; Zhu, X.; Dziegielewska, B.; Ellenberger, T.; Wilson, G.M.; MacKerell, A.D., Jr.; Tomkinson, A.E. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008, 68, 3169–3177. [Google Scholar] [CrossRef]
- Tomkinson, A.E.; Howes, T.R.; Wiest, N.E. DNA ligases as therapeutic targets. Transl. Cancer Res. 2013, 2, 1219. [Google Scholar]
- Caracciolo, D.; Riillo, C.; Arbitrio, M.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Error-prone DNA repair pathways as determinants of immunotherapy activity: An emerging scenario for cancer treatment. Int. J. Cancer 2020, 147, 2658–2668. [Google Scholar] [CrossRef]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Hartlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kroger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Pefani, D.E.; Pateras, I.S.; Trougakos, I.P. Integrating the DNA damage and protein stress responses during cancer development and treatment. J. Pathol. 2018, 246, 12–40. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Hanscom, T.; McVey, M. Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution. Cells 2020, 9, 1657. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef]
- Carvajal-Garcia, J.; Cho, J.E.; Carvajal-Garcia, P.; Feng, W.; Wood, R.D.; Sekelsky, J.; Gupta, G.P.; Roberts, S.A.; Ramsden, D.A. Mechanistic basis for microhomology identification and genome scarring by polymerase theta. Proc. Natl. Acad. Sci. USA 2020, 117, 8476–8485. [Google Scholar] [CrossRef] [PubMed]
- Arbitrio, M.; Scionti, F.; Di Martino, M.T.; Caracciolo, D.; Pensabene, L.; Tassone, P.; Tagliaferri, P. Pharmacogenomics Biomarker Discovery and Validation for Translation in Clinical Practice. Clin. Transl. Sci. 2021, 14, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Gallo Cantafio, M.E.; Grillone, K.; Caracciolo, D.; Scionti, F.; Arbitrio, M.; Barbieri, V.; Pensabene, L.; Guzzi, P.H.; Di Martino, M.T. From Single Level Analysis to Multi-Omics Integrative Approaches: A Powerful Strategy towards the Precision Oncology. High-Throughput 2018, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Nyquist, M.D.; Corella, A.; Coleman, I.; De Sarkar, N.; Kaipainen, A.; Ha, G.; Gulati, R.; Ang, L.; Chatterjee, P.; Lucas, J.; et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep. 2020, 31, 107669. [Google Scholar] [CrossRef]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Ovarian Cancer | Breast Cancer | Pancreatic Cancer | Castration-Resistant Prostate Cancer |
|---|---|---|---|---|
| Olaparib | Treatment: -Germline BRCA 1/2 mutations -Following 3 or more line of therapy (NCT01078662) Maintenance: -Germline or somatic BRCA 1/2 mutations (first-line maintenance) -Also recurrent cancer without BRCA mutations -Advanced cancer with complete or partial response to first-line platinum-based chemotherapy (NCT01874353) | Treatment: -Germline BRCA 1/2 mutations -HER2 negative metastatic patients treated with chemo -Hormone receptor positive cancer treated with endocrine therapy or inappropriate for endocrine therapy (NCT02000622) | Maintenance therapy: -Germline BRCA 1/2 mutations (first-line maintenance) -Metastatic pancreatic adenocarcinoma with no disease progression on at least 16 weeks first-line platinum-based chemotherapy (NCT02184195) | Treatment: -HRR gene mutations -Metastatic castration-resistant prostate cancer who had disease progression while receiving enzalutamide or abiraterone. (NCT02987543) |
| Rucaparib | Treatment: -Germline BRCA 1/2 mutations -Following 2 or more rounds of chemotherapy (NCT01891344) Maintenance: -Recurrent cancer with complete or partial response to platinum-based chemotherapy -Regardless of BRCA mutation (NCT01968213) | Treatment: -adult patients with a deleterious BRCA mutation (germline and/or somatic) -metastatic castration-resistant prostate cancer (mCRPC) who have been treated with androgen receptor-directed therapy and a taxane-based chemotherapy. (NCT02952534) | ||
| Niraparib | Treatment: -Homologous recombination deficiency positive status: BRCA mutation or Genomic instability and progression >6 months after response to the last platinum-based chemotherapy -Advanced cancer treated with 3 or more rounds of chemotherapy (NCT02354586) Maintenance: -Recurrent cancer with complete or partial response to platinum-based chemotherapy Regardless of BRCA mutation (NCT02655016) | |||
| Talazoparib | Treatment: -Germline BRCA1/2 mutations -HER2 negative metastatic cancer patients (NCT01945775) | |||
| Veliparib | Treatment and maintenance: -Veliparib With Carboplatin and Paclitaxel -Newly Diagnosed Stage III or IV, High-grade Serous, Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer (NCT02470585) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers 2021, 13, 1392. https://doi.org/10.3390/cancers13061392
Caracciolo D, Riillo C, Di Martino MT, Tagliaferri P, Tassone P. Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers. 2021; 13(6):1392. https://doi.org/10.3390/cancers13061392
Chicago/Turabian StyleCaracciolo, Daniele, Caterina Riillo, Maria Teresa Di Martino, Pierosandro Tagliaferri, and Pierfrancesco Tassone. 2021. "Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel" Cancers 13, no. 6: 1392. https://doi.org/10.3390/cancers13061392
APA StyleCaracciolo, D., Riillo, C., Di Martino, M. T., Tagliaferri, P., & Tassone, P. (2021). Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers, 13(6), 1392. https://doi.org/10.3390/cancers13061392