
Comprehensive Genomic Characterization of Fifteen Early-Onset Lynch-Like Syndrome Colorectal Cancers

 , , ,
, , ,  , ,
, ,  , , ,
, , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
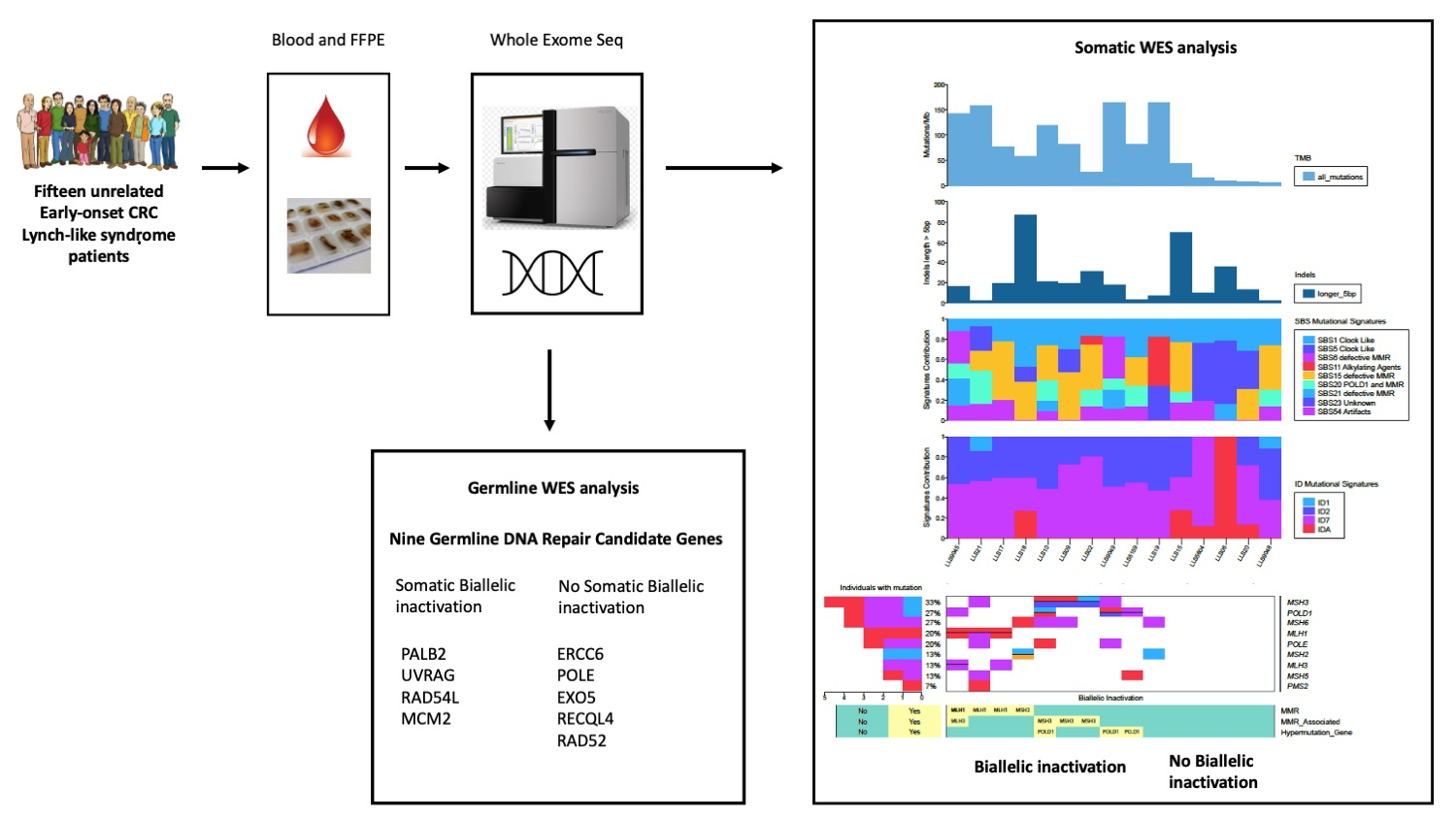
2.1. Patients
2.2. Somatic Biallelic Alterations in MMR Core and MMR-Associated Genes
2.3. Somatic Monoallelic Alterations in MMR Core and MMR-Associated Genes
2.4. Somatic Alterations in Additional Cancer Genes
2.5. MSH3 and MSI Association
2.6. Tumor Mutational Burden (TMB)
2.7. Mutational Signatures
2.8. Germline Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Whole Exome Sequencing
4.3. Mutational Profiling and Mutational Signature Analysis
4.4. Variant Calling and Filtering
4.5. Germline Variant Prioritization and Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Jakubowski, C.D.; Fedewa, S.A.; Davis, A.; Azad, N.S. Colorectal Cancer in the Young: Epidemiology, Prevention, Management. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Young, J.P.; Win, A.K.; Rosty, C.; Flight, I.; Roder, D.; Young, G.P.; Frank, O.; Suthers, G.K.; Hewett, P.J.; Ruszkiewicz, A.; et al. Rising incidence of early-onset colorectal cancer in Australia over two decades: Report and review. J. Gastroenterol. Hepatol. 2015, 30, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Vuik, F.E.; Nieuwenburg, S.A.; Bardou, M.; Lansdorp-Vogelaar, I.; Dinis-Ribeiro, M.; Bento, M.J.; Zadnik, V.; Pellisé, M.; Esteban, L.; Kaminski, M.F.; et al. Increasing incidence of colorectal cancer in young adults in Europe over the last 25 years. Gut 2019, 68, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef]
- Schmeler, K.M.; Lynch, H.T.; Chen, L.-M.; Munsell, M.F.; Soliman, P.T.; Clark, M.B.; Daniels, M.S.; White, K.G.; Boyd-Rogers, S.G.; Conrad, P.G.; et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N. Engl. J. Med. 2006, 354, 261–269. [Google Scholar] [CrossRef]
- Biller, L.H.; Syngal, S.; Yurgelun, M.B. Recent advances in Lynch syndrome. Fam. Cancer 2019, 18, 211–219. [Google Scholar] [CrossRef]
- Antelo, M.; Golubicki, M.; Roca, E.; Mendez, G.; Carballido, M.; Iseas, S.; Cuatrecasas, M.; Moreira, L.; Sanchez, A.; Carballal, S.; et al. Lynch-like syndrome is as frequent as Lynch syndrome in early-onset nonfamilial nonpolyposis colorectal cancer. Int. J. Cancer 2019, 145, 705–713. [Google Scholar] [CrossRef]
- Rodríguez-Soler, M.; Pérez-Carbonell, L.; Guarinos, C.; Zapater, P.; Castillejo, A.; Barberá, V.M.; Juárez, M.; Bessa, X.; Xicola, R.M.; Clofent, J.; et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology 2013, 144, 926–932.e1, quiz e13–e14. [Google Scholar] [CrossRef]
- Morak, M.; Heidenreich, B.; Keller, G.; Hampel, H.; Laner, A.; de la Chapelle, A.; Holinski-Feder, E. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur. J. Hum. Genet. 2014, 22, 1334–1337. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; van den Akker, B.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2015, 23, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.M.; van Wezel, T.; van den Akker, B.E.; Ventayol Garcia, M.; Ruano, D.; Tops, C.M.; Wagner, A.; Letteboer, T.G.; Gómez-García, E.B.; Devilee, P.; et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur. J. Hum. Genet. 2016, 24, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Sourrouille, I.; Coulet, F.; Lefevre, J.H.; Colas, C.; Eyries, M.; Svrcek, M.; Bardier-Dupas, A.; Parc, Y.; Soubrier, F. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam. Cancer 2013, 12, 27–33. [Google Scholar] [CrossRef]
- Mensenkamp, A.R.; Vogelaar, I.P.; van Zelst-Stams, W.A.G.; Goossens, M.; Ouchene, H.; Hendriks-Cornelissen, S.J.B.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J.L. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology 2014, 146, 643–646.e8. [Google Scholar] [CrossRef]
- Geurts-Giele, W.R.R.; Leenen, C.H.M.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.B.M.; Kuipers, E.J.; Goverde, A.; van den Ouweland, A.M.W.; van Lier, M.G.F.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014, 147, 1308–1316.e1. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Golubicki, M.; Bonjoch, L.; Acuña-Ochoa, J.G.; Díaz-Gay, M.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Cisterna, D.; et al. Germline biallelic Mcm8 variants are associated with early-onset Lynch-like syndrome. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.-R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e20. [Google Scholar] [CrossRef]
- Pérez-Carbonell, L.; Ruiz-Ponte, C.; Guarinos, C.; Alenda, C.; Payá, A.; Brea, A.; Egoavil, C.M.; Castillejo, A.; Barberá, V.M.; Bessa, X.; et al. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut 2012, 61, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Pearlman, R.; Beightol, M.; Zhao, W.; Jones, D.; Frankel, W.L.; Goodfellow, P.J.; Yilmaz, A.; Miller, K.; Bacher, J.; et al. Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol. 2018, 4, 806–813. [Google Scholar] [CrossRef]
- Xicola, R.M.; Clark, J.R.; Carroll, T.; Alvikas, J.; Marwaha, P.; Regan, M.R.; Lopez-Giraldez, F.; Choi, J.; Emmadi, R.; Alagiozian-Angelova, V.; et al. Implication of DNA repair genes in Lynch-like syndrome. Fam. Cancer 2019, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Porkka, N.; Lahtinen, L.; Ahtiainen, M.; Böhm, J.P.; Kuopio, T.; Eldfors, S.; Mecklin, J.-P.; Seppälä, T.T.; Peltomäki, P. Epidemiological, clinical and molecular characterization of Lynch-like syndrome: A population-based study. Int. J. Cancer 2019, 145, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, A.; Bowen, N.; Kolodner, R.D. Mispair-specific recruitment of the Mlh1-Pms1 complex identifies repair substrates of the Saccharomyces cerevisiae Msh2-Msh3 complex. J. Biol. Chem. 2014, 289, 9352–9364. [Google Scholar] [CrossRef]
- Haugen, A.C.; Goel, A.; Yamada, K.; Marra, G.; Nguyen, T.-P.; Nagasaka, T.; Kanazawa, S.; Koike, J.; Kikuchi, Y.; Zhong, X.; et al. Genetic instability caused by loss of MutS homologue 3 in human colorectal cancer. Cancer Res. 2008, 68, 8465–8472. [Google Scholar] [CrossRef]
- Plaschke, J.; Preußler, M.; Ziegler, A.; Schackert, H.K. Aberrant protein expression and frequent allelic loss of MSH3 in colorectal cancer with low-level microsatellite instability. Int. J. Colorectal Dis. 2012, 27, 911–919. [Google Scholar] [CrossRef]
- Hile, S.E.; Shabashev, S.; Eckert, K.A. Tumor-specific microsatellite instability: Do distinct mechanisms underlie the MSI-L and EMAST phenotypes? Mutat. Res. 2013, 743–744, 67–77. Mutat. Res. 2013, 743–744, 67–77. [Google Scholar] [CrossRef]
- Edelmann, W.; Umar, A.; Yang, K.; Heyer, J.; Kucherlapati, M.; Lia, M.; Kneitz, B.; Avdievich, E.; Fan, K.; Wong, E.; et al. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res. 2000, 60, 803–807. [Google Scholar]
- Hegan, D.C.; Narayanan, L.; Jirik, F.R.; Edelmann, W.; Liskay, R.M.; Glazer, P.M. Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6. Carcinogenesis 2006, 27, 2402–2408. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.M.; Lea, D.; Rewcastle, E.; Hagland, H.R.; Søreide, K. Elevated microsatellite alterations at selected tetranucleotides in early-stage colorectal cancers with and without high-frequency microsatellite instability: Same, same but different? Cancer Med. 2016, 5, 1580–1587. [Google Scholar] [CrossRef]
- Orimo, H.; Nakajima, E.; Yamamoto, M.; Ikejima, M.; Emi, M.; Shimada, T. Association between single nucleotide polymorphisms in the hMSH3 gene and sporadic colon cancer with microsatellite instability. J. Hum. Genet. 2000, 45, 228–230. [Google Scholar] [CrossRef][Green Version]
- Berndt, S.I.; Platz, E.A.; Fallin, M.D.; Thuita, L.W.; Hoffman, S.C.; Helzlsouer, K.J. Mismatch repair polymorphisms and the risk of colorectal cancer. Int. J. Cancer 2007, 120, 1548–1554. [Google Scholar] [CrossRef]
- Hirata, H.; Hinoda, Y.; Kawamoto, K.; Kikuno, N.; Suehiro, Y.; Okayama, N.; Tanaka, Y.; Dahiya, R. Mismatch repair gene MSH3 polymorphism is associated with the risk of sporadic prostate cancer. J. Urol. 2008, 179, 2020–2024. [Google Scholar] [CrossRef]
- Adam, R.; Spier, I.; Zhao, B.; Kloth, M.; Marquez, J.; Hinrichsen, I.; Kirfel, J.; Tafazzoli, A.; Horpaopan, S.; Uhlhaas, S.; et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am. J. Hum. Genet. 2016, 99, 337–351. [Google Scholar] [CrossRef]
- Mur, P.; García-Mulero, S.; Del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020. [Google Scholar] [CrossRef]
- Salem, M.E.; Weinberg, B.A.; Xiu, J.; El-Deiry, W.S.; Hwang, J.J.; Gatalica, Z.; Philip, P.A.; Shields, A.F.; Lenz, H.-J.; Marshall, J.L. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget 2017, 8, 86356–86368. [Google Scholar] [CrossRef]
- Salem, M.E.; Puccini, A.; Grothey, A.; Raghavan, D.; Goldberg, R.M.; Xiu, J.; Korn, W.M.; Weinberg, B.A.; Hwang, J.J.; Shields, A.F.; et al. Landscape of Tumor Mutation Load, Mismatch Repair Deficiency, and PD-L1 Expression in a Large Patient Cohort of Gastrointestinal Cancers. Mol. Cancer Res. 2018, 16, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.C.; Lin, M.-T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Rigter, L.S.; Snaebjornsson, P.; Rosenberg, E.H.; Atmodimedjo, P.N.; Aleman, B.M.; Ten Hoeve, J.; Geurts-Giele, W.R.; van Ravesteyn, T.W.; Hoeksel, J.; PALGA Group; et al. Double somatic mutations in mismatch repair genes are frequent in colorectal cancer after Hodgkin’s lymphoma treatment. Gut 2018, 67, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Pich, O.; Muiños, F.; Lolkema, M.P.; Steeghs, N.; Gonzalez-Perez, A.; Lopez-Bigas, N. The mutational footprints of cancer therapies. Nat. Genet. 2019, 51, 1732–1740. [Google Scholar] [CrossRef]
- Arora, S.; Yan, H.; Cho, I.; Fan, H.-Y.; Luo, B.; Gai, X.; Bodian, D.L.; Vockley, J.G.; Zhou, Y.; Handorf, E.A.; et al. Genetic Variants That Predispose to DNA Double-Strand Breaks in Lymphocytes From a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology 2015, 149, 1872–1883.e9. [Google Scholar] [CrossRef]
- Kohzaki, M.; Ootsuyama, A.; Sun, L.; Moritake, T.; Okazaki, R. Human RECQL4 represses the RAD52-mediated single-strand annealing pathway after ionizing radiation or cisplatin treatment. Int. J. Cancer 2020, 146, 3098–3113. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Kundu, S.; Laskar, S.; Choudhury, Y.; Ghosh, S.K. Assessment of DNA repair susceptibility genes identified by whole exome sequencing in head and neck cancer. DNA Repair 2018, 66–67, 50–63. DNA Repair 2018, 66–67, 50–63. [Google Scholar] [CrossRef]
- Ui, A.; Chiba, N.; Yasui, A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020, 111, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef]
- Fei, L.; Xu, H. Role of MCM2-7 protein phosphorylation in human cancer cells. Cell Biosci. 2018, 8, 43. [Google Scholar] [CrossRef]
- Mazin, A.V.; Mazina, O.M.; Bugreev, D.V.; Rossi, M.J. Rad54, the motor of homologous recombination. DNA Repair 2010, 9, 286–302. [Google Scholar] [CrossRef]
- Quach, C.; Song, Y.; Guo, H.; Li, S.; Maazi, H.; Fung, M.; Sands, N.; O’Connell, D.; Restrepo-Vassalli, S.; Chai, B.; et al. A truncating mutation in the autophagy gene UVRAG drives inflammation and tumorigenesis in mice. Nat. Commun. 2019, 10, 5681. [Google Scholar] [CrossRef]
- Ali, S.; Zhang, Y.; Zhou, M.; Li, H.; Jin, W.; Zheng, L.; Yu, X.; Stark, J.M.; Weitzel, J.N.; Shen, B. Functional deficiency of DNA repair gene EXO5 results in androgen-induced genomic instability and prostate tumorigenesis. Oncogene 2020, 39, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Li, H.; Li, S.; Chen, L.; Li, S.C. Oviz-Bio: A web-based platform for interactive cancer genomics data visualization. Nucleic Acids Res. 2020, 48, W415–W426. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Jurado, C.; Vila-Casadesús, M.; Garre, P.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Muñoz, J.; Ocaña, T.; Balaguer, F.; López-Cerón, M.; et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med. 2015, 17, 131–142. [Google Scholar] [CrossRef]
- Díaz-Gay, M.; Franch-Expósito, S.; Arnau-Collell, C.; Park, S.; Supek, F.; Muñoz, J.; Bonjoch, L.; Gratacós-Mulleras, A.; Sánchez-Rojas, P.A.; Esteban-Jurado, C.; et al. Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer. Cancers 2019, 11, 362. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Ladabaum, U. What Is Lynch-like Syndrome and How Should We Manage It? Clin. Gastroenterol. Hepatol. 2020, 18, 294–296. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Patient ID | Cohort | Age of Onset (Years) | Sex | Tumor Location | CRC Relation to Splenic Flexure | Stage | Histological Differentiation | Bethesda Clinical Criteria | MSI Status | MMR Protein Loss | Somatic Braf v600e | Biallelic Somatic Alteration Gene | TMB (Mutations/mb) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LLS02 | Udaondo | 12 | male | rectum | Left | IV | well | 1 | MSI | MSH2/MSH6 | WT | MSH3 | 26.7 |
| LLS06 | Udaondo | 30 | female | ascending colon | Right | IIIA | moderately | 1;5 | MSS | MSH6 | WT | no alteration | 9.4 |
| LLS09 | Udaondo | 30 | female | rectum | Left | IIIA | moderately | 1;5 | MSI | MSH2/MSH6 | WT | MSH3 | 81.2 |
| LLS10 | Udaondo | 25 | female | descending colon | Left | IIB | moderately | 1;3 | MSI | MSH2/MSH6 | WT | MSH3; POLD1 | 118 |
| LLS15 | Udaondo | 19 | male | descending colon | Left | IIA | no data | 1;3 | MSI | MLH1/PMS2 | WT | no alteration | 44.7 |
| LLS17 | Udaondo | 40 | male | descending colon | Left | IIIB | poorly | 1;3 | MSI | MLH1/PMS2 | WT | MLH1 | 77 |
| LLS18 | Udaondo | 30 | female | descending colon | Left | IIA | moderately | 1;3 | MSI | MSH2/MSH6 | WT | MSH2 | 57.8 |
| LLS19 | Udaondo | 25 | female | caecum | Right | IIIA | well | 1;2;3 | MSI | MSH2/MSH6 | WT | no alteration | 163.3 |
| LLS20 | Udaondo | 33 | male | ascending colon | Right | IIIA | moderately | 1 | MSI | MSH2/MSH6 | WT | no alteration | 7.6 |
| LLS21 | Udaondo | 36 | female | descending colon | Left | IIIA | moderately | 1 | MSI | MLH1/PMS2 | WT | MLH1 | 157.7 |
| LLS9049 | Clinic | 31 | female | descending colon | Left | IIIB | moderately | 1 | MSI | MSH2/MSH6 | WT | POLD1 | 164.6 |
| LLS9045 | Clinic | 35 | male | rectum | Left | IIIB | moderately | 1 | MSI | MLH1/PMS2 | WT | MLH1 | 142.4 |
| LLS5159 | Clinic | 37 | female | sigma | Left | IV | poorly | 1;3 | MSI | MLH1/PMS2 | WT | POLD1 | 81.4 |
| LLS5604 | Clinic | 33 | male | caecum | Right | IIIB | poorly | 1;3 | MSI | MLH1/PMS2 | WT | no alteration | 16.3 |
| LLS9048 | Clinic | 30 | male | ascending colon | Right | IIA | poorly | 1;3 | MSI | MLH1 | WT | no alteration | 6.8 |
| Sample ID | Gene Name | HGVS.c | HGVS.p | Variant Impact | gnomAD | ClinVar | COSMIC | PolyPhen2 | SIFT | CADD | Tumor AF | Average Sample AF | LOH AF Increase over Average (%) | LOH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biallelic Somatic Variants | ||||||||||||||
| LLS02 | MSH3 | c.423C > A | p.Cys141 * | stop_gained | not present | not present | not present | - | - | 38 | 0.60 | 0.35 | 73 | yes |
| LLS09 | MSH3 | c.1148delA | p.Lys383fs | frameshift_variant | 1.64399 × 10−5 | Pathogenic | COSM1438888 | - | - | - | 0.46 | 0.33 | 40 | yes |
| LLS10 | MSH3 | c.1148delA | p.Lys383fs | frameshift_variant | 1.64399 × 10−5 | Pathogenic | COSM1438888 | - | - | - | 0.61 | 0.35 | 75 | yes |
| LLS10 | POLD1 | c.583C > T | p.Arg195 * | stop_gained | 0.000647 | Conflicting | not present | - | - | 37 | 0.36 | 0.35 | 5 | no |
| LLS10 | POLD1 | c.2959delG | p.Asp987fs | frameshift_variant | 0.000057209 | not present | COSM3686158 | - | - | - | 0.40 | 0.35 | 15 | no |
| LLS17 | MLH1 | c.129dupA | p.Ser44fs | frameshift_variant | not present | Pathogenic | not present | - | - | - | 0.22 | 0.25 | −14 | no |
| LLS17 | MLH1 | c.1831delA | p.Ile611fs | frameshift_variant | not present | not present | not present | - | - | - | 0.33 | 0.25 | 31 | no |
| LLS18 | MSH2 | c.2251G > T | p.Gly751 * | stop_gained | not present | not present | not present | - | - | 48 | 0.15 | 0.17 | −11 | no |
| LLS18 | MSH2 | c.2634+1G > A | - | splice_variant | not present | Likely_pathogenic | not present | - | - | 27.6 | 0.19 | 0.17 | 14 | no |
| LLS21 | MLH1 | c.199G > A | p.Gly67Arg | missense_variant | not present | Pathogenic | COSM1422567 | D | D | 34 | 0.13 | 0.18 | −25 | no |
| LLS21 | MLH1 | c.602delT | p.Val201fs | frameshift_variant | not present | not present | not present | - | - | - | 0.24 | 0.18 | 36 | no |
| LLS5159 | POLD1 | c.1562G > A | p.Arg521Gln | missense_variant | 0.000126296 | VUS | not present | P | D | 31 | 0.22 | 022 | 4 | no |
| LLS5159 | POLD1 | c.3047G > A | p.Arg1016His | missense_variant | not present | VUS | COSM7587416 | P | D | 29.6 | 0.27 | 0.22 | 25 | no |
| LLS9045 | MLH3 | c.3694C > T | p.Arg1232Cys | missense_variant | 3.24934 × 10−5 | VUS | not present | D | D | 31 | 0.28 | 0.28 | 4 | no |
| LLS9045 | MLH3 | c.1924T > C | p.Phe642Leu | missense_variant | not present | not present | not present | B | D | 20.1 | 0.46 | 0.28 | 68 | yes |
| LLS9045 | MLH1 | c.588delA | p.Lys196fs | frameshift_variant | 4.06276 × 10−6 | Pathogenic | not present | - | - | - | 0.22 | 0.28 | −21 | no |
| LLS9045 | MLH1 | c.1489delC | p.Arg497fs | frameshift_variant | 4.06078 × 10−6 | not present | COSM1422596 | - | - | - | 0.21 | 0.28 | −23 | no |
| LLS9049 | POLD1 | c.2959delG | p.Asp987fs | frameshift_variant | 0.000057209 | not present | COSM3686158 | - | - | - | 0.31 | 0.19 | 57 | yes |
| Monoallelic Somatic Variants | ||||||||||||||
| LLS09 | MSH6 | c.3552G > A | p.Met1184Ile | missense_variant | 4.06835 × 10−6 | not present | not present | B | T | 28 | 0.30 | 0.33 | −10 | no |
| LLS10 | MSH6 | c.2875C > T | p.Arg959Cys | missense_variant | 1.22491 × 10−5 | VUS | not present | P | D | 27.2 | 0.34 | 0.35 | −1 | no |
| LLS10 | POLE | c.2091delC | p.Leu698fs | frameshift_variant | 8.20836 × 10−6 | VUS | COSM4612998 | - | - | - | 0.27 | 0.35 | −21 | no |
| LLS18 | MSH6 | c.3261dupC | p.Phe1088fs | frameshift_variant | 0.00003449 | Pathogenic | COSM13394 | - | - | - | 0.13 | 0.17 | −23 | no |
| LLS19 | MSH2 | c.1225C > T | p.Gln409 * | stop_gained | not present | Pathogenic | COSM7508782 | - | - | 41 | 0.16 | 0.15 | 7 | no |
| LLS19 | MSH6 | c.1993G > A | p.Glu665Lys | missense_variant | 4.06792 × 10−6 | not present | not present | P | T | 24.9 | 0.12 | 0.15 | −18 | no |
| LLS21 | MSH3 | c.3356T > C | p.Leu1119Pro | missense_variant | not present | not present | not prensent | D | D | 27.3 | 0.14 | 0.18 | −20 | no |
| LLS21 | PMS2 | c.1239delA | p.Asp414fs | frameshift_variant | not present | not present | COSM150905 | - | - | - | 0.20 | 0.18 | 12 | no |
| LLS21 | POLE | c.1060A > G | p.Thr354Ala | missense_variant | not present | not present | not present | B | T | 22.5 | 0.20 | 0.18 | 11 | no |
| LLS9045 | MUTYH | c.724C > T | p.Arg242Cys | missense_variant | 5.29614 × 10−5 | Pathogenic | COSM6954579 | D | D | 29.5 | 0.36 | 0.28 | 32 | no |
| LLS9045 | POLD1 | c.735G > T | p.Glu245Asp | missense_variant | not present | not present | not present | D | T | 22.9 | 0.27 | 0.28 | −2 | no |
| LLS9049 | MUTYH | c.544C > T | p.Arg182Trp | missense_variant | 1.21817 × 10−5 | not present | COSM6922477 | D | T | 23.3 | 0.13 | 0.19 | −32 | no |
| LLS9049 | MSH3 | c.433G > T | p.Ala145Ser | missense_variant | not present | VUS | not present | P | T | 17.64 | 0.15 | 0.19 | −23 | no |
| LLS9049 | POLE | c.3176G > A | p.Arg1059His | missense_variant | 1.21838 × 10−5 | not present | COSM6965827 | D | D | 35 | 0.20 | 0.19 | 2 | no |
| ID | Germline Candidate Gene | HGVS.c | HGVS.p | Coding Impact | gnomAD | CADD | ClinVar | ACMG Classification |
|---|---|---|---|---|---|---|---|---|
| Patients without biallelic inactivation in MMR core or their associated genes | ||||||||
| LLS06 | ERCC6 | c.1670G > A | R557H (p.Arg557His) | missense | 0.00018 | 27.6 | Not present | VUS(PM1;PM2;PP3;BP1) |
| LLS20 | POLE | c.1847G > A | R616H (p.Arg616His) | missense | 0.0000358 | 26.4 | Not present | VUS (PM2;PP3;BP1) |
| LLS5604 | EXO5 | c.23A > G | E8G (p.Glu8Gly) | missense | Not present | 25.3 | Not present | VUS (PM2;BP4) |
| LLS9048 | RECQL4 | c.1878+1G > C | - | donor_splice_site | Not present | 32 | Not present | Pathogenic (PVS1;PM2;PP3) |
| LLS15 | RAD52 | c.154A > T | I52L (p.Ile52Leu) | missense | Not present | 25.1 | Not present | VUS (PM2) |
| Patients with biallelic inactivation in MMR core or their associated genes | ||||||||
| LLS18 | PALB2 | c.2606C > T | S869F (p.Ser869Phe) | missense | Not present | 29.2 | VUS | VUS (PM1;PM2;PP3) |
| LLS21 | UVRAG | c.937C > G | Q313E (p.Gln313Glu) | missense | Not present | 26.3 | Not present | VUS (PM2) |
| LLS09 | RAD54L | c.767-2A > G | - | splice_acceptor | 2.38663 × 10−5 | 33 | Not present | VUS (PVS1;PP3) |
| LLS10 | RAD54L | c.17C > T | A6V (p.Ala6Val) | missense | 3.98108 × 10−6 | 32 | Not present | VUS (PM2;PP3) |
| LLS02 | MCM2 | c.364C > T | R122W (p.Arg122Trp) | missense | 1.66295 × 10−5 | 25.2 | Not present | VUS (PM2;PP3;BP1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golubicki, M.; Díaz-Gay, M.; Bonjoch, L.; Franch-Expósito, S.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Carballido, M.; et al. Comprehensive Genomic Characterization of Fifteen Early-Onset Lynch-Like Syndrome Colorectal Cancers. Cancers 2021, 13, 1259. https://doi.org/10.3390/cancers13061259
Golubicki M, Díaz-Gay M, Bonjoch L, Franch-Expósito S, Muñoz J, Cuatrecasas M, Ocaña T, Iseas S, Mendez G, Carballido M, et al. Comprehensive Genomic Characterization of Fifteen Early-Onset Lynch-Like Syndrome Colorectal Cancers. Cancers. 2021; 13(6):1259. https://doi.org/10.3390/cancers13061259
Chicago/Turabian StyleGolubicki, Mariano, Marcos Díaz-Gay, Laia Bonjoch, Sebastià Franch-Expósito, Jenifer Muñoz, Miriam Cuatrecasas, Teresa Ocaña, Soledad Iseas, Guillermo Mendez, Marcela Carballido, and et al. 2021. "Comprehensive Genomic Characterization of Fifteen Early-Onset Lynch-Like Syndrome Colorectal Cancers" Cancers 13, no. 6: 1259. https://doi.org/10.3390/cancers13061259
APA StyleGolubicki, M., Díaz-Gay, M., Bonjoch, L., Franch-Expósito, S., Muñoz, J., Cuatrecasas, M., Ocaña, T., Iseas, S., Mendez, G., Carballido, M., Robbio, J., Cisterna, D., Roca, E., Castells, A., Balaguer, F., Castellví-Bel, S., & Antelo, M. (2021). Comprehensive Genomic Characterization of Fifteen Early-Onset Lynch-Like Syndrome Colorectal Cancers. Cancers, 13(6), 1259. https://doi.org/10.3390/cancers13061259