
Pancreatic Ductal Adenocarcinoma Arising in Young and Old Patients Displays Similar Molecular Features

 ,
,  , , ,
, , ,  , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. DNA-Based Analyses
2.3. RNA-Based Analyses
2.4. Proteomic Analyses
2.4.1. One-Dimension Experiments
2.4.2. Two-Dimension Experiments
2.4.3. Western Blot Analysis
2.5. Statistical Analyses
3. Results
3.1. Clinico-Pathological Characteristics of Early-Onset Patients
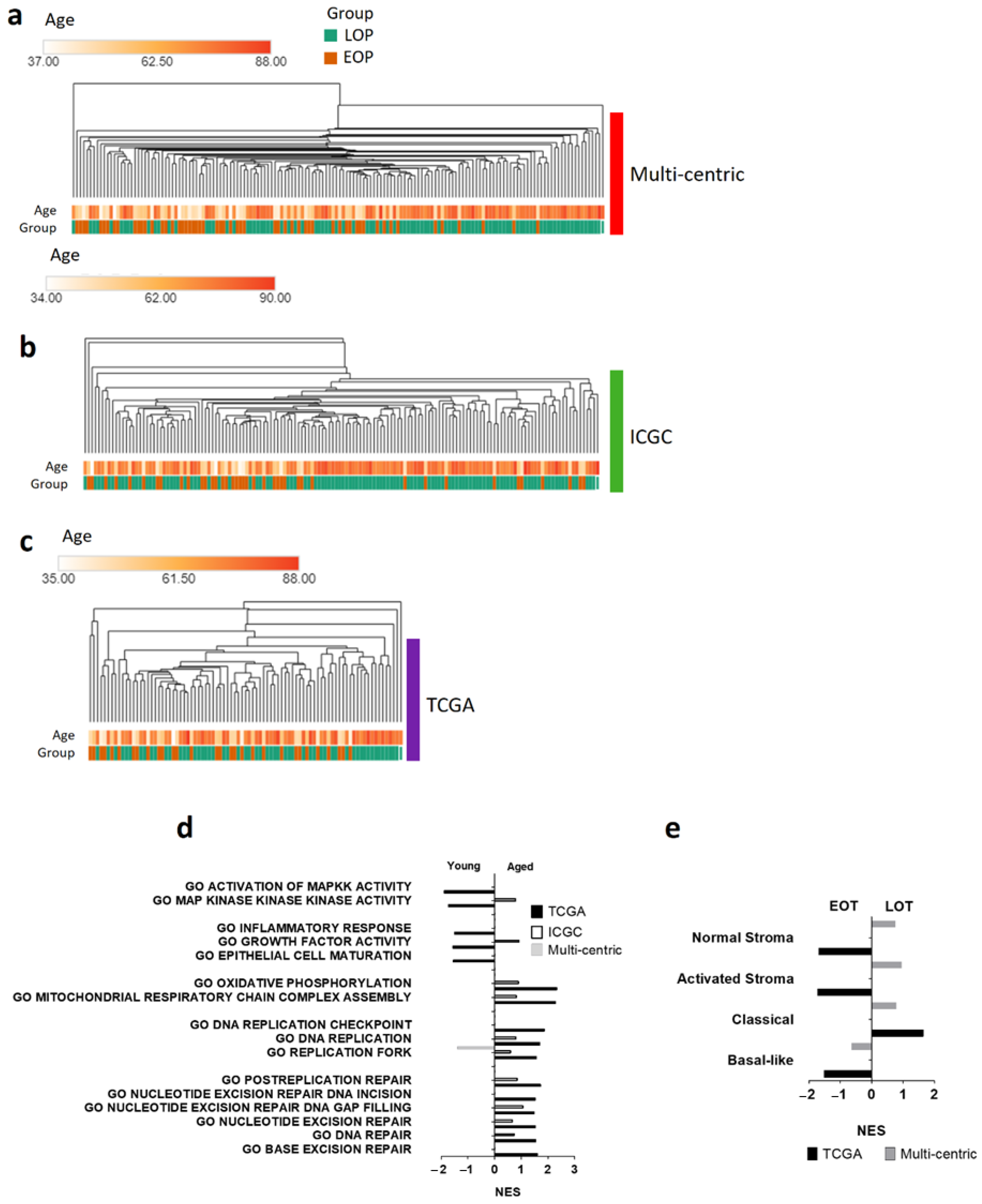
3.2. Genomic and Methylomic Landscape of PDAC in Early-Onset Patients
3.3. Transcriptomic Landscape in Early-Onset Patients
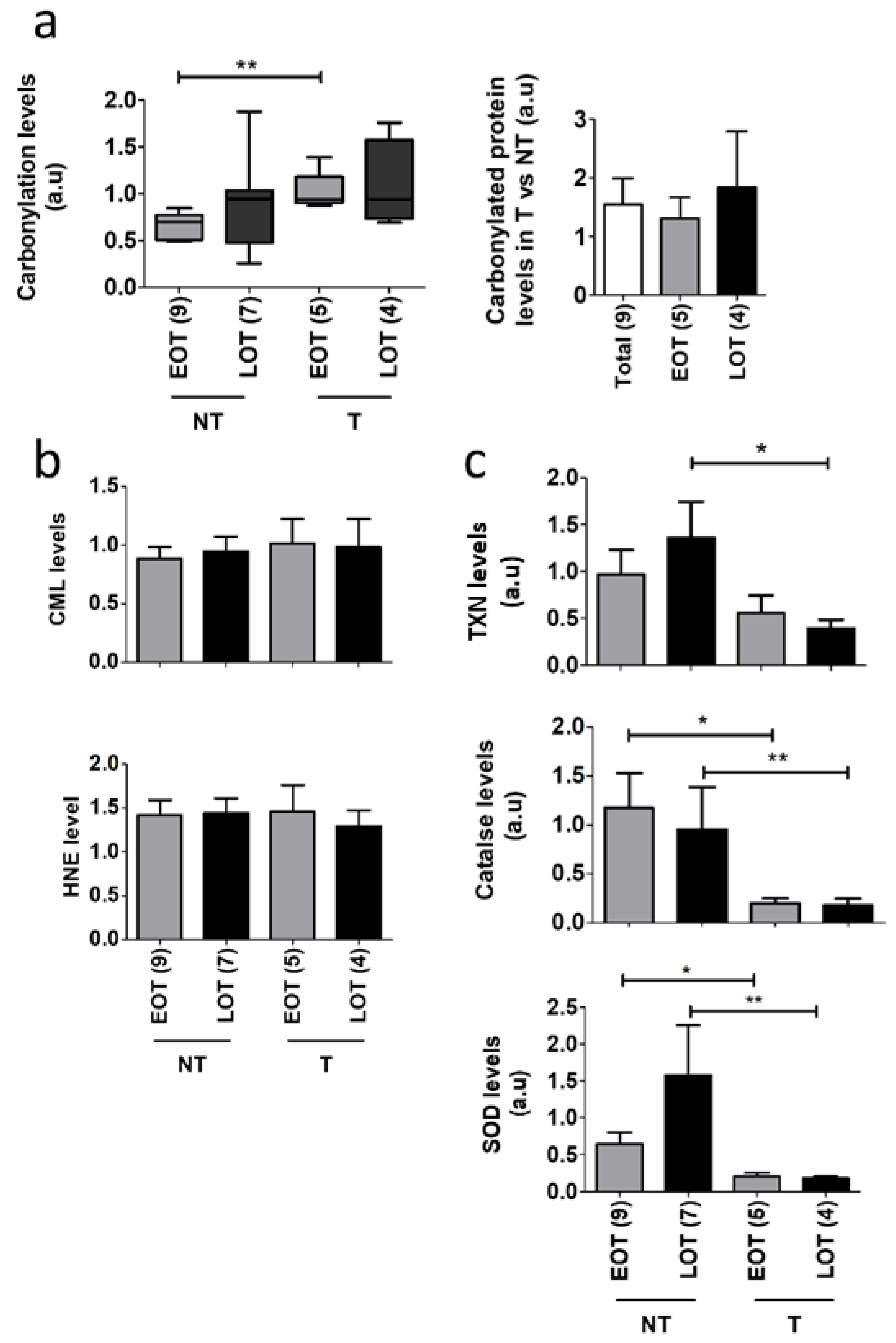
3.4. Oxidative Stress Damages and Defenses in Early-Onset Patients
3.5. Protein Oxidative Damages in Early-Onset Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Miller, K.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 29. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.; Hong, T.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Edil, B.H.; Cameron, J.L.; Schulick, R.D.; Hruban, R.H.; Herman, J.M.; Zheng, L.; Iacobuzio-Donahue, C.; Ahuja, N.; Pawlik, T.M.; et al. Young Patients Undergoing Resection of Pancreatic Cancer Fare Better than their Older Counterparts. J. Gastrointest. Surg. 2013, 17, 339–344. [Google Scholar] [CrossRef]
- Duffy, A.; Capanu, M.; Allen, P.; Kurtz, R.; Olson, S.H.; Ludwig, E.; Klimstra, D.S.; O’Reilly, E.M. Pancreatic adenocarcinoma in a young patient population—12-Year experience at Memorial Sloan Kettering Cancer Center. J. Surg. Oncol. 2009, 100, 8–12. [Google Scholar] [CrossRef]
- Tingstedt, B.; Weitkämper, C.; Andersson, R. Early onset pancreatic cancer: A controlled trial. Ann. Gasteroenterol. 2011, 24, 206–212. [Google Scholar]
- Eguchi, H.; Yamaue, H.; Unno, M.; Mizuma, M.; Hamada, S.; Igarashi, H.; Kuroki, T.; Satoi, S.; Shimizu, Y.; Tani, M.; et al. Clinicopathological Characteristics of Young Patients With Pancreatic Cancer: An Analysis of Data From Pancreatic Cancer Registry of Japan Pancreas Society. Pancreas 2016, 45, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, F.; Aulmann, S.; Wente, M.N.; Penzel, R.; Esposito, I.; Kleeff, J.; Friess, H.; Schirmacher, P. Molecular characterisation of pancreatic ductal adenocarcinoma in patients under 40. J. Clin. Pathol. 2006, 59, 580–584. [Google Scholar] [CrossRef]
- Bannon, S.A.; Montiel, M.F.; Goldstein, J.B.; Dong, W.; Mork, M.E.; Borras, E.; Hasanov, M.; Varadhachary, G.R.; Maitra, A.; Katz, M.H.; et al. High Prevalence of Hereditary Cancer Syndromes and Outcomes in Adults with Early-Onset Pancreatic Cancer. Cancer Prev. Res. 2018, 11, 679–686. [Google Scholar] [CrossRef]
- Ben-Aharon, I.; Elkabets, M.; Pelossof, R.; Yu, K.H.; Iacobuzio-Donahue, C.A.; Leach, S.D.; Lowery, M.A.; Goodman, K.A.; O’Reilly, E.M. Genomic Landscape of Pancreatic Adenocarcinoma in Younger vs Older Patients: Does Age Matter? Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Whittle, M.C.; Izeradjene, K.; Rani, P.G.; Feng, L.; Carlson, M.A.; DelGiorno, K.E.; Wood, L.D.; Goggins, M.; Hruban, R.H.; Chang, A.E.; et al. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell 2015, 161, 1345–1360. [Google Scholar] [CrossRef]
- Jha, P.; Suri, V.; Singh, G.; Jha, P. Characterization of Molecular Genetic Alterations in GBMs Highlights a Distinctive Molecular Profile in Young Adults. Diagn. Mol. Pathol. 2011, 20, 225–232. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harbor Perspect. Biol. 2016, 8, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Protein oxidation and aging. Free Radic. Res. 2006, 40, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef]
- Grune, T.; Jung, T.; Merker, K.; Davies, K.J.A. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and “aggresomes” during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004, 36, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Mehrabi, S.; Partridge, E.E.; Seffens, W.; Yao, X.; Aikhionbare, F.O. Oxidatively modified proteins in the serous subtype of ovarian carcinoma. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Rossner, P.; Terry, M.B.; Gammon, M.D.; Agrawal, M.; Zhang, F.F.; Ferris, J.S.; Teitelbaum, S.L.; Eng, S.M.; Gaudet, M.M.; Neugut, A.I.; et al. Plasma protein carbonyl levels and breast cancer risk. J. Cell. Mol. Med. 2007, 11, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radic. Biol. Med. 2002, 32, 790–796. [Google Scholar] [CrossRef]
- Garcia-Garcia, A.; Rodriguez-Rocha, H.; Madayiputhiya, N.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Biomarkers of protein oxidation in human disease. Curr. Mol. Med. 2012, 12, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal--a one-stop shop for cancer genomics data. Database 2011, 2011, bar026. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Koestler, D.C.; Cheng, C.; Christensen, B.C. Age-related DNA methylation in normal breast tissue and its relationship with invasive breast tumor methylation. Epigenetics 2014, 9, 268–275. [Google Scholar] [CrossRef]
- Lim, U.; Song, M.-A. DNA Methylation as a Biomarker of Aging in Epidemiologic Studies. Cancer Epigenetics Precis. Med. 2018, 219–231. [Google Scholar]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Nash, N.; Sherman, M.; Hanna, A.; Knight, J.; Zehr, C.; Nelson, S.; Norton, D. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 162, 481–491. [Google Scholar] [CrossRef]
- Yamada, S.; Fujii, T.; Shimoyama, Y.; Kanda, M.; Nakayama, G.; Sugimoto, H.; Koike, M.; Nomoto, S.; Fujiwara, M.; Nakao, A.; et al. SMAD4 Expression Predicts Local Spread and Treatment Failure in Resected Pancreatic Cancer. Pancreas 2015, 44, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 Gene Status of the Primary Carcinoma Correlates With Patterns of Failure in Patients With Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Nicolle, R.; Raffenne, J.; Paradis, V.; Couvelard, A.; de Reynies, A.; Blum, Y.; Cros, J. Prognostic Biomarkers in Pancreatic Cancer: Avoiding Errata When Using the TCGA Dataset. Cancers (Basel) 2019, 11, 126. [Google Scholar] [CrossRef]
- Peran, I.; Madhavan, S.; Byers, S.W.; McCoy, M.D. Curation of the Pancreatic Ductal Adenocarcinoma Subset of the Cancer Genome Atlas Is Essential for Accurate Conclusions about Survival-Related Molecular Mechanisms. Clin. Cancer Res. 2018, 24, 3813–3819. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Martín-Guerrero, I.; de Prado, E.; Lopez-Lopez, E.; Ardanaz, M.; Vitoria, J.C.; Parada, L.A.; García-Orad, C.; García-Orad, A. Methylation of the nonhomologous end joining repair pathway genes does not explain the increase of translocations with aging. AGE 2014, 36, 9730. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vyjayanti, V.N.; Swain, U.; Rao, K.S. Age-related decline in DNA polymerase β activity in rat brain and tissues. Neurochem. Res. 2012, 37, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, L.; Rong, S.; Qu, H.; Zhang, Y.; Chang, D.; Pan, H.; Wang, W. Relation between gastric cancer and protein oxidation, DNA damage, and lipid peroxidation. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort Features n = 255 | ||||
|---|---|---|---|---|
| Age Group | EOP < 55 y.o. (106) | LOP > 70 y.o. (149) | p | |
| Sex (n = Men (%)) n = 255 | 46 (43.39%) | 75 (50.33%) | 0.389 ⁱ | |
| Age (median (min-max)) n = 255 | 51 (34–55) | 74 (70–88) | <0.0001 ⁱ | |
| T n = 255 | 0.841 * | |||
| T1 | 4 | 4 | ||
| T2 | 16 | 25 | ||
| T3 | 86 | 120 | ||
| N n = 255 | N0/N1 | 32/74 | 35/141 | 0.292 * |
| Tumor differentiation n = 255 | 0.197 * | |||
| Well | 35 | 68 | ||
| Moderate | 48 | 50 | ||
| Poorly | 20 | 27 | ||
| Unknown | 3 | 4 | ||
| Tumor size n = 238 (mm mean—CI95) | 31.9 (27.8–35) | 33.9 (31.0–36.8) | 0.239 ⁱ | |
| Vascular invasion n = 204 (n (%)) | 57 (53.7) | 58 (38.9) | 0.024 ⁱ | |
| Perinervous invasion n = 237 (n (%)) | 81 (76.6) | 104 (69.8) | 0.346 ⁱ | |
| Cohort features n = 255 | Univariate PFS EOP <55 y.o. (106) | Univariate PFS LOP >70 y.o. (149) | Multivariate PFS LOP >70 y.o. (149) | Univariate OS EOP <55 y.o. (106) | Univariate OS LOP > 70 y.o. (149) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age Group | HR | 95% Ci | p * | HR | 95% Ci | p * | HR | 95%Ci | p * | HR | 95%Ci | p * | HR | 95%Ci | p * |
| Sex (Men) | 0.79 | 0.49–1.27 | 0.335 | 0.841 | 0.56–1.26 | 0.405 | 0.956 | 0.58–1.57 | 0.86 | 0.786 | 0.52-.13 | 0.187 | |||
| Age | 1.023 | 0.97–1.07 | 0.396 | 0.944 | 0.88–1.00 | 0.084 | 1.013 | 0.96–1.06 | 0.638 | 0.985 | 0.93–1.04 | 0.611 | |||
| T | 0.023 | 0.954 | |||||||||||||
| T1 | 0.883 | 0.27–2.81 | 0.834 | 0.2 | 0.02–1.42 | 0.11 | 0.326 | 0.04–2.39 | 0.273 | 1.131 | 0.35–3.62 | 0.837 | 0.266 | 0.03–1.89 | 0.189 |
| T2 | 0.656 | 0.32–1.32 | 0.24 | 0.583 | 0.32–1.04 | 0.072 | 0.706 | 0.38–1.28 | 0.255 | 0.931 | 0.47–1.83 | 0.837 | 0.716 | 0.42–1.22 | 0.223 |
| T3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | - | 1 | 1 | 1 |
| N0/N1 | 1.639 | 0.96–2.79 | 0.071 | 2.697 | 1.52–4.75 | 0.0007 | 2.53 | 1.11–4.54 | 0.002 | 0.571 | 0.32–1.02 | 0.06 | 0.415 | 0.24–0.71 | 0.002 |
| Tumor differentiation n = 255 | 0.015 | ||||||||||||||
| Well | 0.875 | 0.50–1.50 | 0.634 | 1 | 1 | 1 | 1 | 1 | 1 | 0.942 | 0.52–1.68 | 0.841 | 1 | 1 | 1 |
| Moderate | 1 | 1 | 1 | 1.787 | 1.13–2.82 | 0.013 | 1.738 | 1.09–2.76 | 0.02 | 1 | 1 | 1 | 1.18 | 0.76–1.83 | 0.459 |
| Poorly | 1.215 | 0.65–2.26 | 0.542 | 1.966 | 1.14–3.38 | 0.015 | 2.14 | 1.23–3.71 | 0.006 | 1.343 | 0.696–2.59 | 0.382 | 1.65 | 0.96–2.83 | 0.068 |
| Unknown | 0.841 | 0.20–3.49 | 0.813 | 0.451 | 0.06–3.25 | 0.432 | 0.893 | 0.11–6.77 | 0.913 | 1.525 | 0.46–5.00 | 0.488 | 0.728 | 0.17–2.99 | 0.662 |
| Tumor size | 1.005 | 0.99–1.01 | 0.436 | 1.008 | 0.99–1.02 | 0.147 | 1.003 | 0.98–1.02 | 0.747 | 1.008 | 0.99–1.01 | 0.122 | |||
| Vascular invasion | 1.43 | 0.99–2.0 | 0.054 | 1.35- | 0.88–1.45 | 0.322 | 1.508 | 1.03–2.19 | 0.032 | 1.064 | 0.83–1.36 | 0.619 | |||
| Perinervous invasion | 1.246 | 0.72–2.13 | 0.425 | 1.42 | 0.98–2.07 | 0.061 | 0.999 | 0.57–1.75 | 0.999 | 1.295 | 0.90–1.85 | 0.158 | |||
| Mutations | Multi-Centric | ICGC | TCGA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| EOP (53) | LOP (89) | p * | EOP (39) | LOP (110) | p * | EOP (27) | LOP (55) | p * | |
| KRAS n = (%) | 48 (90.5) | 81 (91) | 0.832 | 29 (74.3) | 87 (79.1) | 0.653 | 25 (92.6) | 50 (90.9) | 0.704 |
| TP53 n = (%) | 37 (69.8) | 63 (70.8) | 0.946 | 19 (48.7) | 70 (63.6) | 0.129 | 23 (85.2) | 39 (70.9) | 0.183 |
| CDKN2A n = (%) | 11 (20.7) | 14 (15.7) | 0.594 | 4 (10.2) | 15 (13.6) | 0.781 | 16 (59.2) | 33 (60) | 1.00 |
| SMAD4 n = (%) | 5 (9.4) | 22 (24.7) | 0.043 | 7 (17.9) | 27 (24.5) | 0.507 | 12 (44.4) | 22 (40) | 0.812 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raffenne, J.; Martin, F.A.; Nicolle, R.; Konta, M.; Blum, Y.; Torrisani, J.; Puleo, F.; Bachet, J.B.; Svrcek, M.; Bardier-Dupas, A.; et al. Pancreatic Ductal Adenocarcinoma Arising in Young and Old Patients Displays Similar Molecular Features. Cancers 2021, 13, 1234. https://doi.org/10.3390/cancers13061234
Raffenne J, Martin FA, Nicolle R, Konta M, Blum Y, Torrisani J, Puleo F, Bachet JB, Svrcek M, Bardier-Dupas A, et al. Pancreatic Ductal Adenocarcinoma Arising in Young and Old Patients Displays Similar Molecular Features. Cancers. 2021; 13(6):1234. https://doi.org/10.3390/cancers13061234
Chicago/Turabian StyleRaffenne, Jérôme, Fernando A. Martin, Rémy Nicolle, Marina Konta, Yuna Blum, Jérôme Torrisani, Francesco Puleo, Jean Baptiste Bachet, Magali Svrcek, Armel Bardier-Dupas, and et al. 2021. "Pancreatic Ductal Adenocarcinoma Arising in Young and Old Patients Displays Similar Molecular Features" Cancers 13, no. 6: 1234. https://doi.org/10.3390/cancers13061234
APA StyleRaffenne, J., Martin, F. A., Nicolle, R., Konta, M., Blum, Y., Torrisani, J., Puleo, F., Bachet, J. B., Svrcek, M., Bardier-Dupas, A., Emile, J. F., Demetter, P., Radman, M., Van Laethem, J. L., Hammel, P., Rebours, V., Paradis, V., Couvelard, A., & Cros, J. (2021). Pancreatic Ductal Adenocarcinoma Arising in Young and Old Patients Displays Similar Molecular Features. Cancers, 13(6), 1234. https://doi.org/10.3390/cancers13061234