Simple Summary
Pancreatic cancer is a digestive tumor that is most difficult to treat and carries one of the worst prognoses. The anatomical location of the pancreas makes it very difficult to obtain enough tumor material to establish a molecular diagnosis, so knowing the biology of this tumor and implementing new targeted-therapies is still a pending issue. The use of liquid biopsy, a blood sample test to detect circulating-tumor DNA fragments (ctDNA), is key to overcoming this difficulty and improving the evolution of this tumor. Liquid biopsies are equally representative of the tissue from which they come and allow relevant molecular and diagnostic information to be obtained in a faster and less invasive way. One challenge related to ctDNA is the lack of consistency in the study design. Moreover, ctDNA accounts for only a small percentage of the total cell-free circulating DNA and prior knowledge about particular mutations is usually required. Thus, our aim was to understand the current role and future perspectives of ctDNA in pancreatic cancer using digital-droplet PCR technology.
Abstract
Pancreatic cancer (PC) is one of the most devastating malignant tumors, being the seventh leading cause of cancer-related death worldwide. Researchers and clinicians are endeavoring to develop strategies for the early detection of the disease and the improvement of treatment results. Adequate biopsy is still challenging because of the pancreas’s poor anatomic location. Recently, circulating tumor DNA (ctDNA) could be identified as a liquid biopsy tool with huge potential as a non-invasive biomarker in early diagnosis, prognosis and management of PC. ctDNA is released from apoptotic and necrotic cancer cells, as well as from living tumor cells and even circulating tumor cells, and it can reveal genetic and epigenetic alterations with tumor-specific and individual mutation and methylation profiles. However, ctDNA sensibility remains a limitation and the accuracy of ctDNA as a biomarker for PC is relatively low and cannot be currently used as a screening or diagnostic tool. Increasing evidence suggests that ctDNA is an interesting biomarker for predictive or prognosis studies, evaluating minimal residual disease, longitudinal follow-up and treatment management. Promising results have been published and therefore the objective of our review is to understand the current role and the future perspectives of ctDNA in PC.
1. Introduction
There is a great need to unveil the genetic landscape of pancreatic ductal adenocarcinoma (PDAC), one of the most aggressive human malignancies. This tumor type, although not very frequent, has an increasing incidence and is the seventh leading cause of cancer-related death in both men and women worldwide [1]. Indeed, it is predicted to become the second by 2030 [2]. Globally, 495,773 new cases and 466,003 deaths of pancreatic cancer were reported in 2020 (GLOBOCAN, https://gco.iarc.fr/ (accessed date 4 February 2021)). The overall 5-year survival rate is 9%, and the median survival is still shorter than 6 months, the lowest among all cancer types [3]. This poor survival is mostly attributed to the absence of effective screening methods, late diagnosis due to non-specific symptoms, lack of sensitive or specific biomarkers for early diagnosis, propensity for early metastatic spread, and the limited therapeutic advancements over the last years [4,5]. Consequently, the prognosis of pancreatic cancer remains unimproved. The onset of symptoms and PDAC diagnosis is usually late, at an advanced stage, where most patients have metastatic disease.
Cell-free circulating tumor DNA (ctDNA) is a promising non-invasive blood-based biomarker in cancer management, and an alternative to traditional blood-based protein biomarkers [6,7]. ctDNA has shown benefit in early detection, prognosis estimation, treatment selection, tumor dynamics monitoring, minimal residual disease detection and tumor recurrence during follow up [8,9,10,11,12,13,14,15,16]. ctDNA is composed of short segments of nucleic acids and reflects the genetic and epigenetic makeup of the tumor from which it originates, making it a desirable and highly specific biomarker. ctDNA provides a better representation of the molecular composition of a malignant disease than a single section from a surgical tumor specimen or a tissue biopsy and, therefore, its clinical application in PDAC is extremely important and interesting due to the difficulty of obtaining tumor tissues (Figure 1).
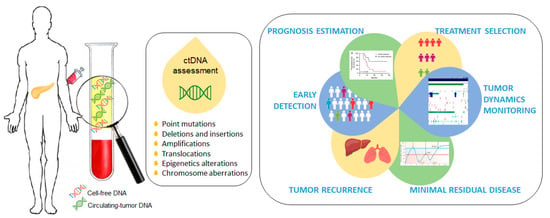
Figure 1.
Circulating-tumor DNA (ctDNA) as a promising non-invasive blood-based biomarker in cancer management. Quantitative detection of ctDNA is based on the identification of various tumor-specific genetic or epigenetic aberrations in plasma cell-free DNA (cfDNA) samples.
One challenge related to ctDNA biology is a lack of consistency in the study design. Thus, in this article, we review current knowledge regarding ctDNA for pancreatic cancer using digital-droplet PCR (ddPCR). The publications cited were selected from the PubMed database (http://www.ncbi.nlm.nih.gov/pubmed (accessed date 27 February 2021)). Key search terms, or aliases, used were ctDNA, cfDNA, liquid biopsy, pancreatic cancer, digital PCR and ddPCR. The eligibility criteria for articles reviewed here included original studies published beyond 2015 in patients with a clinical and histological diagnosis of pancreatic adenocarcinoma that was molecularly characterized in plasma and serum liquid biopsies by digital PCR technology. Studies published in a language other than English were excluded. This paper will focus on the potential application of ddPCR to detect ctDNA for PDAC.
2. Increasing Genetic Knowledge to Improve Clinical Practice
In recent years, better understanding of the molecular pathways that direct tumor progression is leading to the development of what we know as precision medicine. Although to a lesser extent than in other tumors, there have also been advances in this area in PDAC.
PDAC exome sequencing studies suggest a 20-year window opportunity for early detection before any symptoms occur [17,18]. Early identification of both the initial and recurrence disease would therefore improve clinical outcomes. Several common gene mutations are involved in PDAC carcinogenesis. The most important frequent gene mutations include KRAS, CDKN2A, SMAD4 and TP53. However, their widespread use is limited by the difficulty of obtaining tumor tissues. In this context, a diagnostic and prognostic noninvasive blood test for pancreatic cancer would be very valuable. Currently, the only non-invasive blood-based biomarker routinely used in clinical practice is carbohydrate antigen 19-9 (CA 19-9). However, because of its low sensitivity (78%) and specificity (82%), it is unsuitable for screening the general population and its diagnostic utilization to detect early-stage tumors is discouraged unless in combination with other circulating biomarkers [19,20,21]. Thus, novel biomarkers that can reliably identify initial, residual or progressive disease are urgently needed.
Surgical resection is the only radical treatment with a potential chance of cure. However, at the time of diagnosis, only a few patients (10–15%) have localized disease. Molecular tumor profiling has relied on the analysis of tumor tissues obtained from surgical resection specimens in the majority of the tumor types. Unlike many other tumor types, PDAC treatment decisions are not made according to tumor biology, and patients undergo chemotherapy and radiotherapy according to their tumor stage. Until a year ago, nine different chemotherapeutic drugs were approved by the Food and Drug Administration (FDA), and some progress had been made in the management of advanced disease by the administration of multidrug regimens such as FOLFIRINOX and gemcitabine/nab-paclitaxel therapy, without any biomarkers that guide the choice of one or another treatment [22,23,24,25,26]. Fortunately, on December 2019, the FDA approved the first targeted-treatment for pancreatic cancer: olaparib. The randomized phase III POLO trial demonstrated statistically significant benefit in progression free survival (PFS), as maintenance therapy versus placebo, in patients with germline BRCA-mutated (gBRCAm) metastatic pancreatic adenocarcinoma whose disease has not progressed after a first-line platinum-based chemotherapy regimen [27]. Although the results are very promising, the prevalence of gBRCAm in PDAC is only 4–7% [27]. Despite these significant advances, treatment is always with a palliative intent and unfortunately, there are no long-term survivors. Thus, medical management of pancreatic cancer remains a challenge and developing new immunotherapies, and stroma-directed and targeted-therapies is an unmet need.
In 1983, Shapiro et al. first reported the presence of circulating cell-free DNA (cfDNA) in pancreatic cancer [28]. Since then, a research focus on genetic alterations in ctDNA has become mainstream.
Personalized and precision treatments are the novel goal for pancreatic cancer and involve understanding each patient’s driver genes. The analysis of ctDNA represents a unique method to explore the genetic mutation and may be translated into the clinical setting. KRAS represents an important potential biomarker for PDAC. It has been the best-characterized PDAC tumor-related gene due to the following reasons: (i) among human malignancies, PDAC has the highest frequency of KRAS mutations (up to 90%), (ii) the most frequent KRAS point mutations are located in codon 12, and (iii) alterations in this gene appear to occur at an early stage of pancreatic carcinogenesis. Several studies have evaluated the role of KRAS in the diagnosis, prognosis and treatment of PDAC [29]. In addition, the evaluation of KRAS mutation testing in PDAC patients has been discussed in depth for the past 20 years [30].
In 1998, Yamada et al. demonstrated that KRAS mutations in plasma may be clinically useful for evaluating tumor burden and treatment efficacy for pancreatic cancer [31]. In 1999, Castells and colleagues detected, for the first time, circulating mutant KRAS genes in plasma from PDAC patients and reported the association between poor survival and the presence of KRAS mutations in PDAC patients’ plasma [32]. Since then, several research studies have reported the prognosis and predictive significance of KRAS ctDNA detected in metastatic and perioperative settings, as well as their therapeutic evaluation along longitudinal monitoring of the disease [33,34]. However, most studies have focused on the presence of ctDNA mutations in a broad set of genes to highlight tumor heterogeneity and demonstrate clonal evolution over the course of disease progression. Despite different genetic mutations being identified in PDAC, nearly all of them have failed to facilitate a treatment approach. Moreover, PDAC tumor biology is not completely known and new-targeted therapies cannot be implemented.
ctDNA-based assays are confronted with several challenges, such as that ctDNA accounts for only a small percentage of the total cfDNA in the peripheral blood, sometimes less than 0.01%, and that prior knowledge of particular mutations is usually required, which in pancreatic cancer is hard to obtain [35,36]. However, the analysis of ctDNA has evolved since its inception with improvements in the technologies and detection limits [8]. Third-generation sequencing techniques have rapidly advanced and have the potential to expedite extensive application of ctDNA detection for routine patient management.
Currently, high-throughput next-generation sequencing (NGS) and droplet digital PCR (ddPCR) are the most promising techniques for the detection of liquid biopsy mutations. NGS-based methods generate extensive information as they allow the simultaneous evaluation and detection of multiple genetic and epigenetic aberrations over millions of ctDNA molecules. Thus, NGS leads to the discovery of novel mutated variants and presents high multiplexing capabilities. However, it is time consuming, has higher cost, requires powerful informatics support and cannot be readily applied to monitor patients longitudinally [37]. Conventional NGS methods allow a sensitivity higher than 2%. Moreover, whole-exome or whole-genome sequencing approaches usually generate around 30× to 100× average sequencing coverage, which leads to a low sensitivity detection on ctDNA [36]. Several different NGS-based technologies have been developed to enhance ctDNA detection sensitivity and specificity: Safe-Sequencing System (Safe-Seq) [38], Cancer Personalized Profiling by deep Sequencing (CAPP-seq) [39], Integrated digital error suppression-enhanced CAPP-seq (iDES-enhanced CAPP-seq) [40], or Base-Position Error Rate (BPER) [36,41]. Despite sensitivities of improved NGS-based approaches being similar to ddPCR, this paper will focus on the potential application of ddPCR to detect ctDNA for PDAC.
2.1. Digital-Droplet PCR Technology
In 1999, Vogelstein and Kinzler described the digital PCR (dPCR), a new microtiter plate-based technology for rare sequences detection [42]. This dPCR technology allows partitioning and individually testing of target sequences within separate compartments. dPCR sensitivity relies on the number of individual compartments and sequences created and analyzed, along with the false-positive rate of each assay [36].
Digital-droplet PCR (ddPCR) is a novel next-generation technique based on nanoliter-sized water-in-oil emulsion droplet technology. At present, ddPCR is one of the most powerful methods available for the accurate quantification of a scarce amount of circulating nucleic acids in plasma. It is currently being used for absolute quantification, rare mutation detection, copy number variation analysis, DNA methylation and gene rearrangements in different types of clinical samples. Compared to NGS approaches, ddPCR experiments are easier to set up, faster, present higher sensitivity and do not require complex bioinformatics analysis (Figure 2).
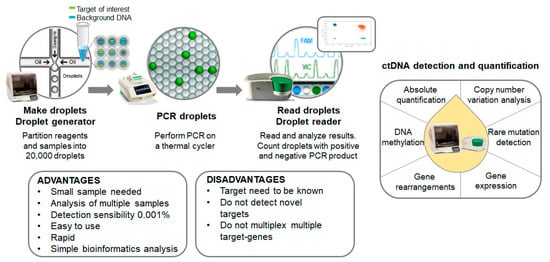
Figure 2.
Overview of the mutation screening process. Summary of both advantages and disadvantages of the technology disclosed and other putative applications.
BEAMing (beads, emulsion, amplification and magnetics) was the first high-throughput droplet-based digital PCR used for the detection and enumeration of genetic variants [43,44]. Despite the low limit of detection achieved, up to 0.0001%, it requires a relatively cumbersome and complicated procedure for routine clinical use [44]. Nowadays, several droplet-based digital PCR platforms are being commercialized, including RaindropTM digital PCR (Raindance Technologies, Lexington, MA, USA), Bio-Rad QX200TM Droplet DigitalTM system (Bio-Rad Laboratories, Hercules, CA, USA) or NaicaTM System (Stilla Technologies, Villejuif, France) [45]. In all of these platforms, ctDNA samples are partitioned in aqueous droplets acting as independent micro-compartments. Ideally, each droplet contains one haploid genome and all the reagents needed to perform the PCR assay. Different fluorescent signals identify mutant and wild-type sequences. The most widely used is the QX200 Droplet digital PCR System (Bio-Rad Laboratories) that generates up to 20,000 nanoliter-sized droplets.
In cancer, ddPCR represents a robust method for quantifying low-abundance point mutations in cfDNA, with high sensitivity ranging from 0.001% to 0.1%, in order to reflect intratumoral heterogeneity and to track the dynamic changes in tumor burden in response to treatment during follow-up. An extensive spectrum of molecular markers has been interrogated in liquid biopsies from different tumor types using ddPCR for diagnosis, predictive and monitoring purposes. Therefore, ddPCR is a particularly promising technology due to its low turnaround time, high scalability and exquisite sensitivity [46]. Different clinical scenarios have been established, where the reliance on precision and sensitivity offered by ddPCR is the highest priority. A description of the most relevant studies of ddPCR ctDNA application in PDAC is provided in Table 1.

Table 1.
Most relevant studies of digital-droplet PCR (ddPCR) ctDNA application in pancreatic cancer.
2.1.1. ddPCR Research Approach
The high detection capability of ddPCR may permit the elucidation of alternative biomarkers for PDAC. ctDNA is found at relatively high concentrations in the circulation of most patients with metastatic disease and at lower, but detectable, concentrations in patients with localized tumors [47,62,63]. Thus, an extremely sensitive method, able to discriminate between the few mutation-associated alleles released into the bloodstream from normal variant background, is needed to follow early-stage tumor development.
Earl et al. reported plasma KRAS (G12D, G12V and G12R) mutant ctDNA detection using ddPCR in 8 of 31 (26%) patients across various PDAC stages. In addition, they found that KRAS mutant detection was significantly correlated with overall survival (OS) (HR = 12.2, p < 0.001) [52]. Lin et al. reported similar plasma KRAS results (31% ctDNA KRAS detection and worse overall survival association) [56]. Similarly, Hadano and collaborators used ddPCR to detect KRAS (G12D, G12V and G12R) mutations in plasma ctDNA from 105 PDAC patients [10]. They reported a cumulative rate of 31% ctDNA detection across stages, with a median survival of 13.6 months vs. 27.6 months in those patients with detectable vs. no detectable ctDNA, respectively, and a significant association with OS (p < 0.0001). They demonstrated that KRAS ctDNA mutations were associated with significantly poorer survival and therefore concluded that the presence of ctDNA in plasma could be an important and powerful survival predictor. Consistently, Cheng and collaborators (2020) found that both KRAS G12D and G12V mutations were associated with poor prognosis [51]. Kinugasa and colleagues (2015) analyzed serum KRAS (G12D, G12V and G12R) ctDNA mutations in 75 pancreatic cancer patients, with previously published KRAS mutations in a development cohort, and in 66 patients in an independent blinded validation cohort. KRAS ctDNA mutations were found in 62.6% and 54.5% of the development cohort and validation set samples respectively. Similarly, they found significantly shorter median survival time in patients with ctDNA KRAS mutations (p = 0.02) [55]. Serum and plasma are good tissues for detecting cancer-specific DNA and the presence of KRAS mutations in blood-derived DNA may be used as a prognostic biomarker for PDAC patients [47]. However, plasma has been generally used as the material for detecting ctDNA, as serum contains genomic DNA released from white blood cells during the clotting process that can interfere with ctDNA detection [64,65,66,67].
Multiplex strategies have been described, allowing one to screen for a pool of RAS/RAF mutations [68]. However, the subsequent identification of particular mutations is needed. Sefrioui and colleagues (2017) used ddPCR in combination with a multiplex assay to screen the seven most common KRAS mutations found in pancreatic cancer. This multiplex assay covers the mutation sites G12A, G12C, G12D, G12R, G12S, G12V and G13D. The authors performed ddPCR blindly from clinical data and 56% of the cases were reported to have a KRAS circulating mutation with a sensitivity and specificity for diagnosis of 65% and 75% respectively. Moreover, the multiplex and simplex assay results were significantly correlated [58]. Similarly, Kim and collaborators used a multiplex KRAS assay and demonstrated that KRAS mutant concentration and fractional abundance in plasma cfDNA were associated with prognosis in PDAC patients [54]. Thus, the authors suggested that cfDNA can serve as a biomarker to aid in determining who will benefit from treatment and which tumors will recur. Woo et al. (2017) also screened multiplex KRAS using ddPCR in a cohort of patients with locally advanced unresectable pancreatic cancer treated with chemoradiotherapy. The authors reported significantly lower concentration of cfDNA after treatment (p < 0.001). However, KRAS mutant concentration and fractional abundance was not significantly different before and after treatment. In addition, and in contrast with previous studies, overall survival and progression free survival were not related to cfDNA concentration, KRAS mutation concentration or fractional abundance [61].
Sugimori and collaborators (2020) detected KRAS mutations by ddPCR using a multiplex probe that screens 16 KRAS mutations (G12A, G12C, G12D, G12F, G12G, G12L, G12R, G12S, G12V and G13A, G13C, G13D, G13G, G13R, G13S and G13V). The authors reported that patients with distant metastases, except peritoneal metastases, showed a significantly higher KRAS mutation detection rate in serum ctDNA compared to those with locally advanced disease or peritoneal metastases. Furthermore, for those patients without a ctDNA KRAS mutation at the time of diagnosis, a KRAS mutation was detected at the time of progression of the disease. Thus, progression free survival analysis revealed that ctDNA KRAS mutation patients undergoing first-line chemotherapy tended to have worse progression free survival than those without a KRAS mutation (median 308.5 vs. 168 days, p = 0.07). Interestingly, they showed that, in those cases with a KRAS mutation in ctDNA at the time of diagnosis, the KRAS mutation disappeared after the initial course of chemotherapy and reappeared concurrently with or earlier than progression of the disease, highlighting the predictive factor for disease progression of PDAC patients [59].
Despite the potential of ddPCR to identify ctDNA KRAS mutations, most studies have analyzed KRAS mutations in ctDNA in order to verify the corresponding mutation in matched tumor tissues [57,69]. Finally, KRAS alleles have been assessed by quantitative ddPCR in a large series of patients with PDAC, pre-neoplastic pancreatic cyst and non-neoplastic pancreatic diseases [48,49]. Quantitative ddPCR found average KRAS MAF to be highest in baseline metastatic samples, followed by localized disease, cystic lesions and finally, non-neoplastic pancreatic diseases.
A similar approach for KRAS mutations has been described in other tumor types. KRAS is an oncogenic driver that appeared mutated in 30% of non-small cell lung carcinoma (NSCLC) and up to 50% of colon tumors. Furthermore, it is associated with a poor prognosis. The detection of KRAS mutations in these tumor types is a predictive biomarker for the anti-EGFR therapy. Wahl et al. reported plasma KRAS (G12A/C/D/S/V and G13D) mutant ctDNA detection using ddPCR in patients across various lung adenocarcinoma stages. They found that 38% of the patients had detectable KRAS mutation in plasma and it was significantly associated with shorter PFS and OS [70]. Similarly, Michaelidou et al. used multiplex ddPCR to quantify KRAS G12/G13 MAF in ctDNA from 114 pre-treated advance NSCLC patients. Again, plasma KRAS G12/G13 status was associated with poor patient outcome in terms of PFS and OS (p < 0.001, respectively) [71]. Finally, Guibert and collaborators detected KRAS mutations in up to 81% of the patients with a sensitivity of 78% [72]. The authors conclude that the presence of a KRAS mutation in cfDNA is correlated with a poor response to treatment. Thus, the detection of KRAS mutations in plasma could also serve as an independent biomarker of unfavorable prognosis in NSCLC patients. ddPCR is a precise and easily feasible technique for ctDNA quantification of KRAS mutations. However, it is important to note the limitations of this research approach. The detection of a single mutation may lead to underestimation of the true circulating tumor burden due to the stochastic nature of circulating nucleic acids. Furthermore, the majority of the studies did not evaluate hotspot mutations in codon 61, which may minimize the true sensitivity. DdPCR requires a priori knowledge of the mutant allele. Different multiplex analyses have been carried out that take into consideration probe concentrations and/or amplicon size [73,74]. Unfortunately, the actual capability is limited to 5–10 multiplex [73].
2.1.2. ddPCR Validation and Monitoring Approach
Tumor mutations are not known a priori in certain liquid biopsy applications, and therefore, all tumor mutations are queried at once. Recent studies confirmed the importance in the pathogenesis of PDAC of different mutations in various genes aside from KRAS, such as TP53, SMAD4 and CDKN2A [75]. NGS approaches have the potential to detect a broad range of molecular targets. Therefore, most studies have focused on the presence of ctDNA mutations in a comprehensive set of genes to highlight tumor heterogeneity and demonstrate clonal evolution over the course of disease progression. In these types of studies, ddPCR technology has been performed in order to validate NGS results and to follow-up the disease. Thus, NGS has been combined with ddPCR for liquid biopsy analysis [50,53,57,76].
Sausen et al. evaluated the utility of using somatic mutations in ctDNA to identify patients likely to recur after surgical intervention. They identified though tumor tissue whole-exome sequencing different somatic mutations likely to be detected in ctDNA of 51 patients. They used ddPCR, focusing on alterations in KRAS, BRAF and PIK3CA, for ctDNA detection prior and after tumor resection in localized PDAC [9]. Alterations were found in 43% of the patients at the time of diagnosis and the analyses revealed that patients with detectable ctDNA in their plasma after surgical resection were more likely to relapse (p = 0.02). Indeed, the authors detected ctDNA at 3.1 months after surgery, on average, compared with 9.6 months when it was clinically detectable on computed tomography scan (p = 0.0004). Based on these results, liquid biopsy could be a useful tool to identify residual or recurrent disease after surgical resection and early detection relapses. Similarly, Cheng and colleagues evaluated the clinical implications of ctDNA detection in metastatic pancreatic cancer patients. Firstly, they screened a panel of 60 genes in cfDNA from 10 metastatic pancreatic cancer patients through exome sequencing. Second, ddPCR was used to identify potential mutations in BRCA2, EGFR, ERBB2, KDR and KRAS, in a cohort of 188 metastatic pancreatic cancer patients [50]. The KRAS mutation rate was 72.3%, whereas BRCA2, KDR, EGFR, ERBB2 exon 17 and ERBB2 exon 27 were 11.7%, 13.8%, 13.3%, 13.3% and 6.4%, respectively. Seufferlein and coworkers analyzed the ctDNA and their corresponding tumor tissue DNA in a cohort of 20 PDAC patients by a targeted NGS and ddPCR gene panel (KRAS, TP53, SMAD4, CDKN2A, APC, ATM and FBXW7). The authors found mutations in up to 75% of the patients with a concordance rate of 80% between ctDNA and tumor tissue. They concluded that 96% of the mutations found in the ctDNA from naïve therapy patients were in KRAS and/or TP53 [69]. Finally, Berger et al. used a seven candidate-gene NGS approach to characterize tumor tissue DNA and ctDNA samples. The mutational status observed was then validated with ddPCR when the KRAS and TP53 NGS results were discordant between paired-samples. KRAS and TP53 mutations in ctDNA were detected but the authors concluded that mutations in other genes, such as SMAD4 or ATM, are rarely observed [77]. Pécuchet et al. reported high sensitivity and specificity for the detection of ctDNA from pancreatic or lung cancer patients using Base-Position Error Rate (BPER) NGS [41]. In a different study, Pietraz et al. evaluated the prognostic value of ctDNA detection with NGS and ddPCR in patients diagnosed with pancreatic adenocarcinoma. According to their results, the presence of ctDNA was strongly correlated with poor overall survival in patients with advanced disease (OS; 6.5 vs. 19.0 months; p < 0.001), and it could be an indicator of shorter disease-free survival when ctDNA is detected after surgery [13].
Watanabe et al. studied the usefulness of monitoring KRAS ctDNA throughout the course of the disease to predict prognosis and response to treatment in PDAC patients. In those cases, without detection of KRAS mutation during the first year after surgery their prognosis was better, regardless of relapse (p < 0.001). Furthermore, in patients receiving first-line chemotherapy in whom KRAS ctDNA was not detected or disappeared within 6 months, it was associated with a statistically significantly better response to treatment (p < 0.001) [60]. The combined strategy of NGS and ddPCR was suggested as a cost-effective and efficient method for analyzing ctDNA in PDAC patients [78].
3. Future Perspectives
PDAC remains a devastating disease. Extensive research efforts have focused on the discovery of early diagnostic biomarkers and efficient therapeutic approaches. Researchers and clinicians are trying to develop novel biomarkers and treatment options.
Current advances in our knowledge of the biology and clinical application of ctDNA have provided evidence that the use of ctDNA as a liquid biopsy can improve cancer diagnosis, monitoring and treatment management. Liquid biopsy is of great importance in PDAC as adequate tumor tissue is scarce. The trouble of obtaining enough tumor sample to carry out molecular studies in PDAC makes it difficult to advance the field of personalized therapy in this tumor type. For this reason, the possibility of performing genetic studies in peripheral blood takes on special relevance in PDAC and would probably stimulate the advancement in precision medicine in these patients. The advancement of liquid biopsy-based cancer research has been largely dependent on parallel advances in oncological genetics and genomics. The detection of KRAS mutations in plasma and serum ctDNA is one of the most frequently utilized liquid biopsy approaches for PDAC [79]. ctDNA is a valued diagnostic PDAC tool. Moreover, ctDNA is believed to play an important role in PDAC prognosis [80]. KRAS ctDNA MAF has been associated with PDAC clinical stage [81]. Although, a KRAS mutation in ctDNA is presumed to be a promising diagnostic biomarker in PDAC.
KRAS is one of the most common human oncogenes, being mutated in around 20–30% of all human cancers. However, KRAS inhibitors have largely failed and therefore, KRAS has earned the title of “undruggable”. Recently, the KRAS G12C inhibitor sotorasib has been approved by the FDA. Responses were observed in KRAS G12C tumor patients of different histologic subtypes, including up to 32% of NSCLC patients, 7% of colorectal patients and one pancreatic tumor patient [82]. The KRAS G12C mutation is the most common KRAS mutation in NSCLC, being around 13%, and it is between 1–3% in colorectal cancer. KRAS G12C mutation is rare in PDAC, being around 1% of all KRAS mutations. However, these results are very encouraging as a previously unapproachable target is now conceivable. The development of KRAS G12C targeted-agents is at an initial stage but provides hope for targeting other KRAS mutations as well as other undruggable targets. The detection of ctDNA KRAS mutations with ddPCR would make it a predictive biomarker.
In line with this, microsatellite instability (MSI) has recently emerged as a predictive pan-tumor biomarker of immunotherapy efficacy. New innovative approaches for MSI marker detection through ctDNA using ddPCR have been developed [83]. This method is straightforward and has the potential to be routinely applied clinically in ddPCR equipped laboratories, aiding in the diagnosis and prognosis of the disease and increasing personalized treatments.
Finally, technology is improving rapidly, and new multiplex strategies are being developed, allowing the simultaneous detection of various genetic alterations and consequently sample-saving material [84,85]. Multi-targeted ddPCR assays with gene hot spot mutations would have broad applicability for both clinical and translational research. ddPCR will become more accessible and clinically useful. However, there are still some challenges that should be addressed so that this technique may ultimately be employed into routine clinical practice.
4. Conclusions
Sequencing technologies develop quickly, and the understanding of ctDNA biology and clinical potential is deepening. Thus, although its application as a predictor biomarker is limited because of its low sensitivity, the eventual use of ctDNA in clinical practice seems to be assured. ddPCR has been recognized as one of the most suitable approaches for rare event detection. Furthermore, it is a cost-effective alternative to the currently used NGS platforms. However, this technology approach is only suitable for analysis when the prior knowledge about the mutation is available, emphasizing a personalized assay design. Additionally, this technology may be used in combination with NGS platforms as both methodologies can provide a robust and accurate quantitative measure of the fraction of mutant alleles.
Author Contributions
Conceptualization of the work, M.H., A.C. and M.I.-V.; acquisition, analysis and interpretation of data: M.H., S.R., L.S., A.F., N.T., D.R., V.G., C.A.-C., M.G.-A., A.C. and M.I.-V.; writing—original draft preparation, M.H., A.C. and M.I.-V.; writing—review and editing: M.H., S.R., L.S., A.F., N.T., D.R., V.G., C.A.-C., M.G.-A., A.C. and M.I.-V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Instituto de Salud Carlos III, grant number PI18/01909 to A.C. and by the Fundación Mutua Madrileña (2019) to M.I.-V.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Maisonneuve, P. Epidemiology and burden of pancreatic cancer. Presse Med. 2019, 48, e113–e123. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA Cancer J. Clin. 2013, 63, 318–348. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Ignatiadis, M.; Lee, M.; Jeffrey, S.S. Circulating Tumor Cells and Circulating Tumor DNA: Challenges and Opportunities on the Path to Clinical Utility. Clin. Cancer Res. 2015, 21, 4786–4800. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Hadano, N.; Murakami, Y.; Uemura, K.; Hashimoto, Y.; Kondo, N.; Nakagawa, N.; Sueda, T.; Hiyama, E. Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br. J. Cancer 2016, 115, 59–65. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Husain, H.; Velculescu, V.E. Cancer DNA in the Circulation: The Liquid Biopsy. JAMA 2017, 318, 1272–1274. [Google Scholar] [CrossRef]
- Pietrasz, D.; Pecuchet, N.; Garlan, F.; Didelot, A.; Dubreuil, O.; Doat, S.; Imbert-Bismut, F.; Karoui, M.; Vaillant, J.C.; Taly, V.; et al. Plasma Circulating Tumor DNA in Pancreatic Cancer Patients Is a Prognostic Marker. Clin. Cancer Res. 2017, 23, 116–123. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Isbell, J.M.; Jones, D.R.; Li, B.T. Circulating tumor DNA: A promising biomarker to guide postoperative treatment and surveillance of non-small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2018, 155, 2628–2631. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: A prospective biomarker study. Gut 2019, 68, 663–671. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A. Genetic evolution of pancreatic cancer: Lessons learnt from the pancreatic cancer genome sequencing project. Gut 2012, 61, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Goonetilleke, K.S.; Siriwardena, A.K. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur. J. Surg. Oncol. 2007, 33, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Gay, D.Z.; Brown, K.; Mulvihill, J.D.; Boucher, K.M.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. The clinical utility of CA 19-9 in pancreatic adenocarcinoma: Diagnostic and prognostic updates. Curr. Mol. Med. 2013, 13, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; Li, C.P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Castillo, C.F.F. A Changing Landscape in Pancreatic Cancer. Ann. Surg. 2018, 268, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Locker, G.Y.; Kindler, H.L. Maintenance Olaparib for Metastatic Pancreatic Cancer. Reply. N. Engl. J. Med. 2019, 381, 1492–1493. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Chakrabarty, M.; Cohn, E.M.; Leon, S.A. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 1983, 51, 2116–2120. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef]
- Yamada, T.; Nakamori, S.; Ohzato, H.; Oshima, S.; Aoki, T.; Higaki, N.; Sugimoto, K.; Akagi, K.; Fujiwara, Y.; Nishisho, I.; et al. Detection of K-ras gene mutations in plasma DNA of patients with pancreatic adenocarcinoma: Correlation with clinicopathological features. Clin. Cancer Res. 1998, 4, 1527–1532. [Google Scholar]
- Castells, A.; Puig, P.; Mora, J.; Boadas, J.; Boix, L.; Urgell, E.; Sole, M.; Capella, G.; Lluis, F.; Fernandez-Cruz, L.; et al. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: Diagnostic utility and prognostic significance. J. Clin. Oncol. 1999, 17, 578–584. [Google Scholar] [CrossRef]
- Tjensvoll, K.; Lapin, M.; Buhl, T.; Oltedal, S.; Steen-Ottosen Berry, K.; Gilje, B.; Soreide, J.A.; Javle, M.; Nordgard, O.; Smaaland, R. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol. Oncol. 2016, 10, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Vivaldi, C.; Rofi, E.; Vasile, E.; Miccoli, M.; Caparello, C.; d’Arienzo, P.D.; Fornaro, L.; Falcone, A.; Danesi, R. Early changes in plasma DNA levels of mutant KRAS as a sensitive marker of response to chemotherapy in pancreatic cancer. Sci. Rep. 2017, 7, 7931. [Google Scholar] [CrossRef]
- Yong, E. Cancer biomarkers: Written in blood. Nature 2014, 511, 524–526. [Google Scholar] [CrossRef]
- Postel, M.; Roosen, A.; Laurent-Puig, P.; Taly, V.; Wang-Renault, S.F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert Rev. Mol. Diagn. 2018, 18, 7–17. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Pecuchet, N.; Rozenholc, Y.; Zonta, E.; Pietrasz, D.; Didelot, A.; Combe, P.; Gibault, L.; Bachet, J.B.; Taly, V.; Fabre, E.; et al. Analysis of Base-Position Error Rate of Next-Generation Sequencing to Detect Tumor Mutations in Circulating DNA. Clin. Chem. 2016, 62, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Diehl, F.; Dressman, D.; Vogelstein, B.; Kinzler, K.W. BEAMing up for detection and quantification of rare sequence variants. Nat. Methods 2006, 3, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Lu, H.; Garlan, F.; Taly, V. Droplet-Based Digital PCR: Application in Cancer Research. Adv. Clin. Chem. 2017, 79, 43–91. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Cheung, M.; Bushell, K.; Arthur, S.E.; Wong, H.L.; Karasinska, J.; Renouf, D.; Schaeffer, D.F.; McNamara, S.; Tertre, M.C.D.; et al. A Novel Multiplex Droplet Digital PCR Assay to Identify and Quantify KRAS Mutations in Clinical Specimens. J. Mol. Diagn. 2019, 21, 214–227. [Google Scholar] [CrossRef]
- Ako, S.; Nouso, K.; Kinugasa, H.; Dohi, C.; Matushita, H.; Mizukawa, S.; Muro, S.; Akimoto, Y.; Uchida, D.; Tomoda, T.; et al. Utility of serum DNA as a marker for KRAS mutations in pancreatic cancer tissue. Pancreatology 2017, 17, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.W.; Schwerdel, D.; Costa, I.G.; Hackert, T.; Strobel, O.; Lam, S.; Barth, T.F.; Schroppel, B.; Meining, A.; Buchler, M.W.; et al. Detection of Hot-Spot Mutations in Circulating Cell-Free DNA From Patients With Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2016, 151, 267–270. [Google Scholar] [CrossRef]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated With Outcomes of Patients With Pancreatic Cancer. Gastroenterology 2019, 156, 108–118. [Google Scholar] [CrossRef]
- Cheng, H.; Liu, C.; Jiang, J.; Luo, G.; Lu, Y.; Jin, K.; Guo, M.; Zhang, Z.; Xu, J.; Liu, L.; et al. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int. J. Cancer 2017, 140, 2344–2350. [Google Scholar] [CrossRef]
- Cheng, H.; Luo, G.; Jin, K.; Fan, Z.; Huang, Q.; Gong, Y.; Xu, J.; Yu, X.; Liu, C. Kras mutation correlating with circulating regulatory T cells predicts the prognosis of advanced pancreatic cancer patients. Cancer Med. 2020, 9, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Earl, J.; Garcia-Nieto, S.; Martinez-Avila, J.C.; Montans, J.; Sanjuanbenito, A.; Rodriguez-Garrote, M.; Lisa, E.; Mendia, E.; Lobo, E.; Malats, N.; et al. Circulating tumor cells (Ctc) and kras mutant circulating free Dna (cfdna) detection in peripheral blood as biomarkers in patients diagnosed with exocrine pancreatic cancer. BMC Cancer 2015, 15, 797. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Shi, X.; Shen, J.; Gao, S.; Wang, H.; Shen, S.; Pan, Y.; Li, B.; Xu, X.; Shao, Z.; et al. Preoperative detection of KRAS G12D mutation in ctDNA is a powerful predictor for early recurrence of resectable PDAC patients. Br. J. Cancer 2020, 122, 857–867. [Google Scholar] [CrossRef]
- Kim, M.K.; Woo, S.M.; Park, B.; Yoon, K.A.; Kim, Y.H.; Joo, J.; Lee, W.J.; Han, S.S.; Park, S.J.; Kong, S.Y. Prognostic Implications of Multiplex Detection of KRAS Mutations in Cell-Free DNA from Patients with Pancreatic Ductal Adenocarcinoma. Clin. Chem. 2018, 64, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, H.; Nouso, K.; Miyahara, K.; Morimoto, Y.; Dohi, C.; Tsutsumi, K.; Kato, H.; Matsubara, T.; Okada, H.; Yamamoto, K. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 2015, 121, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Alnaggar, M.; Liang, S.; Chen, J.; Xu, K.; Dong, S.; Du, D.; Niu, L. Circulating Tumor DNA as a Sensitive Marker in Patients Undergoing Irreversible Electroporation for Pancreatic Cancer. Cell. Physiol. Biochem. 2018, 47, 1556–1564. [Google Scholar] [CrossRef]
- Mohan, S.; Ayub, M.; Rothwell, D.G.; Gulati, S.; Kilerci, B.; Hollebecque, A.; Sun Leong, H.; Smith, N.K.; Sahoo, S.; Descamps, T.; et al. Analysis of circulating cell-free DNA identifies KRAS copy number gain and mutation as a novel prognostic marker in Pancreatic cancer. Sci. Rep. 2019, 9, 11610. [Google Scholar] [CrossRef]
- Sefrioui, D.; Blanchard, F.; Toure, E.; Basile, P.; Beaussire, L.; Dolfus, C.; Perdrix, A.; Paresy, M.; Antonietti, M.; Iwanicki-Caron, I.; et al. Diagnostic value of CA19.9, circulating tumour DNA and circulating tumour cells in patients with solid pancreatic tumours. Br. J. Cancer 2017, 117, 1017–1025. [Google Scholar] [CrossRef]
- Sugimori, M.; Sugimori, K.; Tsuchiya, H.; Suzuki, Y.; Tsuyuki, S.; Kaneta, Y.; Hirotani, A.; Sanga, K.; Tozuka, Y.; Komiyama, S.; et al. Quantitative monitoring of circulating tumor DNA in patients with advanced pancreatic cancer undergoing chemotherapy. Cancer Sci. 2020, 111, 266–278. [Google Scholar] [CrossRef]
- Watanabe, F.; Suzuki, K.; Tamaki, S.; Abe, I.; Endo, Y.; Takayama, Y.; Ishikawa, H.; Kakizawa, N.; Saito, M.; Futsuhara, K.; et al. Longitudinal monitoring of KRAS-mutated circulating tumor DNA enables the prediction of prognosis and therapeutic responses in patients with pancreatic cancer. PLoS ONE 2019, 14, e0227366. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Kim, M.K.; Joo, J.; Yoon, K.A.; Park, B.; Park, S.J.; Han, S.S.; Lee, J.H.; Hong, E.K.; Kim, Y.H.; et al. Induction Chemotherapy with Gemcitabine and Cisplatin Followed by Simultaneous Integrated Boost-Intensity Modulated Radiotherapy with Concurrent Gemcitabine for Locally Advanced Unresectable Pancreatic Cancer: Results from a Feasibility Study. Cancer Res. Treat. 2017, 49, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef]
- Lee, B.; Cohen, J.; Lipton, R.L.; Tie, J.; Javed, A.A.; Li, L.; Goldstein, D.; Cooray, P.; Nagrial, A.; Burge, M.E.; et al. Potential role of circulating tumor DNA (ctDNA) in the early diagnosis and post-operative management of localised pancreatic cancer. J. Clin. Oncol. 2017, 35, 4101. [Google Scholar] [CrossRef]
- Jen, J.; Wu, L.; Sidransky, D. An overview on the isolation and analysis of circulating tumor DNA in plasma and serum. Ann. N. Y. Acad. Sci. 2000, 906, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Montalvo, L.; Chrebtow, V.; Busch, M.P. Quantitation of genomic DNA in plasma and serum samples: Higher concentrations of genomic DNA found in serum than in plasma. Transfusion 2001, 41, 276–282. [Google Scholar] [CrossRef]
- Taback, B.; O’Day, S.J.; Hoon, D.S. Quantification of circulating DNA in the plasma and serum of cancer patients. Ann. N. Y. Acad. Sci. 2004, 1022, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Marcq, M.; Bizieux, A.; Kouri, C.E.; Lacroix, H.; Bennouna, J.; Douillard, J.Y.; Denis, M.G. Plasma is a better source of tumor-derived circulating cell-free DNA than serum for the detection of EGFR alterations in lung tumor patients. Lung Cancer 2013, 82, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.F.; Jakobsen, A. Screening for circulating RAS/RAF mutations by multiplex digital PCR. Clin. Chim. Acta. 2016, 458, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Seufferlein, T.; Berger, A.W.; Schwerdel, D.; Ettrich, T.J.; Schmidt, S.A.; Kleger, A.; Marienfeld, R. Non-invasive diagnosis and tracking of tumor evolution by targeted sequencing of circulating tumor DNA in metastatic pancreatic cancer patients. J. Clin. Oncol. 2017, 35, e15769. [Google Scholar] [CrossRef]
- Wahl, S.G.F.; Dai, H.Y.; Emdal, E.F.; Ottestad, A.L.; Dale, V.G.; Richardsen, E.; Halvorsen, T.O.; Gronberg, B.H. Prognostic value of absolute quantification of mutated KRAS in circulating tumour DNA in lung adenocarcinoma patients prior to therapy. J. Pathol. Clin. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Michaelidou, K.; Koutoulaki, C.; Mavridis, K.; Vorrias, E.; Papadaki, M.A.; Koutsopoulos, A.V.; Mavroudis, D.; Agelaki, S. Detection of KRAS G12/G13 Mutations in Cell Free-DNA by Droplet Digital PCR, Offers Prognostic Information for Patients with Advanced Non-Small Cell Lung Cancer. Cells 2020, 9, 2514. [Google Scholar] [CrossRef]
- Guibert, N.; Pradines, A.; Farella, M.; Casanova, A.; Gouin, S.; Keller, L.; Favre, G.; Mazieres, J. Monitoring KRAS mutations in circulating DNA and tumor cells using digital droplet PCR during treatment of KRAS-mutated lung adenocarcinoma. Lung Cancer 2016, 100, 1–4. [Google Scholar] [CrossRef]
- Zhong, Q.; Bhattacharya, S.; Kotsopoulos, S.; Olson, J.; Taly, V.; Griffiths, A.D.; Link, D.R.; Larson, J.W. Multiplex digital PCR: Breaking the one target per color barrier of quantitative PCR. Lab Chip 2011, 11, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- McDermott, G.P.; Do, D.; Litterst, C.M.; Maar, D.; Hindson, C.M.; Steenblock, E.R.; Legler, T.C.; Jouvenot, Y.; Marrs, S.H.; Bemis, A.; et al. Multiplexed target detection using DNA-binding dye chemistry in droplet digital PCR. Anal. Chem. 2013, 85, 11619–11627. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.; Del Rio Hernandez, A. The Mutational Landscape of Pancreatic and Liver Cancers, as Represented by Circulating Tumor DNA. Front. Oncol. 2019, 9, 952. [Google Scholar] [CrossRef]
- Adamo, P.; Cowley, C.M.; Neal, C.P.; Mistry, V.; Page, K.; Dennison, A.R.; Isherwood, J.; Hastings, R.; Luo, J.; Moore, D.A.; et al. Profiling tumour heterogeneity through circulating tumour DNA in patients with pancreatic cancer. Oncotarget 2017, 8, 87221–87233. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.W.; Schwerdel, D.; Ettrich, T.J.; Hann, A.; Schmidt, S.A.; Kleger, A.; Marienfeld, R.; Seufferlein, T. Targeted deep sequencing of circulating tumor DNA in metastatic pancreatic cancer. Oncotarget 2018, 9, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Takai, E.; Totoki, Y.; Nakamura, H.; Morizane, C.; Nara, S.; Hama, N.; Suzuki, M.; Furukawa, E.; Kato, M.; Hayashi, H.; et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci. Rep. 2015, 5, 18425. [Google Scholar] [CrossRef] [PubMed]
- Buscail, E.; Maulat, C.; Muscari, F.; Chiche, L.; Cordelier, P.; Dabernat, S.; Alix-Panabieres, C.; Buscail, L. Liquid Biopsy Approach for Pancreatic Ductal Adenocarcinoma. Cancers (Basel) 2019, 11, 852. [Google Scholar] [CrossRef]
- Fang, Z.; Meng, Q.; Zhang, B.; Shi, S.; Liu, J.; Liang, C.; Hua, J.; Yu, X.; Xu, J.; Wang, W. Prognostic value of circulating tumor DNA in pancreatic cancer: A systematic review and meta-analysis. Aging (Albany NY) 2020, 12. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Ding, X.Q.; Zhu, H.; Wang, R.X.; Pan, X.R.; Tong, J.H. KRAS Mutant Allele Fraction in Circulating Cell-Free DNA Correlates With Clinical Stage in Pancreatic Cancer Patients. Front. Oncol. 2019, 9, 1295. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Silveira, A.B.; Bidard, F.C.; Kasperek, A.; Melaabi, S.; Tanguy, M.L.; Rodrigues, M.; Bataillon, G.; Cabel, L.; Buecher, B.; Pierga, J.Y.; et al. High-Accuracy Determination of Microsatellite Instability Compatible with Liquid Biopsies. Clin. Chem. 2020, 66, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Hussung, S.; Follo, M.; Klar, R.F.U.; Michalczyk, S.; Fritsch, K.; Nollmann, F.; Hipp, J.; Duyster, J.; Scherer, F.; von Bubnoff, N.; et al. Development and Clinical Validation of Discriminatory Multitarget Digital Droplet PCR Assays for the Detection of Hot Spot KRAS and NRAS Mutations in Cell-Free DNA. J. Mol. Diagn. 2020, 22, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Peitz, C.; Sprussel, A.; Linke, R.B.; Astrahantseff, K.; Grimaldi, M.; Schmelz, K.; Toedling, J.; Schulte, J.H.; Fischer, M.; Messerschmidt, C.; et al. Multiplexed Quantification of Four Neuroblastoma DNA Targets in a Single Droplet Digital PCR Reaction. J. Mol. Diagn. 2020. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).