
Silk Fibroin Nanoparticle Functionalization with Arg-Gly-Asp Cyclopentapeptide Promotes Active Targeting for Tumor Site-Specific Delivery

 ,
,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Silk Fibroin Extraction
2.3. Silk Fibroin Nanoparticle Preparation
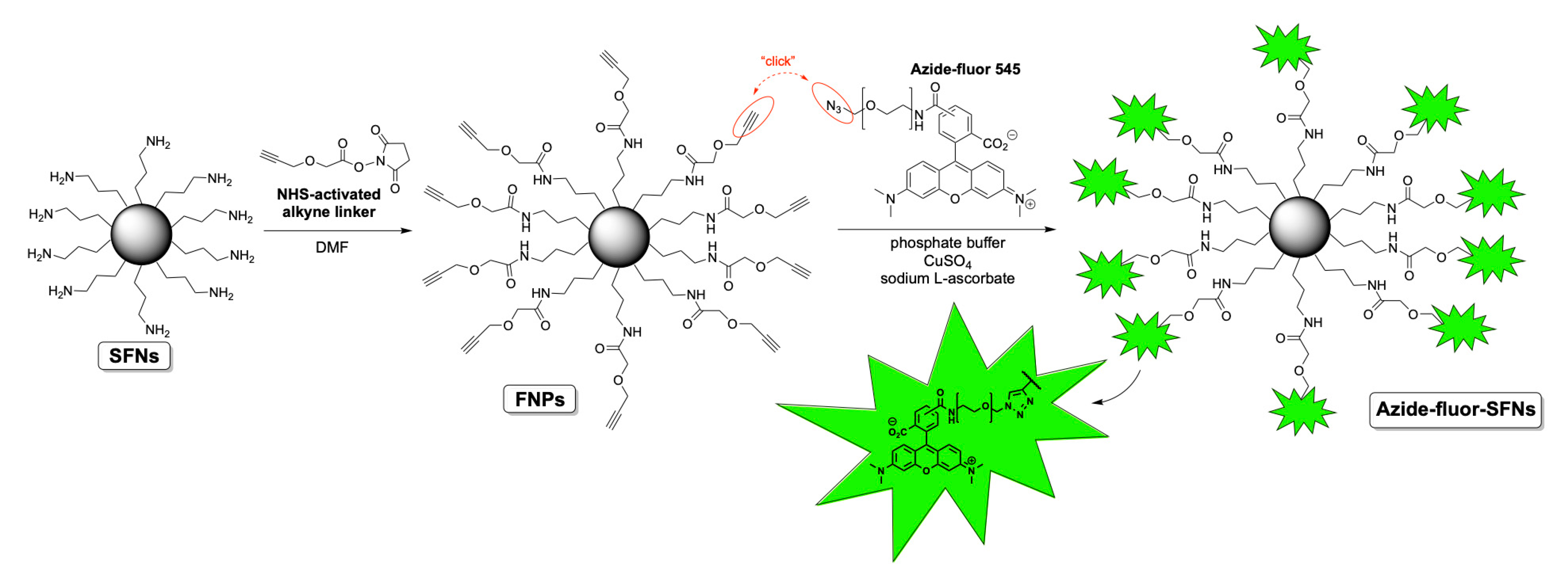
2.4. Functionalization of Silk Fibroin Nanoparticles with Alkyne Linker 4 and Bioconjugation with Azide-Fluor 545
2.5. Determination of Azide-Fluor 545 Conjugated on SFNs
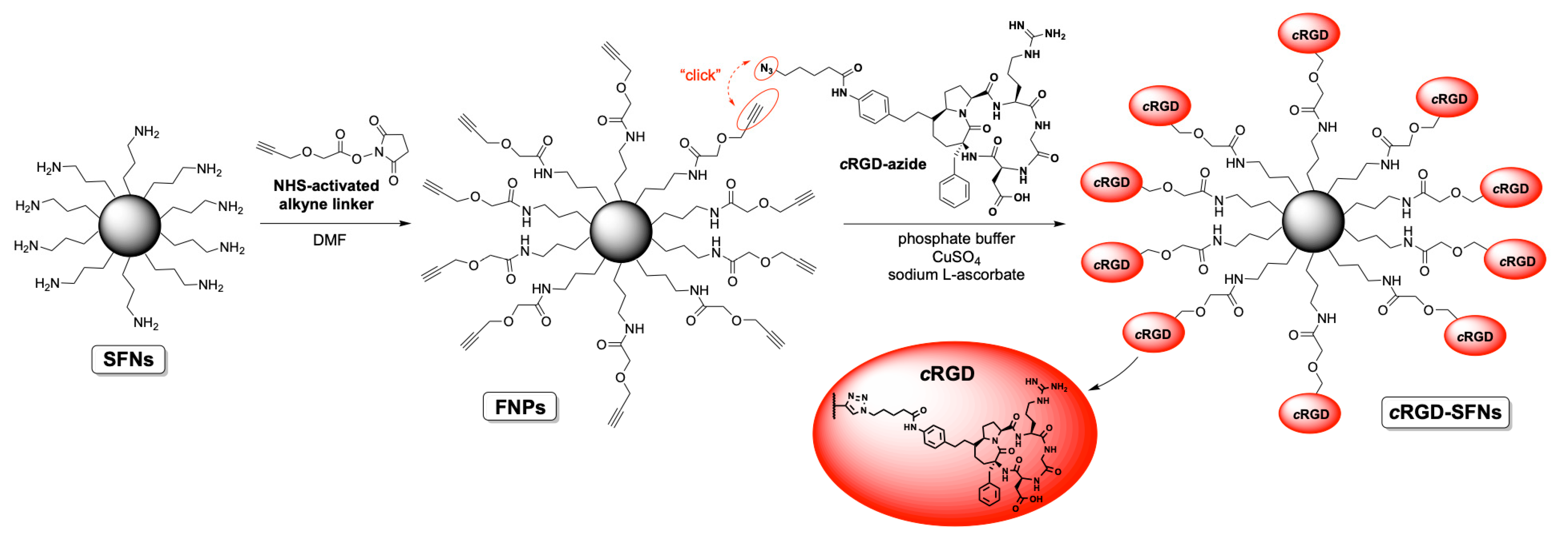
2.6. Silk Fibroin Nanoparticle Functionalization with RGD
2.7. Characterization of Nanoparticles
2.7.1. Production Yield, Drug Loading and Encapsulation Efficiency Evaluation
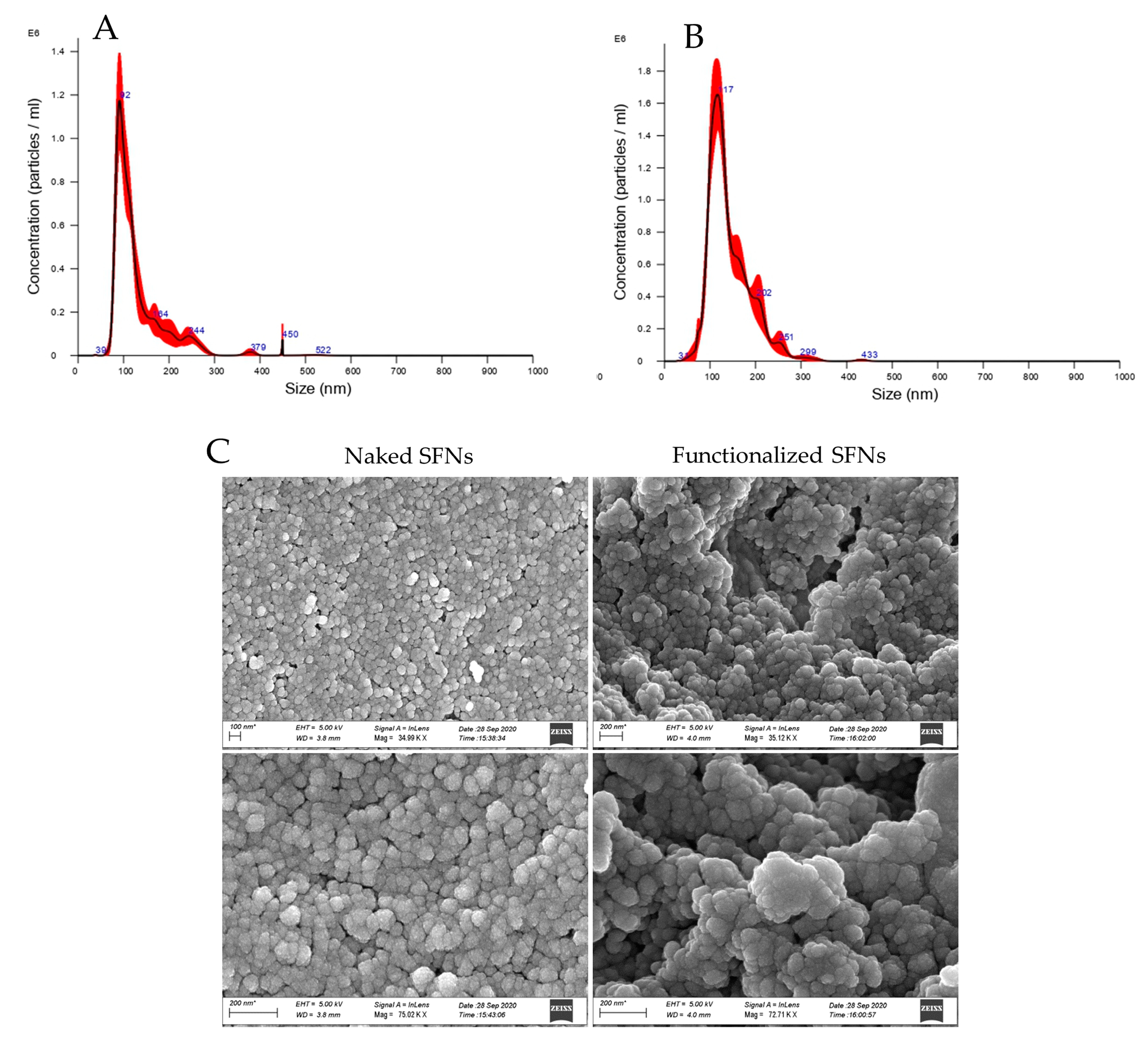
2.7.2. Nanoparticle Size Distribution and Zeta Potential
2.7.3. Morphological Evaluation by Scanning Electron Microscopy (SEM)
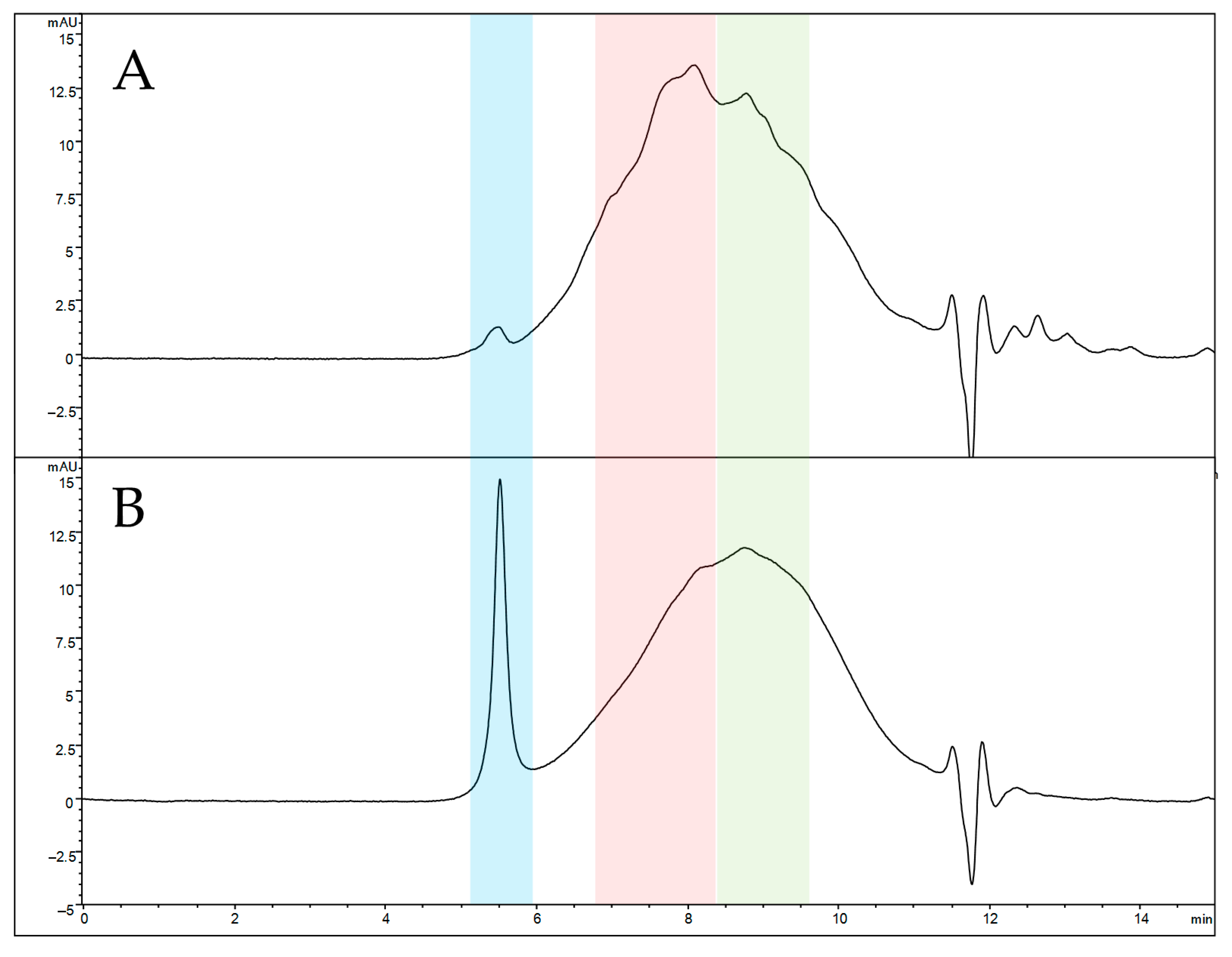
2.7.4. SEC-UV Analysis
2.8. In Vitro Biological Assays
2.8.1. Cell Culture
2.8.2. Real-Time qRT-PCR
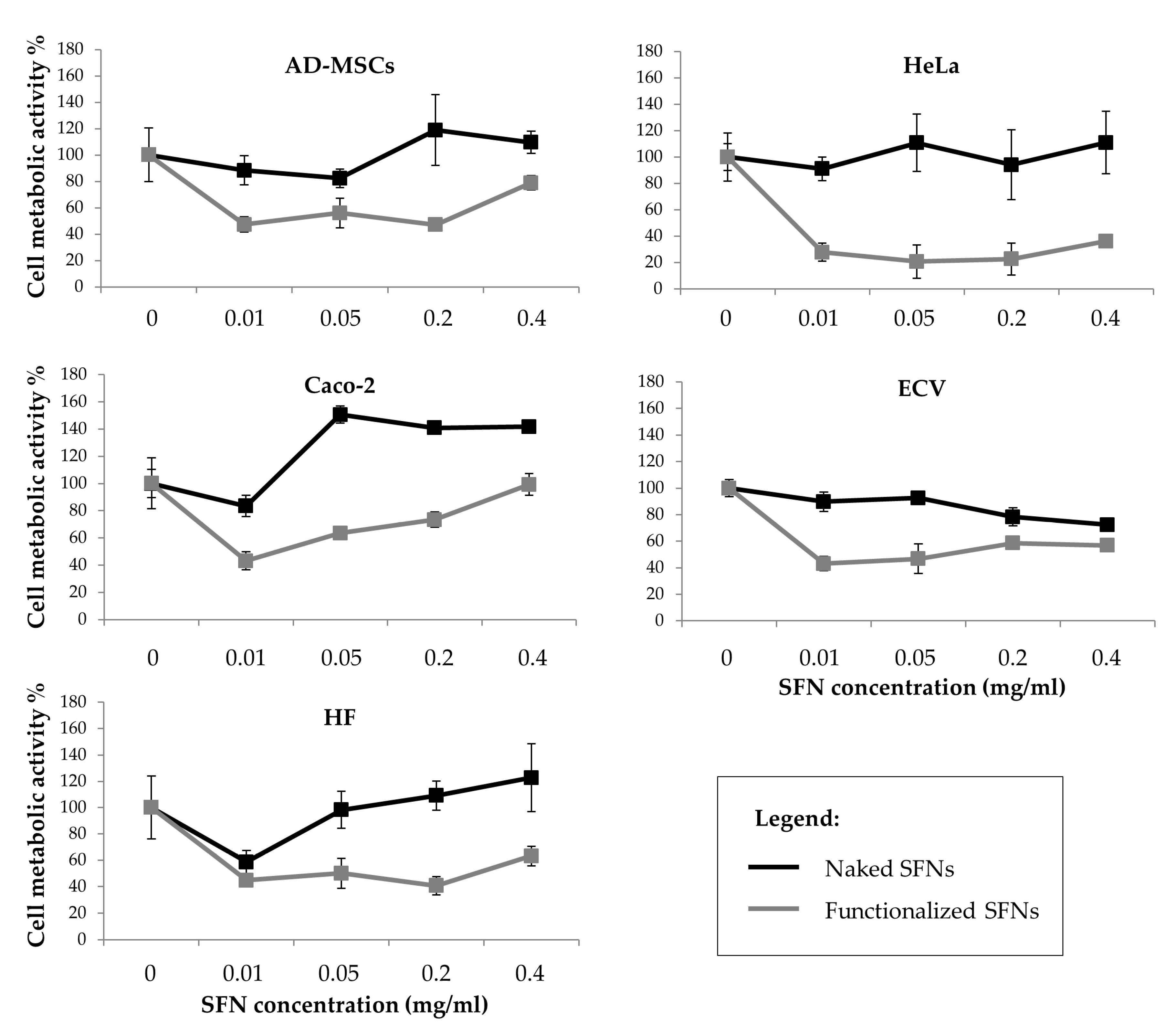
2.8.3. Cell Metabolic Activity Evaluation
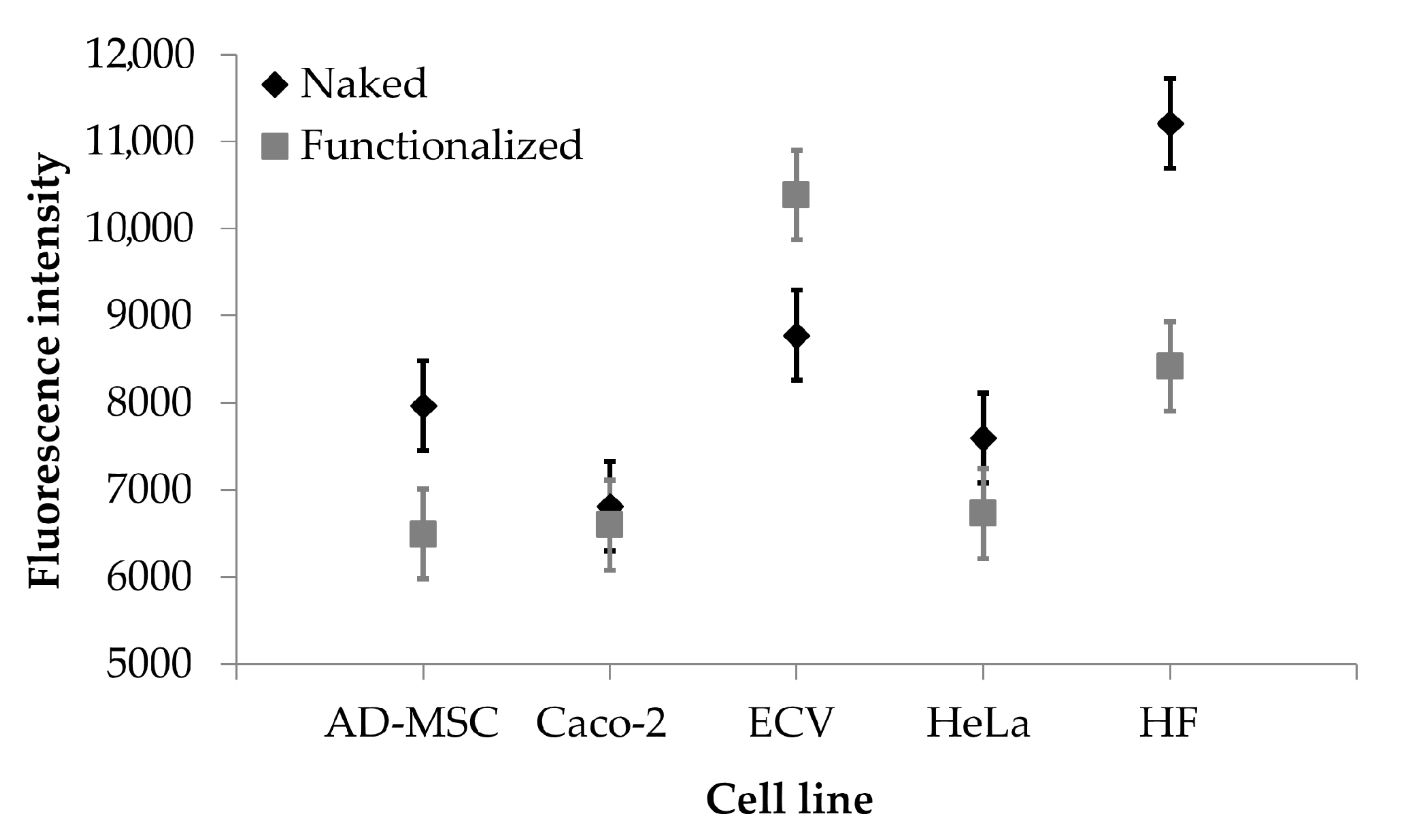
2.8.4. Cell Uptake Evaluation
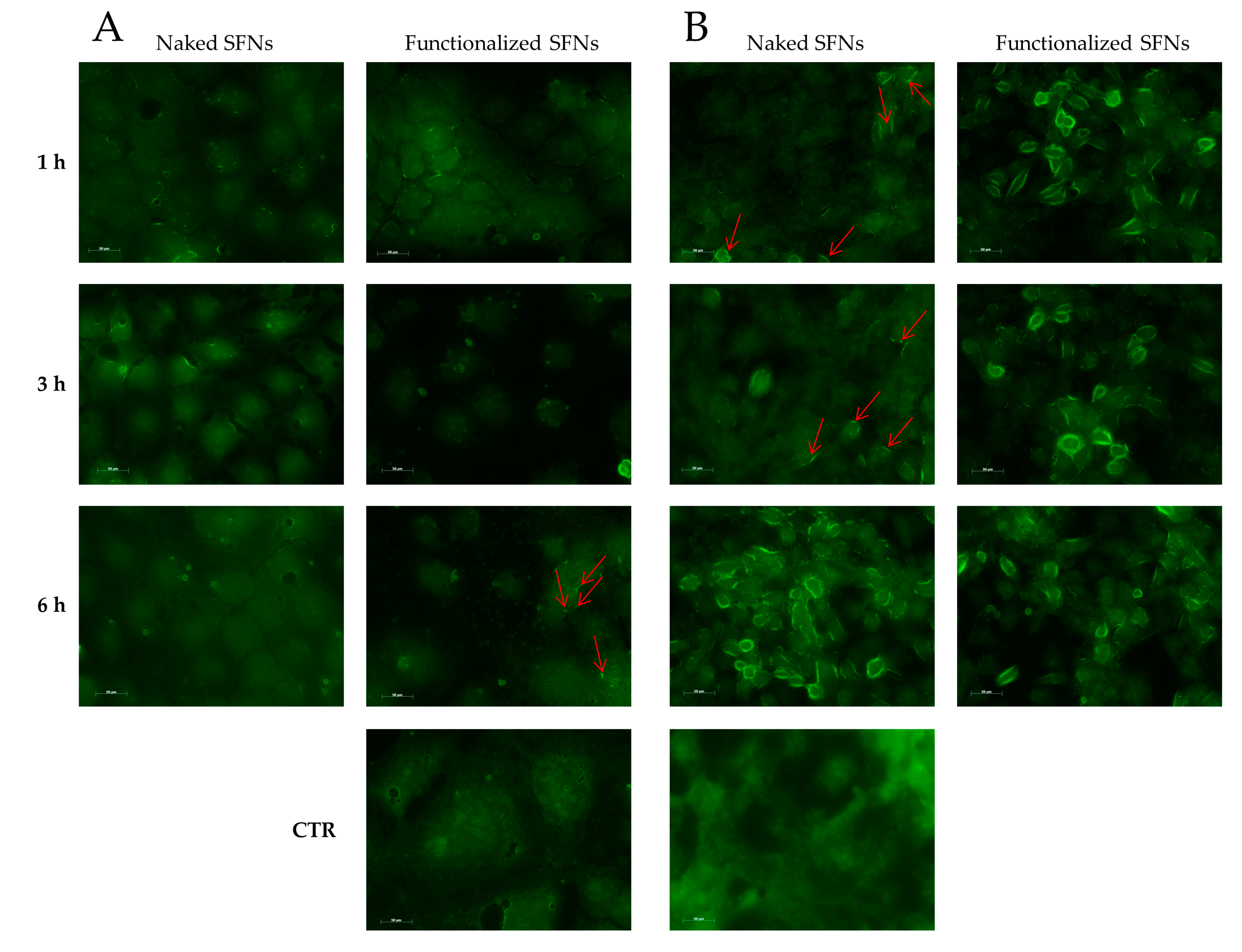
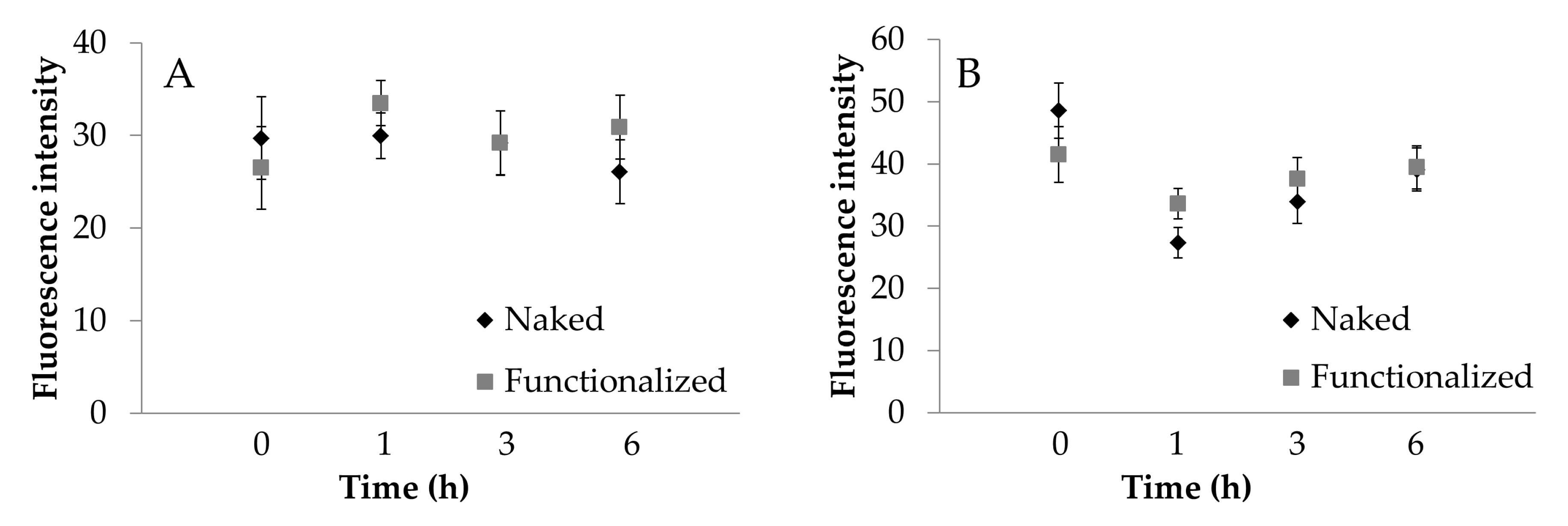
2.8.5. Fluorescence Microscopy Image Analysis
2.9. Statistical Analysis
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, C.-Y.; Cheng, R.; Yang, Z.; Tian, Z.-M. Nanotechnology for Cancer Therapy Based on Chemotherapy. Molecules 2018, 23, 826. [Google Scholar] [CrossRef] [PubMed]
- Markham, M.J.; Wachter, K.; Agarwal, N.; Bertagnolli, M.M.; Chang, S.M.; Dale, W.; Diefenbach, C.S.M.; Rodriguez-Galindo, C.; George, D.J.; Gilligan, T.D.; et al. Clinical Cancer Advances 2020: Annual Report on Progress Against Cancer From the American Society of Clinical Oncology. J. Clin. Oncol. 2020, 38, 1081. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Dal Corso, A.; Neri, D. Targeted Delivery of Cytotoxic Drugs: Challenges, Opportunities and New Developments. Chimia 2017, 71, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Lademann, J.; Richter, H.; Knorr, F.; Patzelt, A.; Darvin, M.E.; Ruehl, E.; Cheung, K.Y.; Lai, K.K.; Renneberg, R.; Mak, W.C. Triggered release of model drug from AuNP-doped BSA nanocarriers in hair follicles using IRA radiation. Acta Biomater. 2016, 30, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Ma, G.; Kampf, N.; Yuan, Z.; Chen, S. Development of Long-Circulating Zwitterionic Cross-Linked Micelles for Active-Targeted Drug Delivery. Biomacromolecules 2016, 17, 2010–2018. [Google Scholar] [CrossRef] [PubMed]
- Llopis-Lorente, A.; Garcia-Fernandez, A.; Murillo-Cremaes, N.; Hortelao, A.C.; Patino, T.; Villalonga, R.; Sancenon, F.; Martinez-Manez, R.; Sanchez, S. Enzyme-Powered Gated Mesoporous Silica Nanomotors for On-Command Intracellular Payload Delivery. ACS Nano 2019, 13, 12171–12183. [Google Scholar] [CrossRef]
- Crivelli, B.; Perteghella, S.; Bari, E.; Sorrenti, M.; Tripodo, G.; Chlapanidas, T.; Torre, M.L. Silk nanoparticles: From inert supports to bioactive natural carriers for drug delivery. Soft Matter 2018, 14, 546–557. [Google Scholar] [CrossRef]
- Perteghella, S.; Crivelli, B.; Catenacci, L.; Sorrenti, M.; Bruni, G.; Necchi, V.; Vigani, B.; Sorlini, M.; Torre, M.L.; Chlapanidas, T. Stem cell-extracellular vesicles as drug delivery systems: New frontiers for silk/curcumin nanoparticles. Int. J. Pharm. 2017, 520, 86–97. [Google Scholar] [CrossRef]
- Crivelli, B.; Bari, E.; Perteghella, S.; Catenacci, L.; Sorrenti, M.; Mocchi, M.; Farago, S.; Tripodo, G.; Prina-Mello, A.; Torre, M.L. Silk fibroin nanoparticles for celecoxib and curcumin delivery: ROS-scavenging and anti-inflammatory activities in an in vitro model of osteoarthritis. Eur. J. Pharm. Biopharm. 2019, 137, 37–45. [Google Scholar] [CrossRef]
- Perteghella, S.; Sottani, C.; Cocce, V.; Negri, S.; Cavicchini, L.; Alessandri, G.; Cottica, D.; Torre, M.L.; Grignani, E.; Pessina, A. Paclitaxel-Loaded Silk Fibroin Nanoparticles: Method Validation by UHPLC-MS/MS to Assess an Exogenous Approach to Load Cytotoxic Drugs. Pharmaceutics 2019, 11, 285. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Wang, Y.; Gao, H.; Wei, G.; Huang, Y.; Yu, H.; Gan, Y.; Wang, Y.; Mei, L.; et al. Recent progress in drug delivery. Acta Pharm. Sin. B 2019, 9, 1145–1162. [Google Scholar] [CrossRef]
- Danhier, F.; Feron, O.; Preat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Paolillo, M.; Russo, M.A.; Serra, M.; Colombo, L.; Schinelli, S. Small Molecule Integrin Antagonists in Cancer Therapy. Mini-Rev. Med. Chem. 2009, 9, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Schinelli, S. Integrins and Exosomes, a Dangerous Liaison in Cancer Progression. Cancers 2017, 9, 95. [Google Scholar] [CrossRef]
- Paolillo, M.; Serra, M.; Schinelli, S. Integrins in glioblastoma: Still an attractive target? Pharmacol. Res. 2016, 113, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Nogales, A.; Algieri, F.; De Matteis, L.; Lozano-Perez, A.A.; Garrido-Mesa, J.; Vezza, T.; de la Fuente, J.M.; Cenis, J.L.; Galvez, J.; Rodriguez-Cabezas, M.E. Intestinal anti-inflammatory effects of RGD-functionalized silk fibroin nanoparticles in trinitrobenzenesulfonic acid-induced experimental colitis in rats. Int. J. Nanomed. 2016, 11, 5945–5958. [Google Scholar] [CrossRef]
- Mao, B.; Liu, C.; Zheng, W.; Li, X.; Ge, R.; Shen, H.; Guo, X.; Lian, Q.; Shen, X.; Li, C. Cyclic cRGDfk peptide and Chlorin e6 functionalized silk fibroin nanoparticles for targeted drug delivery and photodynamic therapy. Biomaterials 2018, 161, 306–320. [Google Scholar] [CrossRef]
- Serra, M.; Tambini, S.M.; Di Giacomo, M.; Peviani, E.G.; Belvisi, L.; Colombo, L. Synthesis of Easy-to-Functionalize Azabicycloalkane Scaffolds as Dipeptide Turn Mimics en Route to cRGD-Based Bioconjugates. Eur. J. Org. Chem. 2015, 2015, 7557–7570. [Google Scholar] [CrossRef]
- Serra, M.; Peviani, E.G.; Bernardi, E.; Colombo, L. Synthesis of Variously Functionalized Azabicycloalkane Scaffolds by Domino Metathesis Reactions. J. Org. Chem. 2017, 82, 11091–11101. [Google Scholar] [CrossRef]
- Serra, M.; Bernardi, E.; De Lorenzi, E.; Colombo, L. Synthesis of Functionalized 6,5-and 7,5-Azabicycloalkane Amino Acids by Metathesis Reactions. J. Org. Chem. 2019, 84, 15726–15734. [Google Scholar] [CrossRef]
- Arosio, D.; Belvisi, L.; Colombo, L.; Colombo, M.; Invernizzi, D.; Manzoni, L.; Potenza, D.; Serra, M.; Castorina, M.; Pisano, C.; et al. A Potent Integrin Antagonist from a Small Library of Cyclic RGD Pentapeptide Mimics Including Benzyl-Substituted Azabicycloalkane Amino Acids. ChemMedChem 2008, 3, 1589–1603. [Google Scholar] [CrossRef]
- Russo, M.A.; Paolillo, M.; Sanchez-Hernandez, Y.; Curti, D.; Ciusani, E.; Serra, M.; Colombo, L.; Schinelli, S. A small-molecule RGD-integrin antagonist inhibits cell adhesion, cell migration and induces anoikis in glioblastoma cells. Int. J. Oncol. 2013, 42, 83–92. [Google Scholar] [CrossRef]
- Paolillo, M.; Galiazzo, M.C.; Daga, A.; Ciusani, E.; Serra, M.; Colombo, L.; Schinelli, S. An RGD small-molecule integrin antagonist induces detachment-mediated anoikis in glioma cancer stem cells. Int. J. Oncol. 2018, 53, 2683–2694. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Colombo, R.; Serra, M.; Belvisi, L.; Papetti, A.; Ciusani, E.; Comincini, S.; Schinelli, S. Stem-Like Cancer Cells in a Dynamic 3D Culture System: A Model to Study Metastatic Cell Adhesion and Anti-Cancer Drugs. Cells 2019, 8, 1434. [Google Scholar] [CrossRef] [PubMed]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, D.N.; Preda, R.C.; Yucel, T.; Wang, X.; Lovett, M.L.; Kaplan, D.L. Materials fabrication from Bombyx mori silk fibroin. Nat. Protoc. 2011, 6, 1612–1631. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.M.; Drummy, L.F.; Conrady, D.G.; Fox, D.M.; Naik, R.R.; Stone, M.O.; Trulove, P.C.; De Long, H.C.; Mantz, R.A. Dissolution and regeneration of Bombyx mori Silk fibroin using ionic liquids. J. Am. Chem. Soc. 2004, 126, 14350–14351. [Google Scholar] [CrossRef]
- Tengattini, S.; Orlandi, G.; Perteghella, S.; Bari, E.; Amadio, M.; Calleri, E.; Massolini, G.; Torre, M.L.; Temporini, C. Chromatographic profiling of silk sericin for biomedical and cosmetic use by complementary hydrophylic, reversed phase and size exclusion chromatographic methods. J. Pharm. Biomed. Anal. 2020, 186, 113291. [Google Scholar] [CrossRef]
- Bari, E.; Perteghella, S.; Di Silvestre, D.; Sorlini, M.; Catenacci, L.; Sorrenti, M.; Marrubini, G.; Rossi, R.; Tripodo, G.; Mauri, P.; et al. Pilot Production of Mesenchymal Stem/Stromal Freeze-Dried Secretome for Cell-Free Regenerative Nanomedicine: A Validated GMP-Compliant Process. Cells 2018, 7, 190. [Google Scholar] [CrossRef]
- Bari, E.; Ferrarotti, I.; Di Silvestre, D.; Grisoli, P.; Barzon, V.; Balderacchi, A.; Torre, M.L.; Rossi, R.; Mauri, P.; Corsico, A.G.; et al. Adipose Mesenchymal Extracellular Vesicles as Alpha-1-Antitrypsin Physiological Delivery Systems for Lung Regeneration. Cells 2019, 8, 965. [Google Scholar] [CrossRef]
- Bari, E.; Perteghella, S.; Catenacci, L.; Sorlini, M.; Croce, S.; Mantelli, M.; Avanzini, M.A.; Sorrenti, M.; Torre, M.L. Freeze-dried and GMP-compliant pharmaceuticals containing exosomes for acellular mesenchymal stromal cell immunomodulant therapy. Nanomedicine 2019. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Corti, M.; Calleri, E.; Perteghella, S.; Ferrara, A.; Tamma, R.; Milanese, C.; Mandracchia, D.; Brusotti, G.; Torre, M.L.; Ribatti, D.; et al. Polyacrylate/polyacrylate-PEG biomaterials obtained by high internal phase emulsions (HIPEs) with tailorable drug release and effective mechanical and biological properties. Mater. Sci. Eng. C-Mater. Biol. Appl. 2019, 105. [Google Scholar] [CrossRef]
- Collins, T.J. ImageJ for microscopy. Biotechniques 2007, 43, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Li, Y.; Piccinini, F.; Csucs, G.; Balazs, C.; Bevilacqua, A.; Horvath, P. CIDRE: An illumination-correction method for optical microscopy. Nat. Methods 2015, 12, 404–406. [Google Scholar] [CrossRef]
- 2021_BariElia_Collection1. Available online: https://doi.org/10.6084/m9.figshare.c.5270543 (accessed on 1 January 2021).
- Crivelli, B.; Chlapanidas, T.; Perteghella, S.; Lucarelli, E.; Pascucci, L.; Brini, A.T.; Ferrero, I.; Marazzi, M.; Pessina, A.; Torre, M.L.; et al. Mesenchymal stem/stromal cell extracellular vesicles: From active principle to next generation drug delivery system. J. Control. Release 2017, 262, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Wang, B.; Dutta, P. Nanoparticle processing: Understanding and controlling aggregation. Adv. Colloid Interface Sci. 2020, 279. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.; Chung, Y.-I.; Kim, Y.H.; Taeb, G.; Kundu, S.C. Silk fibroin nanoparticles for cellular uptake and control release. Int. J. Pharm. 2010, 388, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.L.; Urban, D.A.; Rodriguez-Lorenzo, L.; Milosevic, A.; Crippa, F.; Spuch-Calvar, M.; Balog, S.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle administration method in cell culture alters particle-cell interaction. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Yazdanian, M.; Audus, K.L. Microparticulate uptake mechanisms of in-vitro cell culture models of the respiratory epithelium. J. Pharm. Pharmacol. 2001, 53, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Osaka, T.; Nakanishi, T.; Shanmugam, S.; Takahama, S.; Zhang, H. Effect of surface charge of magnetite nanoparticles on their internalization into breast cancer and umbilical vein endothelial cells. Colloids Surf. B-Biointerfaces 2009, 71, 325–330. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primer Sequence |
|---|---|---|
| v | NM_002210 | F: actggcttaagagagggctgtg |
| R: tgccttacaaaaatcgctga | ||
| 3 | NM_000212 | R: tcctcaggaaaggtccaatg |
| R: tcctcaggaaaggtccaatg | ||
| 5 | NM_002213 | F: agcctatctccacgcacac |
| R: cctcggagaaggaaacatca | ||
| 5 | NM_002205 | F: cctgctgtccaccatgtcta |
| R: ttaatggggtgattggtggt | ||
| 1 | NM_133376 | F: tccaatggcttaatttgtgg |
| R: cgttgctggcttcacaagta | ||
| GAPDH | NM_002046.5 | R: cagcaagagcacaagaggaag |
| F: caactgtgaggaggggagatt |
| Integrin Subunit | ECV | MSCs | Caco-2 | HeLa |
|---|---|---|---|---|
| αv | 1.27 | 1.65 | 0.13 | 0.2 |
| α5 | 0.4 | 2.55 | 0.23 | 0.83 |
| β1 | 2.85 | 4.03 | 0.34 | 0.28 |
| β3 | 15.89 | 3.07 | 0.015 | 0.014 |
| β5 | 0.47 | 2.57 | 0.07 | 0.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bari, E.; Serra, M.; Paolillo, M.; Bernardi, E.; Tengattini, S.; Piccinini, F.; Lanni, C.; Sorlini, M.; Bisbano, G.; Calleri, E.; et al. Silk Fibroin Nanoparticle Functionalization with Arg-Gly-Asp Cyclopentapeptide Promotes Active Targeting for Tumor Site-Specific Delivery. Cancers 2021, 13, 1185. https://doi.org/10.3390/cancers13051185
Bari E, Serra M, Paolillo M, Bernardi E, Tengattini S, Piccinini F, Lanni C, Sorlini M, Bisbano G, Calleri E, et al. Silk Fibroin Nanoparticle Functionalization with Arg-Gly-Asp Cyclopentapeptide Promotes Active Targeting for Tumor Site-Specific Delivery. Cancers. 2021; 13(5):1185. https://doi.org/10.3390/cancers13051185
Chicago/Turabian StyleBari, Elia, Massimo Serra, Mayra Paolillo, Eric Bernardi, Sara Tengattini, Filippo Piccinini, Cristina Lanni, Marzio Sorlini, Giovanni Bisbano, Enrica Calleri, and et al. 2021. "Silk Fibroin Nanoparticle Functionalization with Arg-Gly-Asp Cyclopentapeptide Promotes Active Targeting for Tumor Site-Specific Delivery" Cancers 13, no. 5: 1185. https://doi.org/10.3390/cancers13051185
APA StyleBari, E., Serra, M., Paolillo, M., Bernardi, E., Tengattini, S., Piccinini, F., Lanni, C., Sorlini, M., Bisbano, G., Calleri, E., Torre, M. L., & Perteghella, S. (2021). Silk Fibroin Nanoparticle Functionalization with Arg-Gly-Asp Cyclopentapeptide Promotes Active Targeting for Tumor Site-Specific Delivery. Cancers, 13(5), 1185. https://doi.org/10.3390/cancers13051185