
Advanced Pediatric Diffuse Pontine Glioma Murine Models Pave the Way towards Precision Medicine

Abstract
Simple Summary
Abstract
1. Introduction
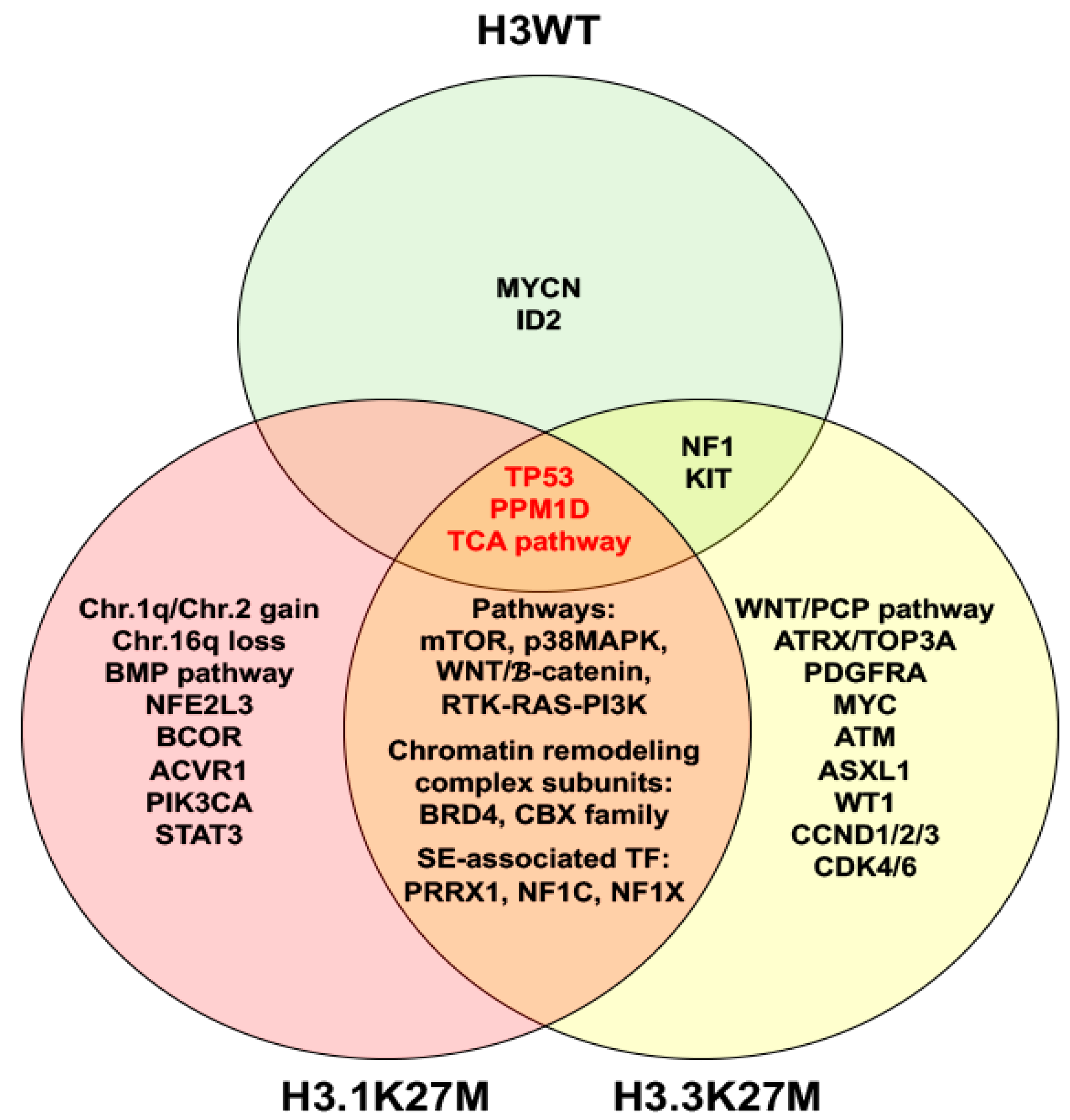
2. Molecular Characteristics of DIPG
3. Murine Models of DIPG
3.1. Syngeneic Brainstem Glioma Models
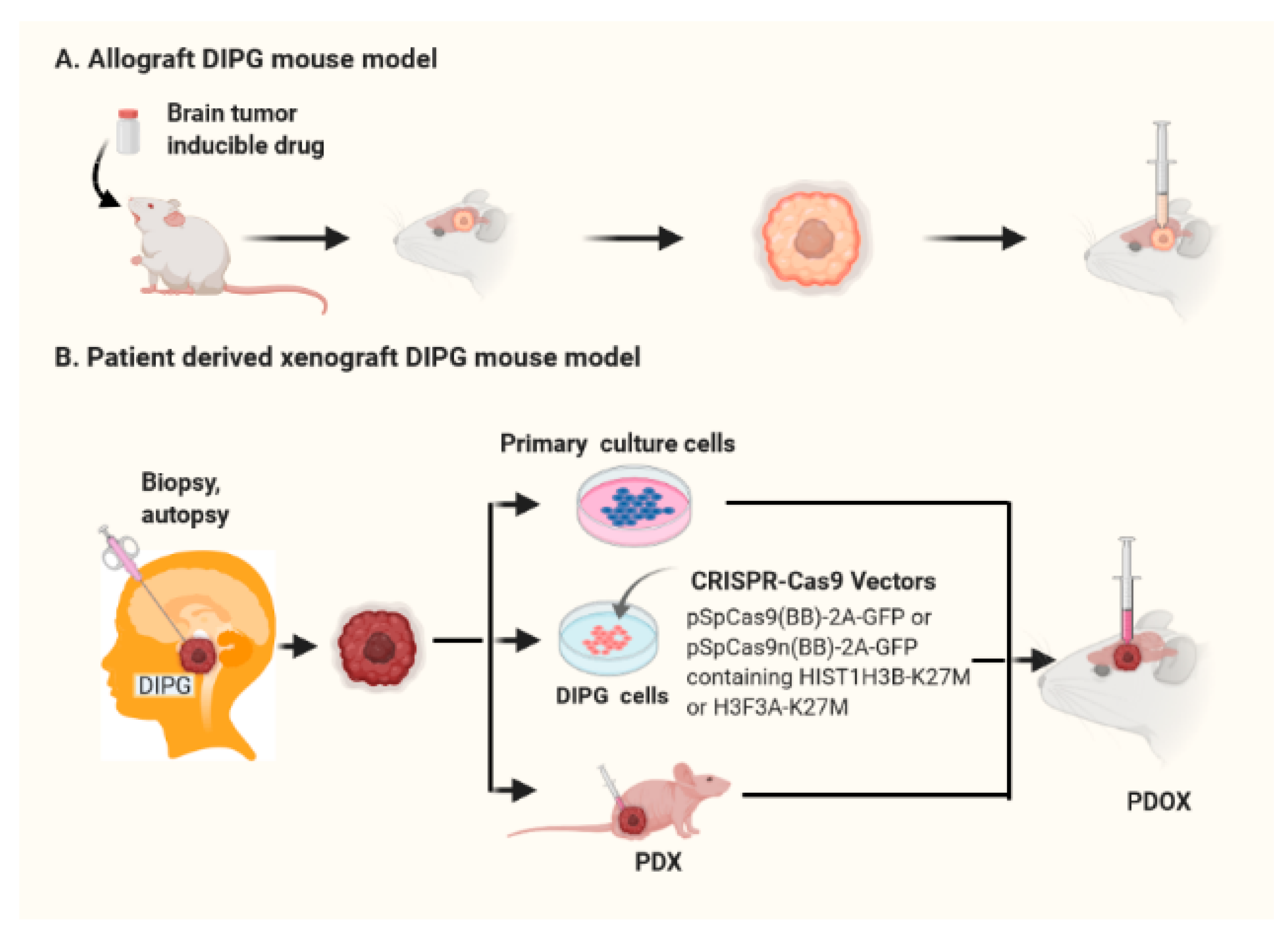
3.2. Patient-Derived Orthotopic Xenograft (PDOX) DIPG Mouse Models
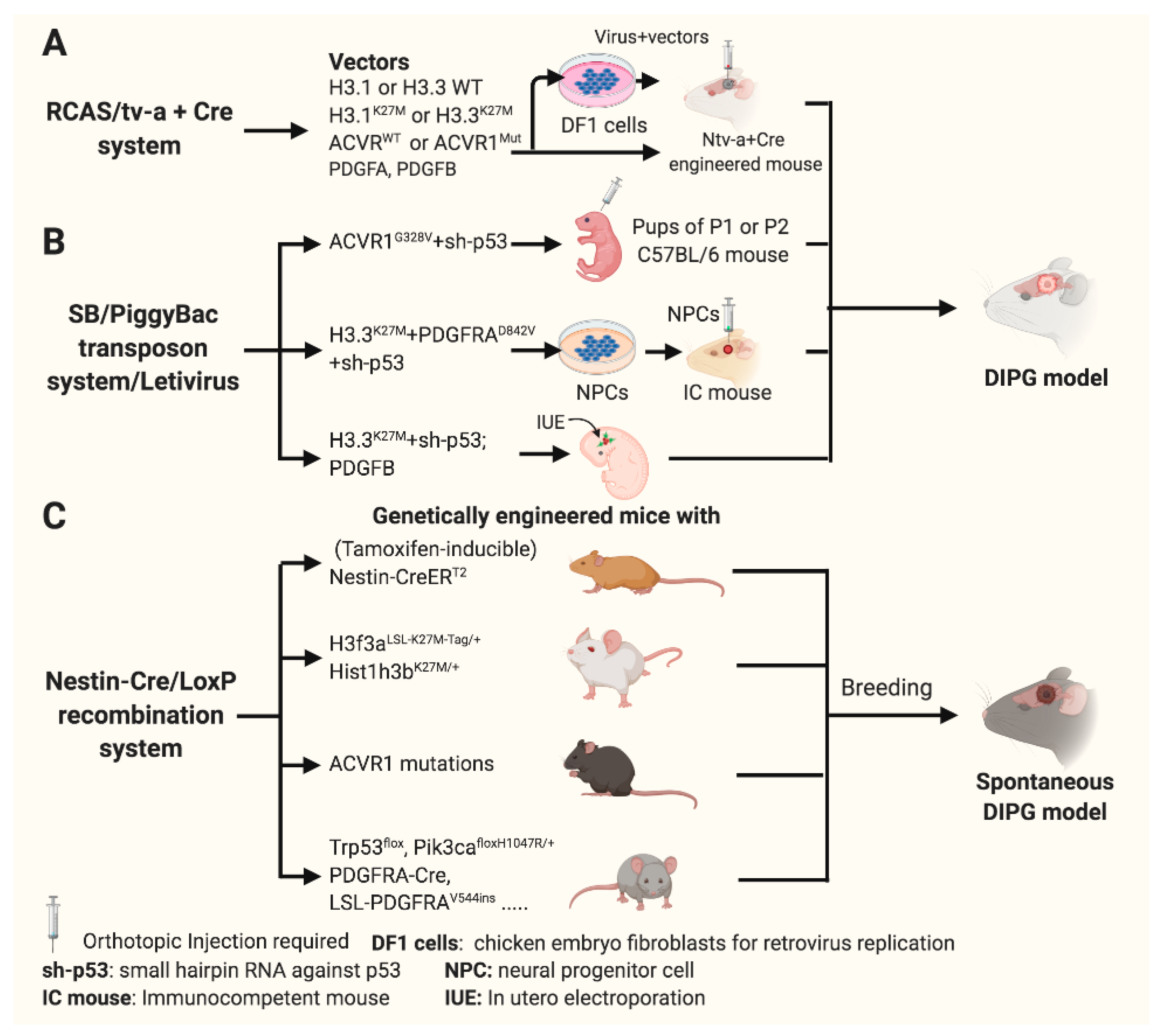
3.3. Genetically Engineered Mouse Models (GEMM) for DIPG
3.4. Fidelity of PDOX and Recapitulation GEMMs of Human DIPG
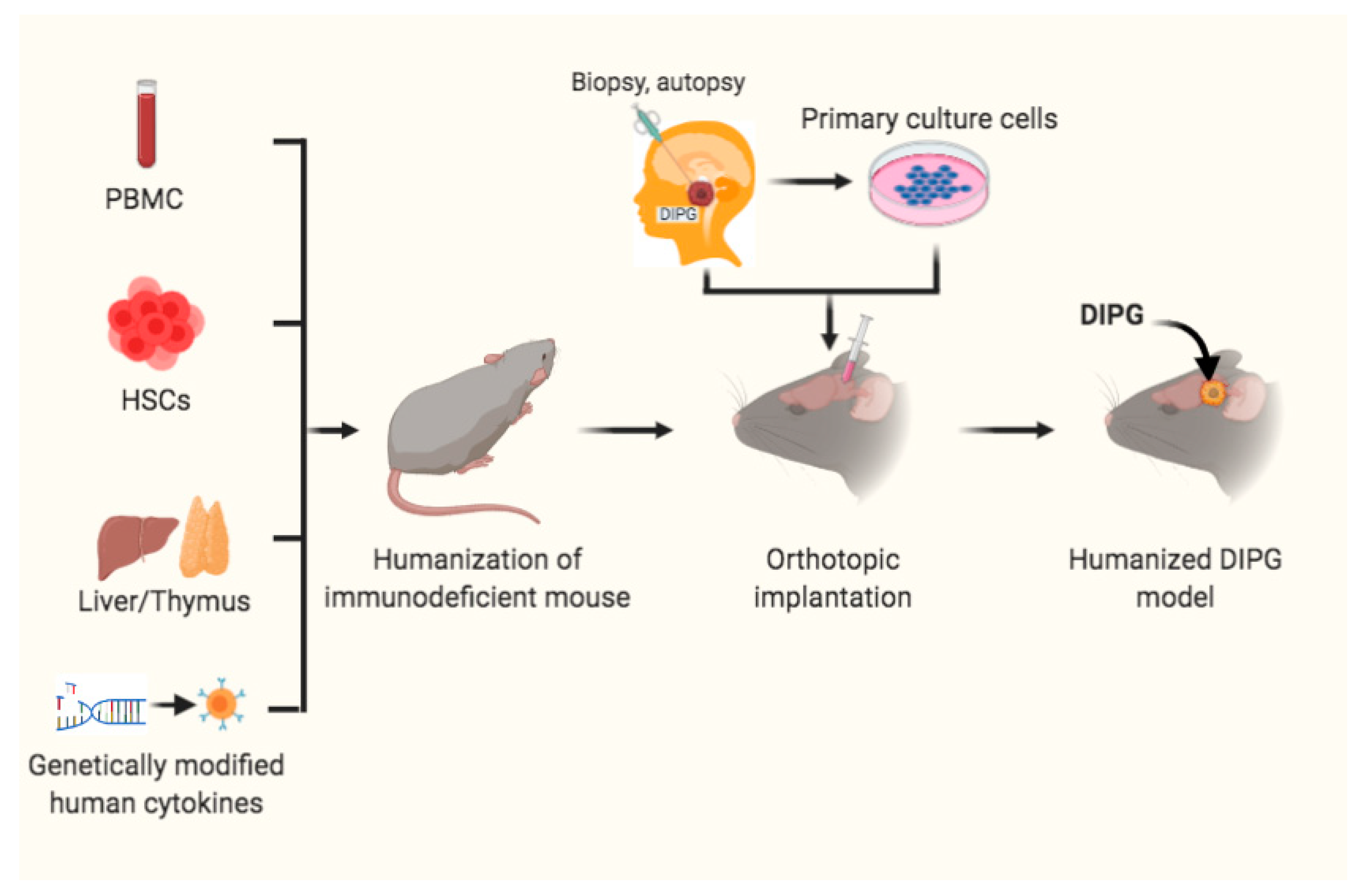
4. Humanized Mouse Models for DIPG Precision Medicine
5. Summary and Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Cooney, T.; Lane, A.; Bartels, U.; Bouffet, E.; Goldman, S.; Leary, S.E.S.; Foreman, N.K.; Packer, R.J.; Broniscer, A.; E Minturn, J.; et al. Contemporary survival endpoints: An International Diffuse Intrinsic Pontine Glioma Registry study. Neuro-Oncology 2017, 19, 1279–1280. [Google Scholar] [CrossRef] [PubMed]
- Hargrave, D.; Bartels, U.; Bouffet, E. Diffuse brainstem glioma in children: Critical review of clinical trials. Lancet Oncol. 2006, 7, 241–248. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Khuong-Quang, D.-A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Paugh, B.S.; Broniscer, A.; Qu, C.; Miller, C.P.; Zhang, J.; Tatevossian, R.G.; Olson, J.M.; Geyer, J.R.; Chi, S.N.; Da Silva, N.S.; et al. Genome-Wide Analyses Identify Recurrent Amplifications of Receptor Tyrosine Kinases and Cell-Cycle Regulatory Genes in Diffuse Intrinsic Pontine Glioma. J. Clin. Oncol. 2011, 29, 3999–4006. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, W.; Yun, S.; Kim, S.P.; Kim, K.H.; Kim, J.; Kim, S.; Wang, K.; Lee, J.Y. STAT3 is a key molecule in the oncogenic behavior of diffuse intrinsic pontine glioma. Oncol. Lett. 2020, 20, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, C.; Diplas, B.H.; Moure, C.J.; Chen, C.-P.J.; Chen, L.H.; Du, C.; Zhu, H.; Greer, P.K.; Zhang, L.; et al. Targeting Mutant PPM1D Sensitizes Diffuse Intrinsic Pontine Glioma Cells to the PARP Inhibitor Olaparib. Mol. Cancer Res. 2020, 18, 968–980. [Google Scholar] [CrossRef]
- Li, G.; Mitra, S.S.; Monje, M.; Henrich, K.N.; Bangs, C.D.; Nitta, R.T.; Wong, A.J. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J. Neuro-Oncol. 2012, 108, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.J.; Perry, A. Diffuse Midline Gliomas with Histone H3-K27M Mutation: A Series of 47 Cases Assessing the Spectrum of Morphologic Variation and Associated Genetic Alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro-Oncology 2013, 16, 352–360. [Google Scholar] [CrossRef]
- Paugh, B.S.; Zhu, X.; Qu, C.; Endersby, R.; Diaz, A.K.; Zhang, J.; Bax, D.A.; Carvalho, D.; Reis, R.M.; Onar-Thomas, A.; et al. Novel Oncogenic PDGFRA Mutations in Pediatric High-Grade Gliomas. Cancer Res. 2013, 73, 6219–6229. [Google Scholar] [CrossRef]
- Fortin, J.; Tian, R.; Zarrabi, I.; Hill, G.; Williams, E.; Sanchez-Duffhues, G.; Thorikay, M.; Ramachandran, P.; Siddaway, R.; Wong, J.F.; et al. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 2020, 37, 308–323.e12. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.S.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Hoeman, C.M.; Cordero, F.J.; Hu, G.; Misuraca, K.; Romero, M.M.; Cardona, H.J.; Nazarian, J.; Hashizume, R.; McLendon, R.; Yu, P.; et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat. Commun. 2019, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Jin, M.; Gao, M.; Zhou, H.; Tao, Y.J.; Skolnick, J. Differential kinase activity of ACVR1 G328V and R206H mutations with implications to possible TβRI cross-talk in diffuse intrinsic pontine glioma. Sci. Rep. 2020, 10, 6140. [Google Scholar] [CrossRef] [PubMed]
- Taylor, I.C.; Hütt-Cabezas, M.; Brandt, W.D.; Kambhampati, M.; Nazarian, J.; Chang, H.T.; Warren, K.E.; Eberhart, C.G.; Raabe, E.H. Disrupting NOTCH Slows Diffuse Intrinsic Pontine Glioma Growth, Enhances Radiation Sensitivity, and Shows Combinatorial Efficacy with Bromodomain Inhibition. J. Neuropathol. Exp. Neurol. 2015, 74, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.-J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020, 38, 334–349.e9. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kambhampati, M.; Yadavilli, S.; Gordish-Dressman, H.; Santi, M.; Cruz, C.R.; Packer, R.J.; Almira-Suarez, M.I.; Hwang, E.I.; Nazarian, J. Differential Expression of Wilms’ Tumor Protein in Diffuse Intrinsic Pontine Glioma. J. Neuropathol. Exp. Neurol. 2019, 78, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Caretti, V.; Hiddingh, L.; Lagerweij, T.; Schellen, P.; Koken, P.W.; Hulleman, E.; Van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J.L.; Noske, D.P.; et al. WEE1 Kinase Inhibition Enhances the Radiation Response of Diffuse Intrinsic Pontine Gliomas. Mol. Cancer Ther. 2013, 12, 141–150. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zanazzi, G.; Liechty, B.L.; Pendrick, D.; Krasnozhen-Ratush, O.; Snuderl, M.; Allen, J.C.; Garvin, J.H.; Mansukhani, M.M.; Roth, K.A.; Hsiao, S.J. Diffuse midline glioma with novel, potentially targetable, FGFR2–VPS35 fusion. Mol. Case Stud. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; McLone, D.G.; Naidich, T.P. Brain stem gliomas in childhood. J. Neuro-Oncol. 1984, 2, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Rajaram, V.; Mania-Farnell, B.; Mayanil, C.S.; Soares, M.B.; Tomita, T.; Goldman, S. Efficacy of vincristine administered via convection-enhanced delivery in a rodent brainstem tumor model documented by bioluminescence imaging. Child’s Nerv. Syst. 2012, 28, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Roujeau, T.; Machado, G.; Garnett, M.R.; Miquel, C.; Puget, S.; Geoerger, B.; Grill, J.; Boddaert, N.; Di Rocco, F.; Zerah, M.; et al. Stereotactic biopsy of diffuse pontine lesions in children. J. Neurosurg. Pediatr. 2007, 107, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R. Patient-derived Tumor Models for Diffuse Intrinsic Pontine Gliomas. Curr. Neuropharmacol. 2016, 15, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Caretti, V.; Zondervan, I.; Meijer, D.H.; Idema, S.; Vos, W.; Hamans, B.; Bugiani, M.; Hulleman, E.; Wesseling, P.; Vandertop, W.P.; et al. Monitoring of Tumor Growth and Post-Irradiation Recurrence in a Diffuse Intrinsic Pontine Glioma Mouse Model. Brain Pathol. 2010, 21, 441–451. [Google Scholar] [CrossRef]
- Misuraca, K.L.; Cordero, F.J.; Becher, O.J. Pre-Clinical Models of Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2015, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro-oncology 2018, 21, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.; Newcomb, W.D. A Case of Pontine Glioma, with Special Reference to the Paths of Gustatory Sensation. Proc. R. Soc. Med. 1926, 19, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.W.; Richter, E.O.; Yachnis, A.T.; Bennett, J.; Bhatti, M.T.; Smith, A. Brainstem stereotactic biopsy sampling in children. J. Neurosurg. Pediatr. 2006, 104, 108–114. [Google Scholar] [CrossRef]
- Broniscer, A.; Baker, J.N.; Baker, S.J.; Chi, S.N.; Geyer, J.R.; Morris, E.B.; Gajjar, A. Prospective collection of tissue samples at autopsy in children with diffuse intrinsic pontine glioma. Cancer 2010, 116, 4632–4637. [Google Scholar] [CrossRef] [PubMed]
- Zarghooni, M.; Bartels, U.; Lee, E.; Buczkowicz, P.; Morrison, A.; Huang, A.; Bouffet, E.; Hawkins, C. Whole-Genome Profiling of Pediatric Diffuse Intrinsic Pontine Gliomas Highlights Platelet-Derived Growth Factor Receptor α and Poly (ADP-ribose) Polymerase as Potential Therapeutic Targets. J. Clin. Oncol. 2010, 28, 1337–1344. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.-Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016, 7, 11185. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Mondal, G.; Lee, J.C.; Ravindranathan, A.; Villanueva-Meyer, J.E.; Tran, Q.T.; Allen, S.J.; Barreto, J.; Gupta, R.; Doo, P.; Van Ziffle, J.; et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020, 139, 1071–1088. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.J.; Jabado, N.; Grill, J. Diffuse intrinsic pontine gliomas—Current management and new biologic insights. Is there a glimmer of hope? Neuro-Oncology 2017, 19, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef]
- Bechet, D.; Gielen, G.G.H.; Korshunov, A.; Pfister, S.M.; Rousso, C.; Faury, D.; Fiset, P.-O.; Benlimane, N.; Lewis, P.W.; Lu, C.; et al. Specific detection of methionine 27 mutation in histone 3 variants (H3K27M) in fixed tissue from high-grade astrocytomas. Acta Neuropathol. 2014, 128, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.-H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Philippe, C.; Kergrohen, T.; Sill, M.; Merlevede, J.; Barret, E.; Puget, S.; Sainte-Rose, C.; Kramm, C.M.; Jones, C.; et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652.e6. [Google Scholar] [CrossRef]
- Nagaraja, S.; Quezada, M.A.; Gillespie, S.M.; Arzt, M.; Lennon, J.J.; Woo, P.J.; Hovestadt, V.; Kambhampati, M.; Filbin, M.G.; Suva, M.L.; et al. Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol. Cell 2019, 76, 965–980.e12. [Google Scholar] [CrossRef]
- Bailey, C.P.; Figueroa, M.; Gangadharan, A.; Yang, Y.; Romero, M.M.; A Kennis, B.; Yadavilli, S.; Henry, V.; Collier, T.; Monje, M.; et al. Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro-Oncology 2020, 22, 1302–1314. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.-H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155.e7. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Camarillo, J.M.; Huang, T.Y.-T.; Li, D.; Morris, J.A.; Zoltek, M.A.; Qi, J.; Behbahani, M.; Kambhampati, M.; Kelleher, N.L.; et al. Histone tail analysis reveals H3K36me2 and H4K16ac as epigenetic signatures of diffuse intrinsic pontine glioma. J. Exp. Clin. Cancer Res. 2020, 39, 261. [Google Scholar] [CrossRef]
- Pedersen, H.; Schmiegelow, K.; Hamerlik, P. Radio-Resistance and DNA Repair in Pediatric Diffuse Midline Gliomas. Cancers 2020, 12, 2813. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Eden, C.J.; Ju, B.; Murugesan, M.; Phoenix, T.N.; Nimmervoll, B.; Tong, Y.; Ellison, D.W.; Finkelstein, D.; Wright, K.; Boulos, N.; et al. Orthotopic models of pediatric brain tumors in zebrafish. Oncogene 2015, 34, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Sho, A.; Kondo, S.; Kamitani, H.; Otake, M.; Watanabe, T. Establishment of experimental glioma models at the intrinsic brainstem region of the rats. Neurol. Res. 2007, 29, 36–42. [Google Scholar] [CrossRef]
- Barth, R.F.; Kaur, B. Rat brain tumor models in experimental neuro-oncology: The C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J. Neuro-Oncol. 2009, 94, 299–312. [Google Scholar] [CrossRef]
- Wu, Q.; Tyler, B.; Sukay, L.; Rhines, L.; DiMeco, F.; Clatterbuck, R.E.; Guarnieri, M.; Carson, B.S.; Carson, S.B.S. Experimental Rodent Models of Brainstem Tumors. Veter. Pathol. 2002, 39, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Ozawa, T.; Dinca, E.B.; Banerjee, A.; Prados, M.D.; James, C.D.; Gupta, N. A human brainstem glioma xenograft model enabled for bioluminescence imaging. J. Neuro-Oncol. 2010, 96, 151–159. [Google Scholar] [CrossRef]
- Aoki, Y.; Hashizume, R.; Ozawa, T.; Banerjee, A.; Prados, M.; James, C.D.; Gupta, N. An experimental xenograft mouse model of diffuse pontine glioma designed for therapeutic testing. J. Neuro-Oncol. 2012, 108, 29–35. [Google Scholar] [CrossRef]
- Klopp, L.S.; Simpson, S.T.; Sorjonen, D.C.; Lenz, S.D. Ventral surgical approach to the caudal brain stem in dogs. Veter. Surg. 2000, 29, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.W.; Zimmer, S.G.; Oeltgen, J.; Markesbery, W.R. Invasiveness in Primary Intracranial Tumors: Part 1 An Experimental Model Using Cloned SV40 Virus-produced Hamster Brain Tumors. Neurosurgery 1986, 19, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Nishimoto, A.; Matsumoto, K.; Satoh, T.; Nakasone, S.; Fujiwara, T.; Ogura, H. Establishment of a brain-tumor model in adult monkeys. J. Neurosurg. 1985, 63, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Walbridge, S.; Vortmeyer, A.O.; Pack, S.D.; Nguyen, T.T.; Gogate, N.; Olson, J.J.; Akbasak, A.; Bobo, R.H.; Goffman, T.; et al. Induction of glioblastoma multiforme in nonhuman primates after therapeutic doses of fractionated whole-brain radiation therapy. J. Neurosurg. 2002, 97, 1378–1389. [Google Scholar] [CrossRef]
- Jallo, G.I.; Penno, M.; Sukay, L.; Liu, J.Y.; Tyler, B.; Lee, J.; Carson, B.S.; Guarnieri, M. Experimental models of brainstem tumors: Development of a neonatal rat model. Child’s Nerv. Syst. 2005, 21, 399–403. [Google Scholar] [CrossRef]
- Lee, J.; Jallo, G.I.; Guarnieri, M.; Carson, B.S.; Penno, M.B. A novel brainstem tumor model: Guide screw technology with functional, radiological, and histopathological characterization. Neurosurg. Focus 2005, 18, 1–6. [Google Scholar] [CrossRef]
- Kondo, A.; Goldman, S.; Vanin, E.F.; Sredni, S.T.; Rajaram, V.; Soares, M.B.; Tomita, T. An experimental brainstem tumor model using in vivo bioluminescence imaging in rat. Child’s Nerv. Syst. 2009, 25, 527–533. [Google Scholar] [CrossRef]
- Schuelke, M.R.; Wongthida, P.; Thompson, J.; Kottke, T.; Driscoll, C.B.; Huff, A.L.; Shim, K.G.; Coffey, M.; Pulido, J.; Evgin, L.; et al. Diverse immunotherapies can effectively treat syngeneic brainstem tumors in the absence of overt toxicity. J. Immunother. Cancer 2019, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Smirnov, I.; Liu, S.; Phillips, J.J.; Hyer, J.; McKnight, T.R.; Wendland, M.; Prados, M.; Banerjee, A.; Nicolaides, T.; et al. Characterization of a diffuse intrinsic pontine glioma cell line: Implications for future investigations and treatment. J. Neuro-Oncol. 2012, 110, 305–313. [Google Scholar] [CrossRef]
- Huillard, E.; Hashizume, R.; Phillips, J.J.; Griveau, A.; Ihrie, R.A.; Aoki, Y.; Nicolaides, T.; Perry, A.; Waldman, T.; McMahon, M.; et al. Cooperative interactions of BRAFV600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc. Natl. Acad. Sci. USA 2012, 109, 8710–8715. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Howman, A.; Wheatley, K.; Wherton, D.; Boota, N.; Pizer, B.; Fisher, D.; Kearns, P.; Picton, S.; Saran, F.; et al. Diffuse intrinsic pontine glioma treated with prolonged temozolomide and radiotherapy—Results of a United Kingdom phase II trial (CNS 2007 04). Eur. J. Cancer 2013, 49, 3856–3862. [Google Scholar] [CrossRef] [PubMed]
- DeWire, M.; Fuller, C.; Hummel, T.R.; Chow, L.M.L.; Salloum, R.; De Blank, P.; Pater, L.; Lawson, S.; Zhu, X.; Dexheimer, P.; et al. A phase I/II study of ribociclib following radiation therapy in children with newly diagnosed diffuse intrinsic pontine glioma (DIPG). J. Neuro-Oncol. 2020, 149, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.; Mitra, S.S.; Freret, M.E.; Raveh, T.B.; Kim, J.; Masek, M.; Attema, J.L.; Li, G.; Haddix, T.; Edwards, M.S.B.; et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl. Acad. Sci. USA 2011, 108, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.L.; Wilson, K.M.; Ceribelli, M.; Stanton, B.Z.; Woo, P.J.; Kreimer, S.; Qin, E.Y.; Zhang, X.; Lennon, J.; Nagaraja, S.; et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci. Transl. Med. 2019, 11, eaaw0064. [Google Scholar] [CrossRef]
- Meel, M.H.; De Gooijer, M.C.; Navarro, M.G.; Waranecki, P.; Breur, M.; Buil, L.C.; Wedekind, L.E.; Twisk, J.W.; Koster, J.; Hashizume, R.; et al. MELK Inhibition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2018, 24, 5645–5657. [Google Scholar] [CrossRef] [PubMed]
- Chornenkyy, Y.; Agnihotri, S.; Yu, M.; Buczkowicz, P.; Rakopoulos, P.; Golbourn, B.; Garzia, L.; Siddaway, R.; Leung, S.; Rutka, J.T.; et al. Poly-ADP-Ribose Polymerase as a Therapeutic Target in Pediatric Diffuse Intrinsic Pontine Glioma and Pediatric High-Grade Astrocytoma. Mol. Cancer Ther. 2015, 14, 2560–2568. [Google Scholar] [CrossRef] [PubMed]
- Fons, N.R.; Sundaram, R.K.; Breuer, G.A.; Peng, S.; McLean, R.L.; Kalathil, A.N.; Schmidt, M.S.; Carvalho, D.M.; Mackay, A.; Jones, C.; et al. PPM1D mutations silence NAPRT gene expression and confer NAMPT inhibitor sensitivity in glioma. Nat. Commun. 2019, 10, 3790. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.; Taylor, K.R.; Olaciregui, N.G.; Molinari, V.; Clarke, M.; Mackay, A.; Ruddle, R.; Henley, A.; Valenti, M.; Hayes, A.; et al. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun. Biol. 2019, 2, 156. [Google Scholar] [CrossRef]
- Anastas, J.N.; Zee, B.M.; Kalin, J.H.; Kim, M.; Guo, R.; Alexandrescu, S.; Blanco, M.A.; Giera, S.; Gillespie, S.M.; Das, J.; et al. Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 2019, 36, 528–544.e10. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.-A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Erratum: Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 827. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.S.; Sengupta, S.; Lee, K.; Hura, N.; Fuller, C.; DeWire, M.; Stevenson, C.B.; Fouladi, M.; Drissi, R. BMI-1 is a potential therapeutic target in diffuse intrinsic pontine glioma. Oncotarget 2017, 8, 62962–62975. [Google Scholar] [CrossRef]
- Martínez-Vélez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Pérez-Larraya, J.G.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10, 2235. [Google Scholar] [CrossRef]
- Abe, H.; Natsumeda, M.; Okada, M.; Watanabe, J.; Tsukamoto, Y.; Kanemaru, Y.; Yoshimura, J.; Oishi, M.; Hashizume, R.; Kakita, A.; et al. MGMT Expression Contributes to Temozolomide Resistance in H3K27M-Mutant Diffuse Midline Gliomas. Front. Oncol. 2020, 9, 1568. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.B.; Kasper, L.H.; Fan, Y.; Jin, H.; Wu, G.; Shaw, T.I.; Zhu, X.; Larson, J.D.; Easton, J.; Shao, Y.; et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 2019, 137, 637–655. [Google Scholar] [CrossRef]
- Sengupta, S.; Sobo, M.; Lee, K.; Kumar, S.S.; White, A.R.; Mender, I.; Fuller, C.; Chow, L.M.; Fouladi, M.; Shay, J.W.; et al. Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Mol. Cancer Ther. 2018, 17, 1504–1514. [Google Scholar] [CrossRef]
- Meel, M.H.; De Gooijer, M.C.; Metselaar, D.S.; Sewing, A.C.P.; Zwaan, K.; Waranecki, P.; Breur, M.; Buil, L.C.; Lagerweij, T.; Wedekind, L.E.; et al. Combined Therapy of AXL and HDAC Inhibition Reverses Mesenchymal Transition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2020, 26, 3319–3332. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, N.R.B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kozono, D.; Yang, X.; Fendler, W.; Fitts, W.; Ni, J.; Alberta, J.A.; Zhao, J.; Liu, K.X.; Bian, J.; et al. Dual HDAC and PI3K Inhibition Abrogates NFκB- and FOXM1-Mediated DNA Damage Response to Radiosensitize Pediatric High-Grade Gliomas. Cancer Res. 2018, 78, 4007–4021. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef]
- Hermans, E.; Hulleman, E. Patient-Derived Orthotopic Xenograft Models of Pediatric Brain Tumors: In a Mature Phase or Still in Its Infancy? Front. Oncol. 2020, 9. [Google Scholar] [CrossRef]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Pre-Clinical Study of Panobinostat in Xenograft and Genetically Engineered Murine Diffuse Intrinsic Pontine Glioma Models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef]
- Jansen, M.H.A.; Lagerweij, T.; Sewing, A.C.P.; Vugts, D.J.; Van Vuurden, D.G.; Molthoff, C.F.M.; Caretti, V.; Veringa, S.J.E.; Petersen, N.; Carcaboso, A.M.; et al. Bevacizumab Targeting Diffuse Intrinsic Pontine Glioma: Results of 89Zr-Bevacizumab PET Imaging in Brain Tumor Models. Mol. Cancer Ther. 2016, 15, 2166–2174. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.S.; Kumar, R.; Moore, K.A.; Smith, L.G.F.; Tinkle, C.L.; Chiang, J.; Patay, Z.; Gajjar, A.; Choudhri, A.F.; Lee-Diaz, J.A.; et al. Safety and efficacy of brainstem biopsy in children and young adults. J. Neurosurg. Pediatr. 2020, 26, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.R.; Young, C.C.; Vitanza, N.A.; McGrath, M.; Feroze, A.H.; Browd, S.R.; Hauptman, J.S. Progress in diffuse intrinsic pontine glioma: Advocating for stereotactic biopsy in the standard of care. Neurosurg. Focus 2020, 48, E4. [Google Scholar] [CrossRef]
- Lindquist, R.A.; Guinto, C.D.; Rodas-Rodriguez, J.L.; Fuentealba, L.C.; Tate, M.C.; Rowitch, D.H.; Alvarez-Buylla, A. Identification of proliferative progenitors associated with prominent postnatal growth of the pons. Nat. Commun. 2016, 7, 11628. [Google Scholar] [CrossRef]
- Misuraca, K.L.; Hu, G.; Barton, K.L.; Chung, A.; Becher, O.J. A Novel Mouse Model of Diffuse Intrinsic Pontine Glioma Initiated in Pax3-Expressing Cells. Neoplasia 2016, 18, 60–70. [Google Scholar] [CrossRef]
- Barton, K.L.; Misuraca, K.; Cordero, F.; Dobrikova, E.; Min, H.D.; Gromeier, M.; Kirsch, D.G.; Becher, O.J. PD-0332991, a CDK4/6 Inhibitor, Significantly Prolongs Survival in a Genetically Engineered Mouse Model of Brainstem Glioma. PLoS ONE 2013, 8, e77639. [Google Scholar] [CrossRef]
- Becher, O.J.; Hambardzumyan, D.; Walker, T.R.; Helmy, K.; Nazarian, J.; Albrecht, S.; Hiner, R.L.; Gall, S.; Huse, J.T.; Jabado, N.; et al. Preclinical Evaluation of Radiation and Perifosine in a Genetically and Histologically Accurate Model of Brainstem Glioma. Cancer Res. 2010, 70, 2548–2557. [Google Scholar] [CrossRef]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3K27M Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017, 32, 684–700.e9. [Google Scholar] [CrossRef] [PubMed]
- Halvorson, K.G.; Barton, K.L.; Schroeder, K.; Misuraca, K.L.; Hoeman, C.; Chung, A.; Crabtree, N.M.; Cordero, F.J.; Singh, R.; Spasojevic, I.; et al. A High-Throughput In Vitro Drug Screen in a Genetically Engineered Mouse Model of Diffuse Intrinsic Pontine Glioma Identifies BMS-754807 as a Promising Therapeutic Agent. PLoS ONE 2015, 10, e0118926. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689.e9. [Google Scholar] [CrossRef] [PubMed]
- Brabetz, S.; Leary, S.E.S.; Gröbner, S.N.; Nakamoto, M.W.; Şeker-Cin, H.; Girard, E.J.; Cole, B.; Strand, A.D.; Bloom, K.L.; Hovestadt, V.; et al. A biobank of patient-derived pediatric brain tumor models. Nat. Med. 2018, 24, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Kambhampati, M.; Panditharatna, E.; Yadavilli, S.; Saoud, K.; Lee, S.; Eze, A.; Almira-Suarez, M.I.; Hancock, L.; Bonner, E.R.; Gittens, J.; et al. Harmonization of postmortem donations for pediatric brain tumors and molecular characterization of diffuse midline gliomas. Sci. Rep. 2020, 10, 10954. [Google Scholar] [CrossRef]
- Tsoli, M.; Shen, H.; Mayoh, C.; Franshaw, L.; Ehteda, A.; Upton, D.; Carvalho, D.; Vinci, M.; Meel, M.H.; Van Vuurden, D.; et al. International experience in the development of patient-derived xenograft models of diffuse intrinsic pontine glioma. J. Neuro-Oncol. 2019, 141, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.L.; Monje, M. A Protocol for Rapid Post-mortem Cell Culture of Diffuse Intrinsic Pontine Glioma (DIPG). J. Vis. Exp. 2017, 7, e55360. [Google Scholar] [CrossRef]
- He, C.; Xu, K.; Zhu, X.; Dunphy, P.S.; Gudenas, B.; Lin, W.; Nathaniel, T.; Laura, D.H.; Chang-Hyuk, K.; Lawryn, H.; et al. Patient-Derived Orthotopic Xenografts and Cell Lines from Pediatric High-Grade Glioma Recapitulate the Heterogeneity of Histopathology, Molecular Signatures, and Drug Response. bioRxiv 2020. [Google Scholar] [CrossRef]
- Welby, J.P.; Kaptzan, T.; Wohl, A.; Peterson, T.E.; Raghunathan, A.; Brown, D.A.; Gupta, S.K.; Zhang, L.; Daniels, D.J. Current Murine Models and New Developments in H3K27M Diffuse Midline Gliomas. Front. Oncol. 2019, 9, 92. [Google Scholar] [CrossRef]
- Grabovska, Y.; Mackay, A.; O’Hare, P.; Crosier, S.; Finetti, M.; Schwalbe, E.C.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Cockle, J.; et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 2020, 11, 4324. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, A.; Lauing, K.L.; Zhai, L.; Ladomersky, E.; Raman, P.; Rathi, K.; Lulla, R.R.; Hashizume, R.; A Wainwright, D. Novel RNA-targeting strategy for treating T cell-driven immunosuppression in human diffuse intrinsic pontine glioma. Neuro-oncology 2019, 21, ii92–ii93. [Google Scholar] [CrossRef]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Takahashi, T.; Katano, I.; Ito, M. Current advances in humanized mouse models. Cell. Mol. Immunol. 2012, 9, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Baiocchi, R.A.; Khatri, V.P.; Lindemann, M.J.; Ross, M.E.; Papoff, G.; Caprio, A.J.; Caprio, T.V.; Fenstermaker, R.; Ruberti, G.; Bernstein, Z.P.; et al. Phenotypic and Functional Analysis of Fas (CD95) Expression in Primary Central Nervous System Lymphoma of Patients with Acquired Immunodeficiency Syndrome. Blood 1997, 90, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef]
- Biancotti, J.-C.; Town, T. Increasing Hematopoietic Stem Cell Yield to Develop Mice with Human Immune Systems. BioMed Res. Int. 2013, 2013, 740892. [Google Scholar] [CrossRef]
- Meraz, I.M.; Majidi, M.; Meng, F.; Shao, R.; Ha, M.J.; Neri, S.; Fang, B.; Lin, S.H.; Tinkey, P.T.; Shpall, E.J.; et al. An Improved Patient-Derived Xenograft Humanized Mouse Model for Evaluation of Lung Cancer Immune Responses. Cancer Immunol. Res. 2019, 7, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Source | H3 Mutation | Name of Cell Line | Novel Therapeutic Compounds Tested |
|---|---|---|---|
| Autopsy | H3WT | VUMC-DIPG-10 [76], DIPGM(T) [74,77] | OTSSP167 [76], veliparib, olaparib, niraparib [77] |
| H3.1K27M | SU-DIPG-IV [17,45,77,78,79,80,81,82] | LDN212854 [17], JQ1 [45], Panobinostat, GSK-J4 [81], veliparib, olaparib, niraparib [77], Corin [80], PTC-209 [82] | |
| H3.3K27M | JHH-DIPG1 (T) [19,54,76,81,83,84] | Delta-24-RGD [83], TMZ [84] *, GSK-J4 [81], Panobinostat, OTSSP167 [76], TAK228 [54], MRK003 [19] | |
| SU-DIPG-VI (T) [85], XIII, XVII [19,54,78,79,80,81,82,86,87] | LDN-193189, LDN-214117, LDN-212854 [79], TAK228 [54], 6-thio-Dg [86], BGB324 [87], Panobinostat [81], GSK-J4 [81], Corin [80], MRK003 [19], PTC-209 [82] | ||
| Biopsy | H3WT | CCHMC-DIPG-1 (T) [82,86] | PTC-209 [82], 6-thio-Dg [86] |
| H3.1K27M | HSJD-DIPG-018 [79] | GSK343 [88], EPZ6438 [88] | |
| VUMC-DIPG-B [81] | Panobinostat, GSK-J4 [81] | ||
| H3.3K27M | SF8628 (T) [13,17,45,89], SF7761(T) [19,45,54,84,87,90,91] | TAK228 [54], MK-1775 [13], JQ1 [45], TMZ [84], Panobinostat, GSK-J4 [81,90], BGB324 [87], CUDC-907 [89], MRK003 [19], GSK343 [88], EPZ6438 [88] | |
| HSJD-DIPG-007 (T) [79], 008, 012, 014 [78,79,80,87,90,92,93] | Bevacizumab [93], OTSSP167 [76], BGB324 [87], Panobinostat [92], LDN-193189, LDN-214117, LDN-212854 [79], GSK343, EPZ6438 [88], Corin [80] | ||
| VUMC-DIPG-A (T) [81,87] [76], F(T) [88,91] | OTSSP167 [76], BGB324 [87], Panobinostat [81] | ||
| TP54, 80 (T) [83], TP83, 84 [83] | Delta-24-RGD [83] | ||
| NEM 157, 163, 165, 168 [81] | Panobinostat, GSK-J4 [81], Delta-24-RGD [83] * | ||
| QCTB-R059(T) [79,91], CHRU-TC68 [16] | LDN-193189, LDN-214117, LDN-212854 [16,79] | ||
| CCHMC-DIPG-2 [82] | PTC-209 [82] |
| Model | Cell Resource or Genetic Engineering | Histology | Molecular Analysis | Fidelity | Reference |
|---|---|---|---|---|---|
| PDOX | Biopsy: DIPG tissue, PED17 | HE; IHC | RNA-seq, WGS, WES-seq DNA methylation-seq | High | [103,105,108] |
| Autopsy: DIPG tissue | [73] | ||||
| Cultured DIPG cells, PED17 | [108,109] | ||||
| GEMM | H3.3K27M + TP53 loss +PDGFRA activation | IF | RNA- & ChIP-seq | High | [99] |
| Pax3+; PDGFB+ Pax3+; p53−; PDGFB+ H3.3K27M+; Pax3+; p53−; PDGFB+ | IHC; IF | N/A | Low Moderate High | [96] | |
| ACVRR206H; H3.1K27M ACVRR206H/G328V; H3.1K27M; p53− ACVRR206H/G328V; H3.1K27M; p53−; PDGFA+ | IHC | RNA-seq | Low Moderate High | [17] | |
| ACVRG328V; PIK3CAH1047; Oligo2+ ACVRG328V; HIST1H3BK27M; PIK3CAH1047; Oligo2+ | IHC | RNA-seq | Low High | [15] | |
| H3.3WT; PDGFRA; p53cKO H3.3K27M; PDGFRA; p53cKO | IHC | RNA- & ChIP-seq | Low High | [101] | |
| H3.3WT; PDGFAWT; Trp53− H3.3K27M; PDGFA+; Trp53− | IHC | RNA- & ChIP-seq, WES | Low High | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Peng, P.; Zhang, X.; Mania-Farnell, B.; Xi, G.; Wan, F. Advanced Pediatric Diffuse Pontine Glioma Murine Models Pave the Way towards Precision Medicine. Cancers 2021, 13, 1114. https://doi.org/10.3390/cancers13051114
Chen Z, Peng P, Zhang X, Mania-Farnell B, Xi G, Wan F. Advanced Pediatric Diffuse Pontine Glioma Murine Models Pave the Way towards Precision Medicine. Cancers. 2021; 13(5):1114. https://doi.org/10.3390/cancers13051114
Chicago/Turabian StyleChen, Zirong, Peng Peng, Xiaolin Zhang, Barbara Mania-Farnell, Guifa Xi, and Feng Wan. 2021. "Advanced Pediatric Diffuse Pontine Glioma Murine Models Pave the Way towards Precision Medicine" Cancers 13, no. 5: 1114. https://doi.org/10.3390/cancers13051114
APA StyleChen, Z., Peng, P., Zhang, X., Mania-Farnell, B., Xi, G., & Wan, F. (2021). Advanced Pediatric Diffuse Pontine Glioma Murine Models Pave the Way towards Precision Medicine. Cancers, 13(5), 1114. https://doi.org/10.3390/cancers13051114