
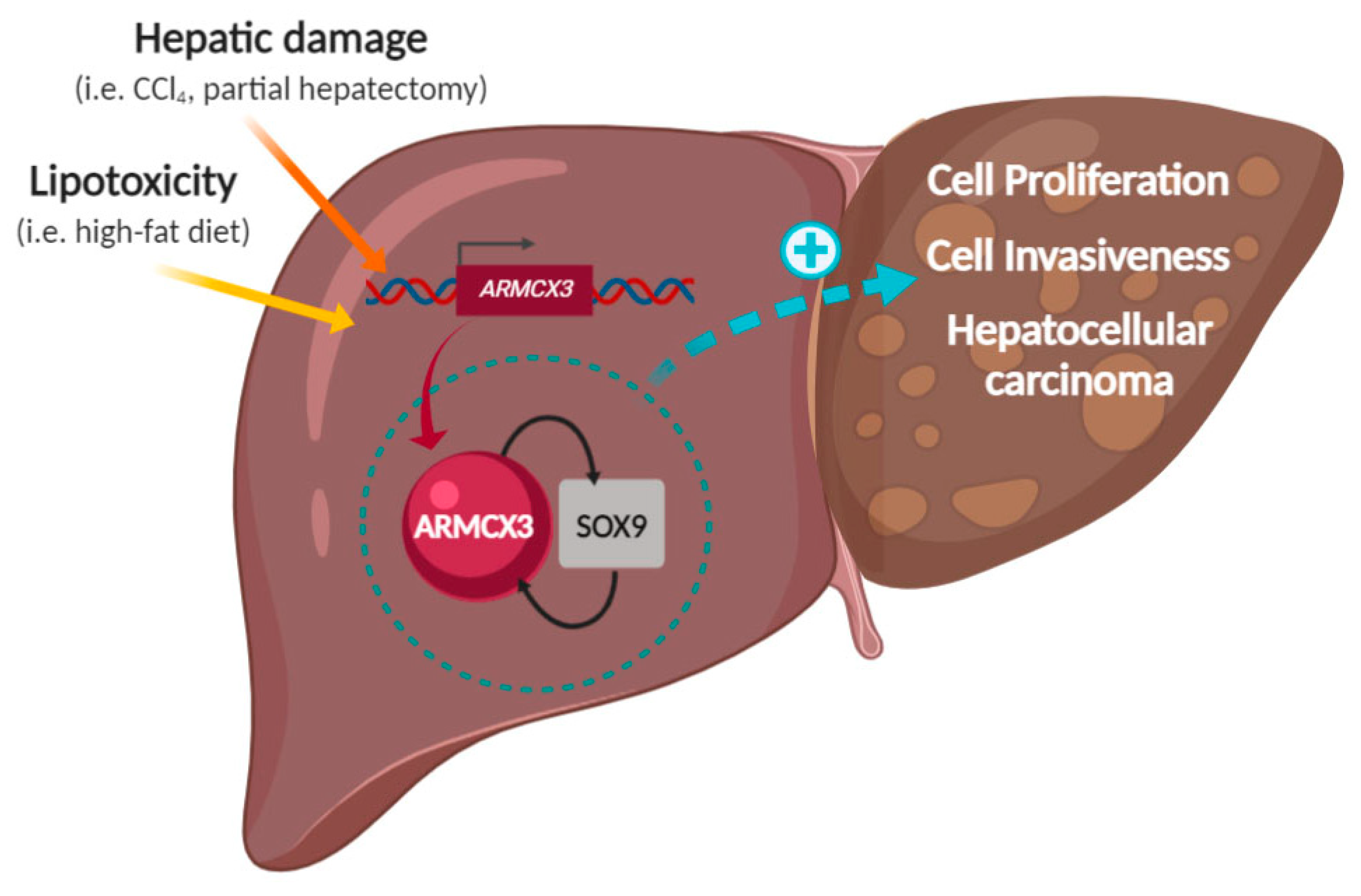
ARMCX3 Mediates Susceptibility to Hepatic Tumorigenesis Promoted by Dietary Lipotoxicity

 , , , , , , ,
, , , , , , ,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
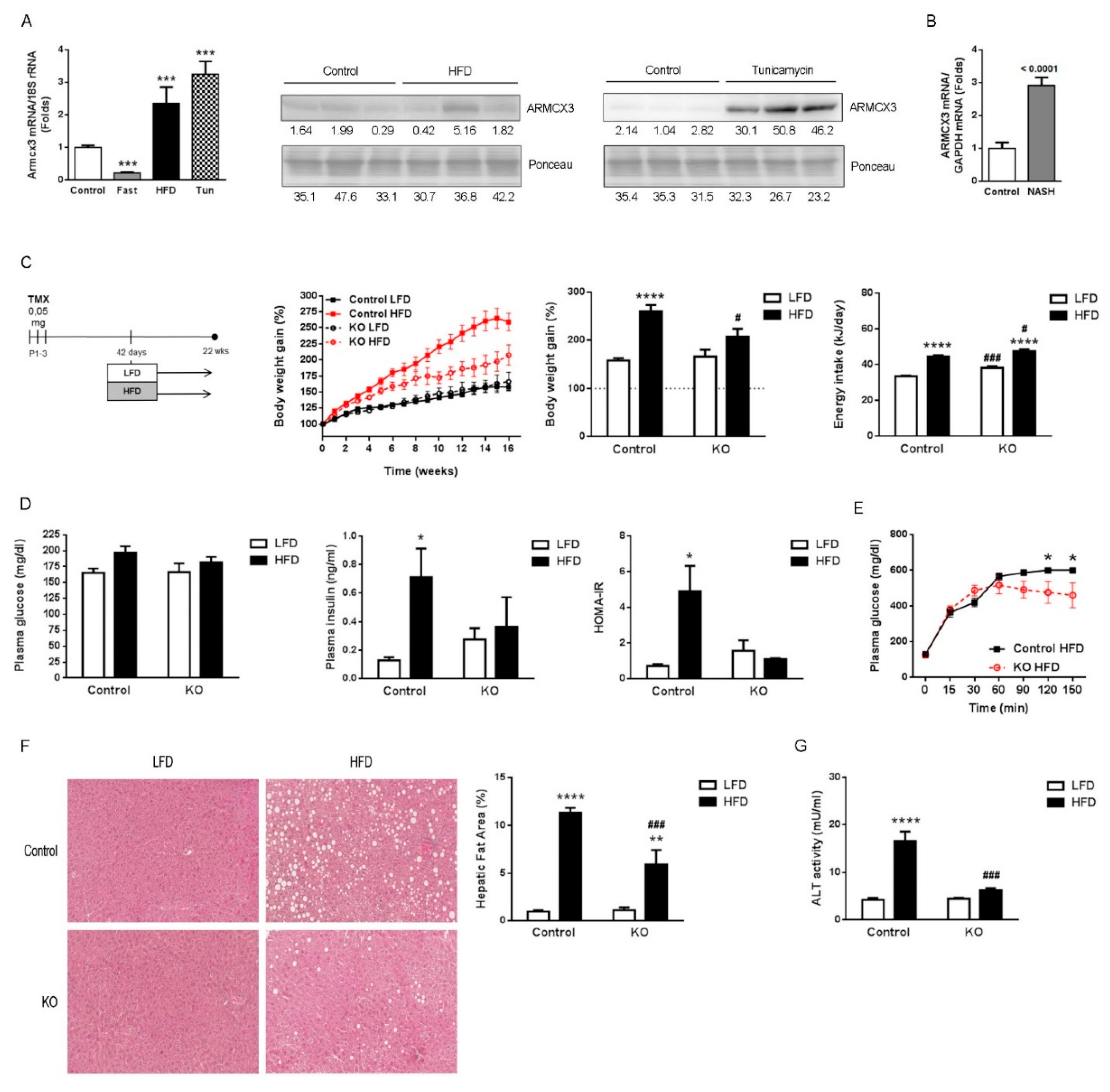
2.1. Hepatic Expression of Armcx3 Is Modulated in Response to Nutritional Challenges
2.2. Gene Invalidation of Armcx3 Ameliorates HFD-Induced Metabolic Alterations and Protects against NAFLD
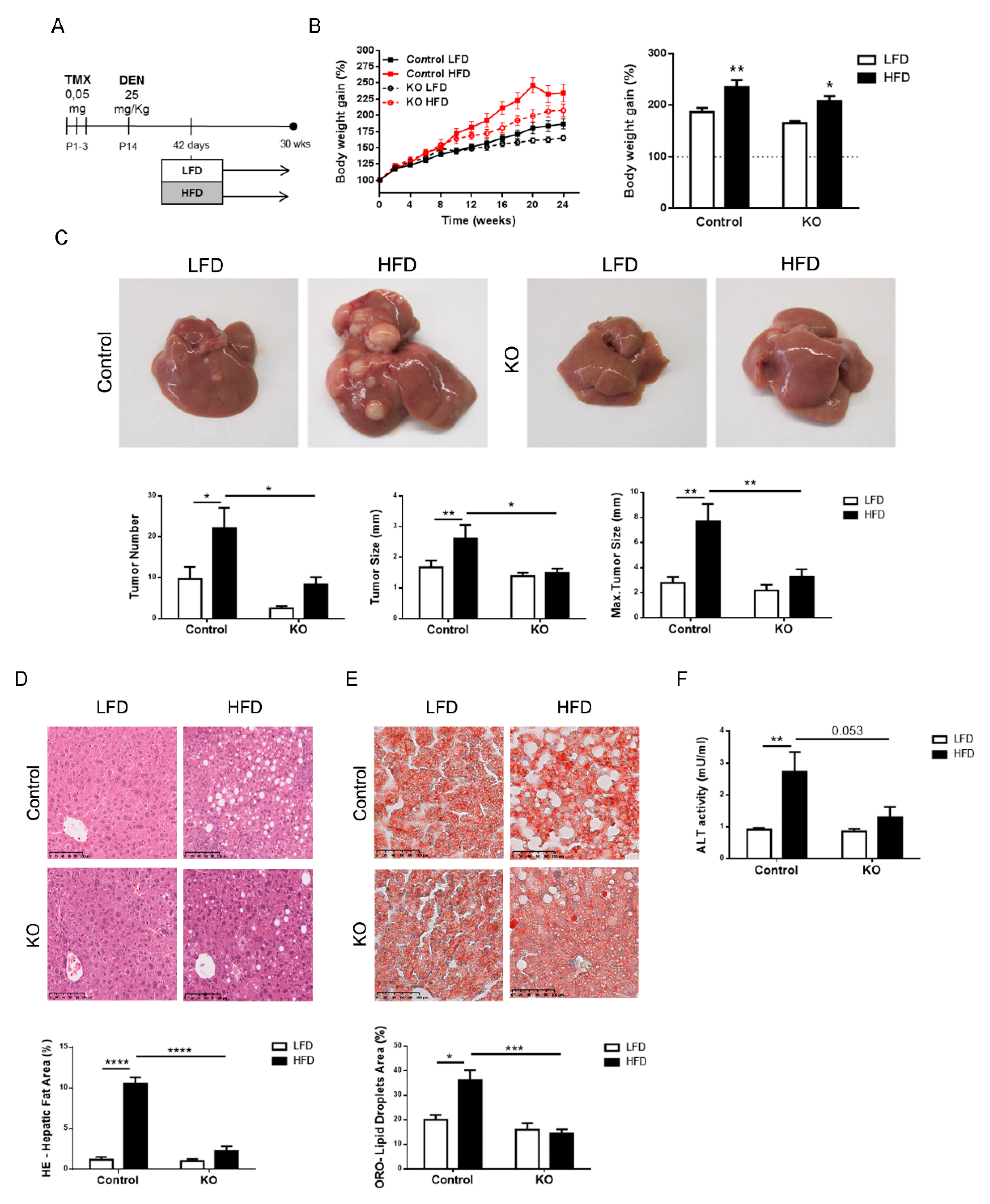
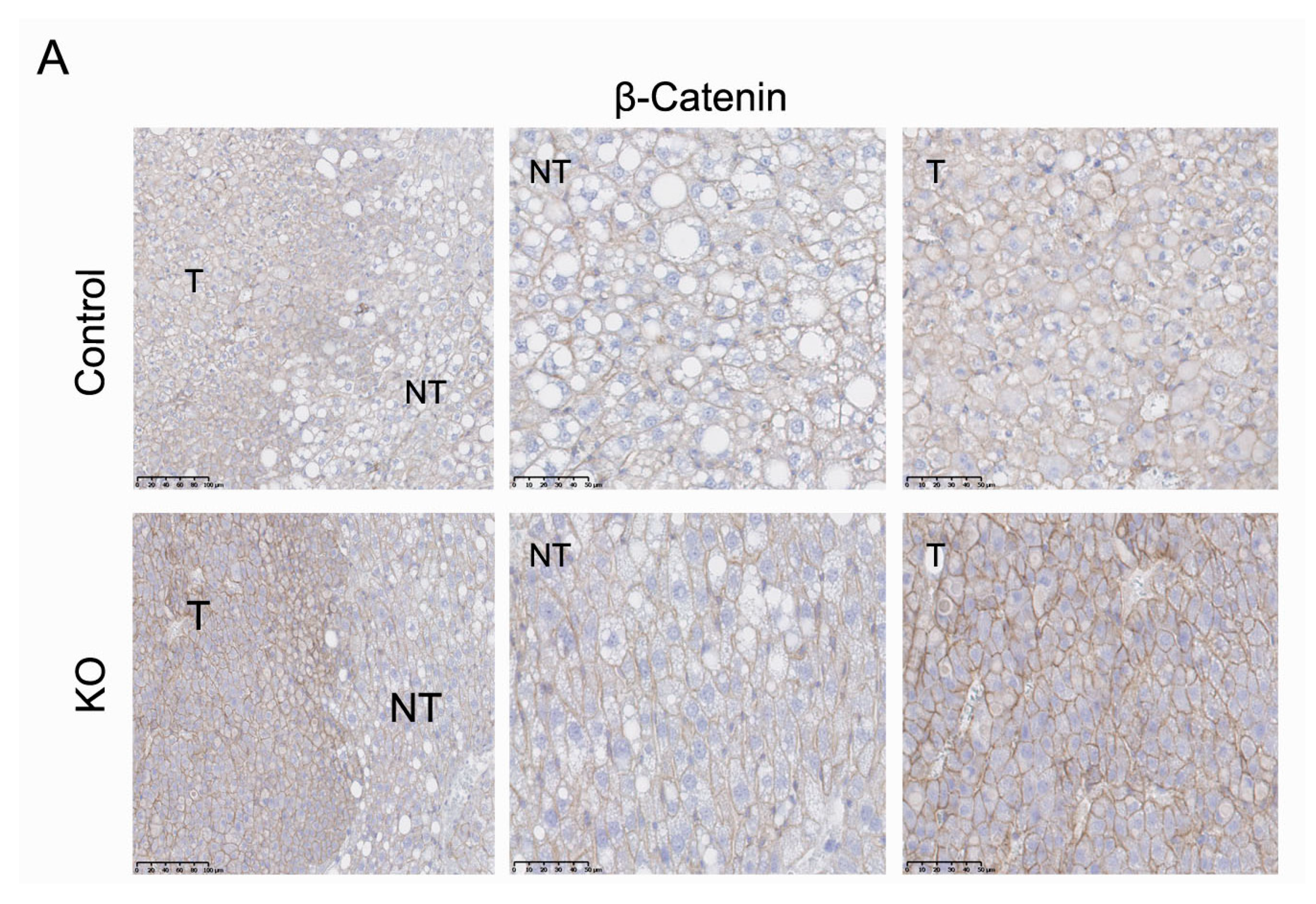
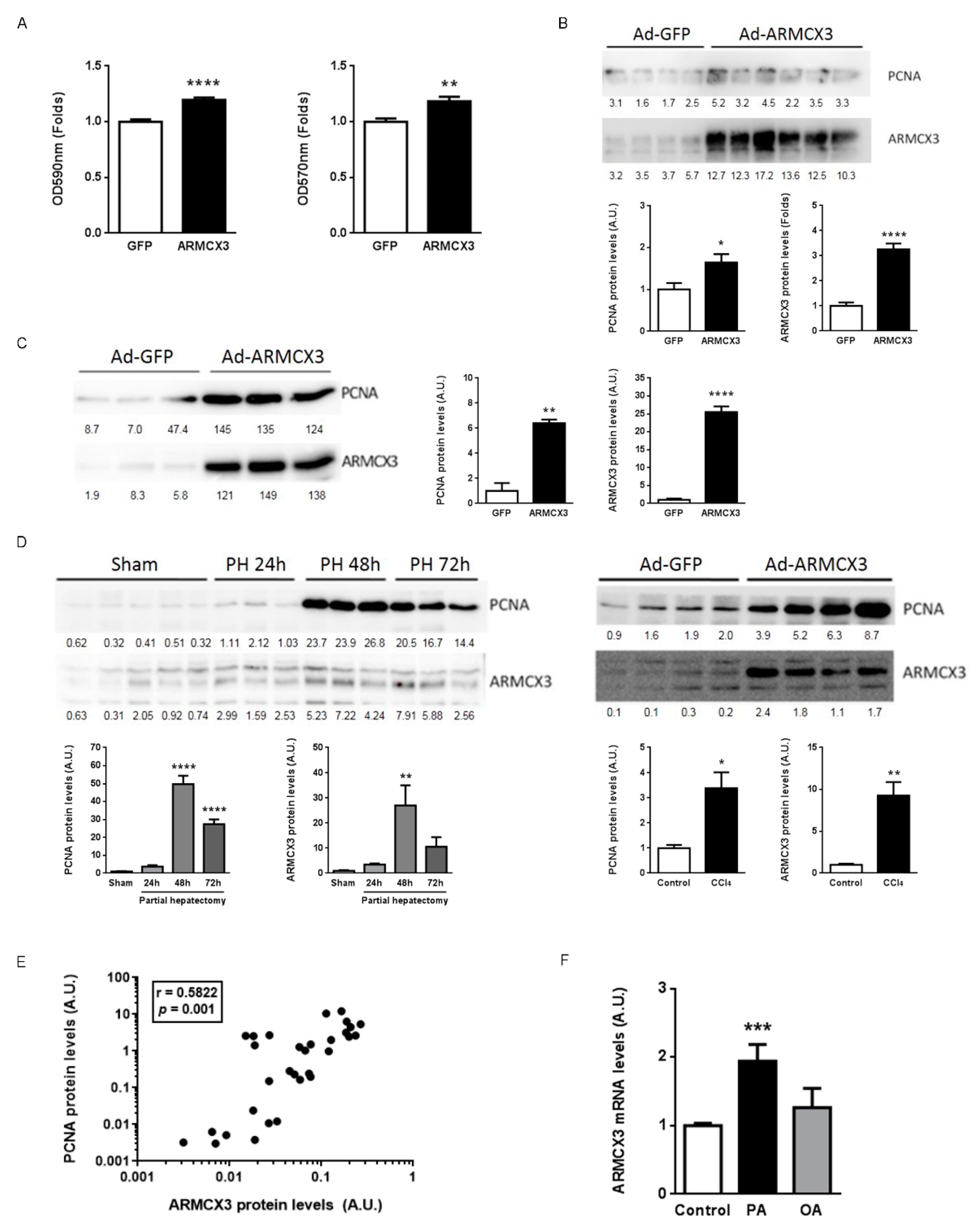
2.3. Inactivation of the Armcx3 Gene Protects Against Hepatic Carcinogenesis
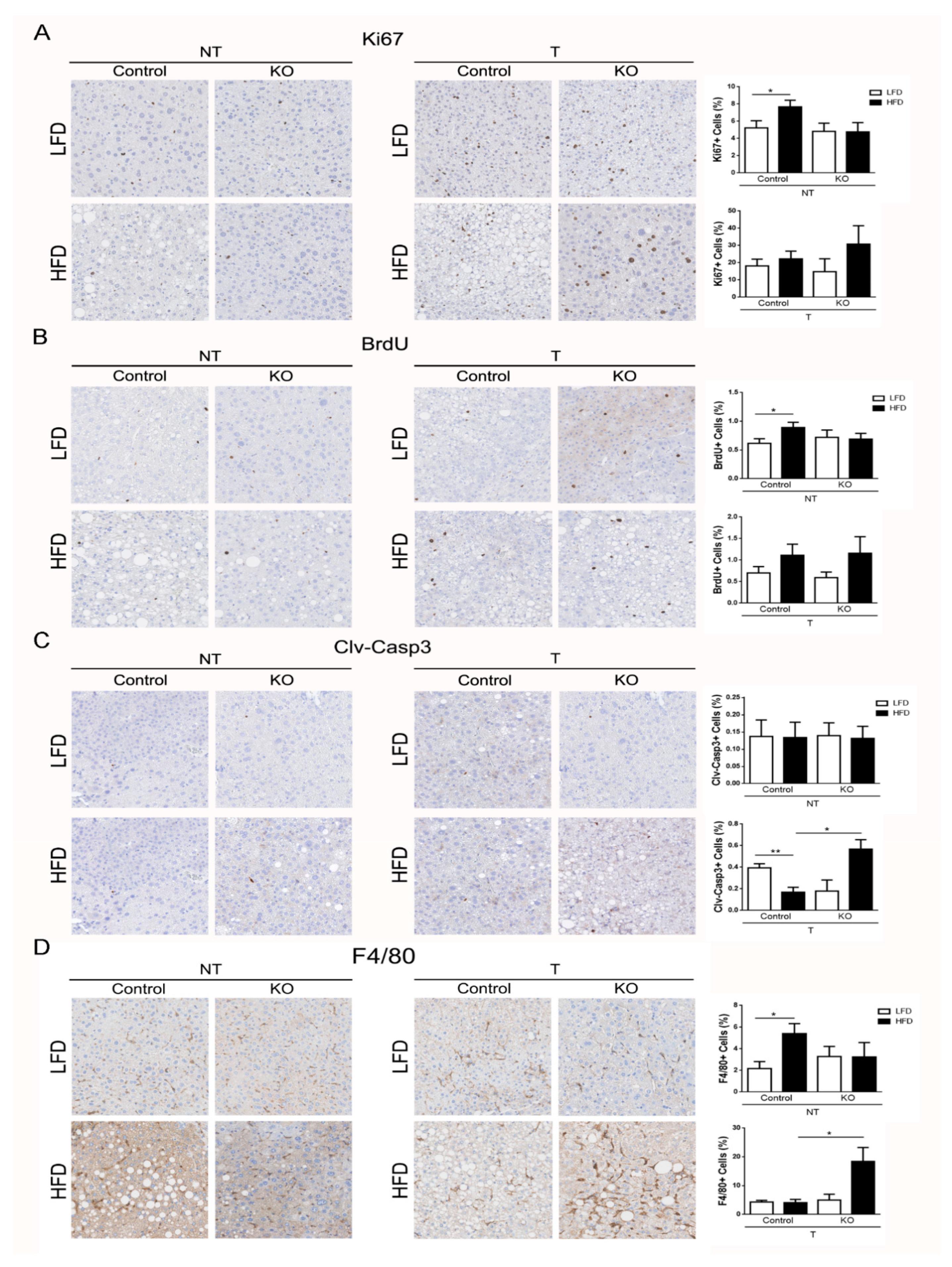
2.4. ARMCX3 Regulates DEN-Induced Apoptotic Death and Macrophage Infiltration
2.5. ARMCX3 Invalidation Does Not Affect Hepatic Mitochondria
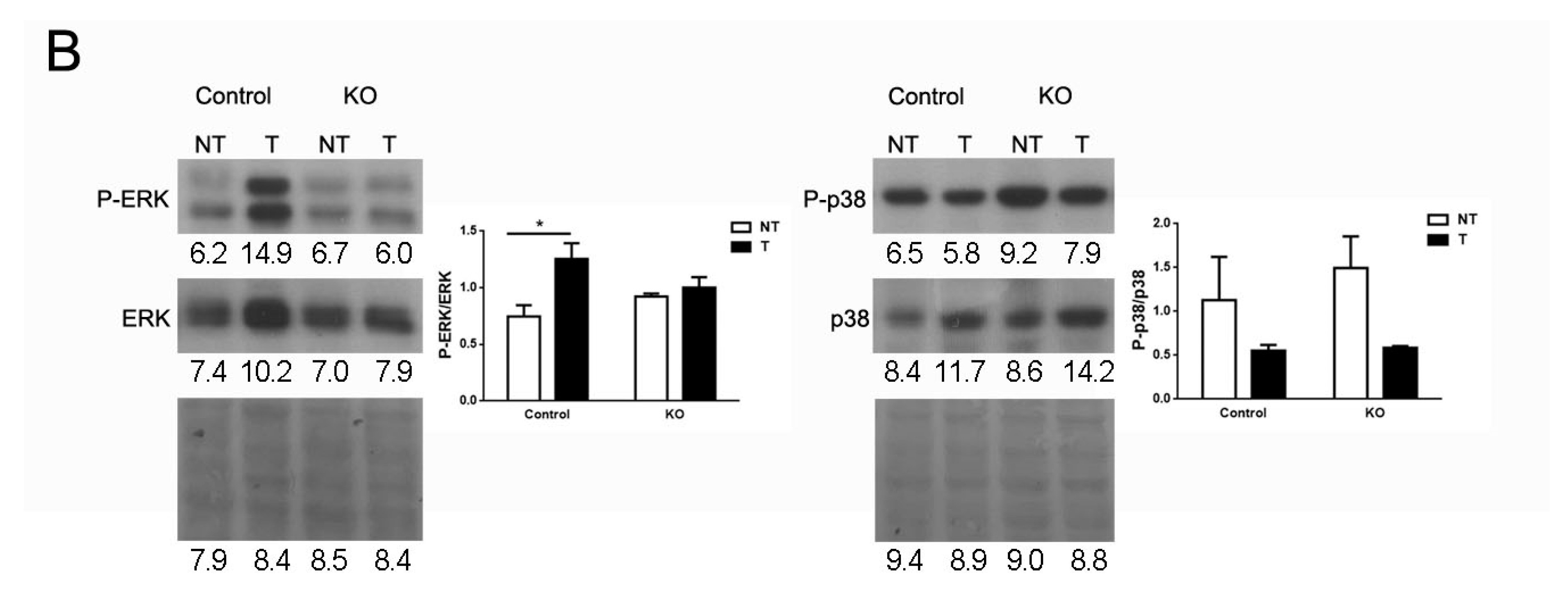
2.6. ARMCX3 Affects ERK Signaling in DEN-Induced Tumors from HFD-Fed Mice
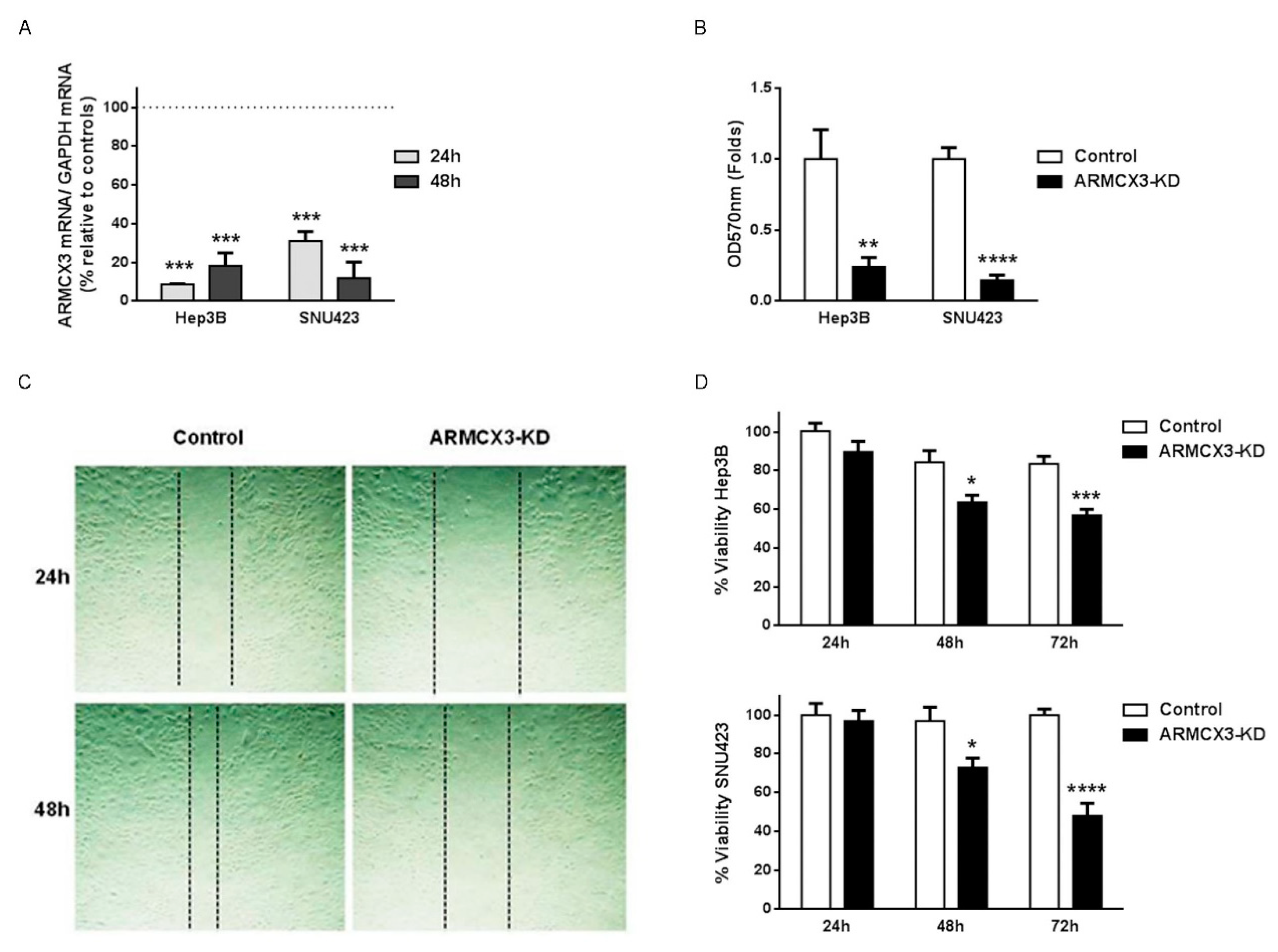
2.7. ARMCX3 Invalidation Reduces Cell Viability, Clonality and Migration in Hepatocellular Carcinoma Cell Lines
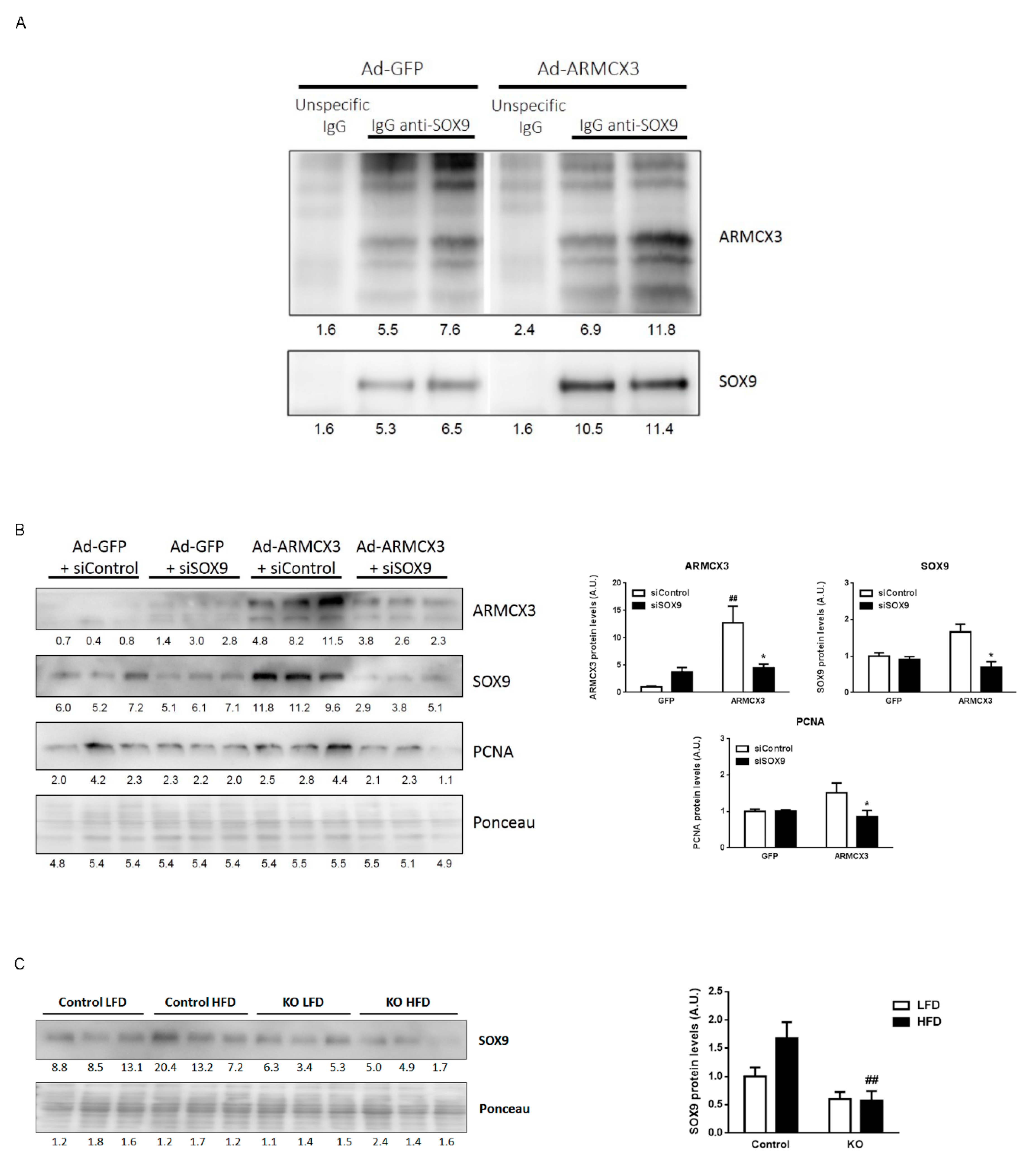
2.8. ARMCX3 Overexpression In Vitro Induces Hepatocellular Carcinoma Cells Proliferation in a SOX9-Dependent Mechanism
3. Discussion
4. Materials and Methods
4.1. Mice Care
4.2. Human Samples
4.3. ARMCX3-KO Mouse Generation
- Armcx3 S1F: GGGGCGGTGGGCAGGATGACAGC
- Armcx3 S4F: AAGTTCTAGGAATCGAGAGCC
- Armcx3 S: ATCATTTCCCCTTGACTCTGG
- Forward Wt-Cre: CTAGGCCACGAATTGAAAGATCT
- Reverse Wt-Cre: GTAGGTGGAAATTCTAGCATCATCC
- Forward Cre: GCGGTCTGGCAGTAAAAACTATC
- Reverse Cre: GTGAAACAGCATTGCTGTCACTT
4.4. Metabolism-Related Studies
4.5. DEN-Induced Hepatocarcinogenesis
4.6. Mouse Partial Hepatectomy and Acute CCl4 Administration
4.7. Mouse Primary Hepatocytes
4.8. Histology and Immunohistochemistry
4.9. Cell Lines and Culture Conditions
4.10. Transfection of siRNAs
4.11. Armcx3 Overexpression and SOX9 Knockdown In Vitro
4.12. Wound Healing Assay
4.13. Cell Viability Assay
4.14. Clonogenic Assay
4.15. Palmitic Acid and Oleic Acid Treatments
4.16. Western Blotting
4.17. RNA Isolation, cDNA Synthesis, and Real-Time PCR
4.18. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopez-Domenech, G.; Serrat, R.; Mirra, S.; D’Aniello, S.; Somorjai, I.; Abad, A.; Vitureira, N.; García-Arumí, E.; Alonso, M.T.; Rodriguez-Prados, M.; et al. The Eutherian Armcx genes regulate mitochondrial trafficking in neurons and interact with Miro and Trak2. Nat. Commun. 2012, 3, 814. [Google Scholar] [CrossRef]
- Serrat, R.; Lopez-Domenech, G.; Mirra, S.; Quevedo, M.; Garcia-Fernandez, J.; Burgaya, F.; Soriano, E. The non-canonical Wnt/PKC pathway regulates mitochondrial dynamics through degradation of the arm-like domain-containing protein ARMCX3. PLoS ONE 2013, 8, e67773. [Google Scholar] [CrossRef]
- Mirra, S.; Ulloa, F.; Gutierrez-Vallejo, I.; Marti, E.; Soriano, E. Function of Armcx3 and Armc10/SVH Genes in the Regulation of Progenitor Proliferation and Neural Differentiation in the Chicken Spinal Cord. Front. Cell. Neurosci. 2016, 10, 47. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Yonemitsu, N.; Funahashi, S.I.; Nomura, H. ALEX1, a novel human armadillo repeat protein that is expressed differentially in normal tissues and carcinomas. Biochem. Biophys. Res. Commun. 2001, 280, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Rohrbeck, A.; Borlak, J. Cancer genomics identifies regulatory gene networks associated with the transition from dysplasia to advanced lung adenocarcinomas induced by c-Raf-1. PLoS ONE 2009, 4, e7315. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Reynoso, M.A.; Ochoa-Hernandez, A.B.; Aguilar-Lemarroy, A.; Jave-Suarez, L.F.; Troyo-Sanroman, R.; Barros-Nunez, P. Gene expression profiling identifies WNT7A as a possible candidate gene for decreased cancer risk in fragile X syndrome patients. Arch. Med. Res. 2010, 41, 110–118. [Google Scholar] [CrossRef]
- Iseki, H.; Takeda, A.; Andoh, T.; Kuwabara, K.; Takahashi, N.; Kurochkin, I.V.; Ishida, H.; Okazaki, Y.; Koyama, I. ALEX1 suppresses colony formation ability of human colorectal carcinoma cell lines. Cancer Sci. 2012, 103, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- Gloss, B.S.; Patterson, K.I.; Barton, C.A.; Gonzalez, M.; Scurry, J.P.; Hacker, N.F.; Sutherland, R.L.; O’Brien, P.M.; Clark, S.J. Integrative genome-wide expression and promoter DNA methylation profiling identifies a potential novel panel of ovarian cancer epigenetic biomarkers. Cancer Lett. 2012, 318, 76–85. [Google Scholar] [CrossRef]
- Du, J.; Zhang, X.; Zhou, H.; Miao, Y.; Han, Y.; Han, Q.; Wang, E. Alex3 suppresses non-small cell lung cancer invasion via AKT/Slug/E-cadherin pathway. Tumour Biol. 2017, 39, 1010428317701441. [Google Scholar] [CrossRef]
- Kusama, Y.; Takayanagi, S.; Tategu, M.; Yoshida, K. Expression and tissue distribution of human X-linked armadillo repeat containing-6. Exp. Ther. Med. 2010, 1, 395–399. [Google Scholar] [CrossRef]
- Huang, R.; Xing, Z.; Luan, Z.; Wu, T.; Wu, X.; Hu, G. A specific splicing variant of SVH, a novel human armadillo repeat protein, is up-regulated in hepatocellular carcinomas. Cancer Res. 2003, 63, 3775–3782. [Google Scholar]
- Younossi, Z.; Henry, L. Contribution of Alcoholic and Nonalcoholic Fatty Liver Disease to the Burden of Liver-Related Morbidity and Mortality. Gastroenterology 2016, 150, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Mokdad, A.H. Epidemiology of obesity in the Western Hemisphere. J. Clin. Endocrinol. Metab. 2008, 93 (Suppl. 1), S1–S8. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef]
- Jung, T.W.; Hong, H.C.; Hwang, H.J.; Yoo, H.J.; Baik, S.H.; Choi, K.M. C1q/TNF-Related Protein 9 (CTRP9) attenuates hepatic steatosis via the autophagy-mediated inhibition of endoplasmic reticulum stress. Mol. Cell. Endocrinol. 2015, 417, 131–140. [Google Scholar] [CrossRef]
- Ennulat, D.; Magid-Slav, M.; Rehm, S.; Tatsuoka, K.S. Diagnostic performance of traditional hepatobiliary biomarkers of drug-induced liver injury in the rat. Toxicol. Sci. 2010, 116, 397–412. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.; He, G.; Ali, S.R.; Holzer, R.G.; Österreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Park, D.H.; Shin, J.W.; Park, S.K.; Seo, J.N.; Li, L.; Jang, J.J.; Lee, M.J. Diethylnitrosamine (DEN) induces irreversible hepatocellular carcinogenesis through overexpression of G1/S-phase regulatory proteins in rat. Toxicol. Lett. 2009, 191, 321–326. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef]
- Mou, Z.; Tapper, A.R.; Gardner, P.D. The armadillo repeat-containing protein, ARMCX3, physically and functionally interacts with the developmental regulatory factor Sox10. J. Biol. Chem. 2009, 284, 13629–13640. [Google Scholar] [CrossRef]
- Taniguchi, K.; Yamachika, S.; He, F.; Karin, M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016, 590, 2375–2397. [Google Scholar] [CrossRef]
- Hui, L.; Bakiri, L.; Stepniak, E.; Wagner, E.F. p38alpha: A suppressor of cell proliferation and tumorigenesis. Cell Cycle 2007, 6, 2429–2433. [Google Scholar] [CrossRef]
- Sakurai, T.; Itoh, K.; Higashitsuji, H.; Nonoguchi, K.; Liu, Y.; Watanabe, H.; Nakano, T.; Fukumoto, M.; Chiba, T.; Fujita, J. Cirp protects against tumor necrosis factor-alpha-induced apoptosis via activation of extracellular signal-regulated kinase. Biochim. Biophys. Acta 2006, 1763, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Brûle, S.; Um, S.H.; Li, Y.; Masuda, K.; Roden, M.; Sun, X.J.; Krebs, M.; Polakiewicz, R.D.; Thomas, G.; et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 14056–14061. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Sonenberg, N.; Gores, G.J. The mTOR pathway in hepatic malignancies. Hepatology 2013, 58, 810–818. [Google Scholar] [CrossRef]
- Lindroos, P.M.; Zarnegar, R.; Michalopoulos, G.K. Hepatocyte growth factor (hepatopoietin A) rapidly increases in plasma before DNA synthesis and liver regeneration stimulated by partial hepatectomy and carbon tetrachloride administration. Hepatology 1991, 13, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.O.; Mak, W.N.; Kai, A.K.; Chan, K.S.; Lee, T.K.; Ng, I.O.; Lo, R.C. Sox9 confers stemness properties in hepatocellular carcinoma through Frizzled-7 mediated Wnt/beta-catenin signaling. Oncotarget 2016, 7, 29371–29386. [Google Scholar] [CrossRef]
- Fassio, E.; Alvarez, E.; Dominguez, N.; Landeira, G.; Longo, C. Natural history of nonalcoholic steatohepatitis: A longitudinal study of repeat liver biopsies. Hepatology 2004, 40, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; Yang, W.S.; Lee, T.H.; Lee, L.T.; Chen, C.Y.; Huang, K.C. Bright liver and alanine aminotransferase are associated with metabolic syndrome in adults. Obes. Res. 2005, 13, 1238–1245. [Google Scholar] [CrossRef]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Hellerbrand, C. Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: Mechanisms and implications. Gut 2010, 59, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Tummala, K.S.; Brandt, M.; Teijeiro, A.; Graña, O.; Schwabe, R.F.; Perna, C.; Djouder, N. Hepatocellular Carcinomas Originate Predominantly from Hepatocytes and Benign Lesions from Hepatic Progenitor Cells. Cell Rep. 2017, 19, 584–600. [Google Scholar] [CrossRef]
- Guguen-Guillouzo, C.; Guillouzo, A. General review on in vitro hepatocyte models and their applications. Methods Mol. Biol. 2010, 640, 1–40. [Google Scholar] [CrossRef]
- Bi, L.; Chiang, J.Y.; Ding, W.X.; Dunn, W.; Roberts, B.; Li, T. Saturated fatty acids activate ERK signaling to downregulate hepatic sortilin 1 in obese and diabetic mice. J. Lipid Res. 2013, 54, 2754–2762. [Google Scholar] [CrossRef]
- Zhu, C.; Kim, K.J.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef]
- Ling, S.; Chang, X.; Schultz, L.; Lee, T.K.; Chaux, A.; Marchionni, L.; Netto, G.J.; Sidranski, D.; Berman, D.M. An EGFR-ERK-SOX9 signaling cascade links urothelial development and regeneration to cancer. Cancer Res. 2011, 71, 3812–3821. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Bedossa, P.; Poynard, T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology 1996, 24, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Jenkins, N.A.; Court, D.L. Recombineering: A powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001, 2, 769–779. [Google Scholar] [CrossRef]
- Uriarte, I.; Fernandez-Barrena, M.G.; Monte, M.J.; Latasa, M.U.; Chang, H.C.Y.; Carotti, S.; Vespasiani-Gentilucci, U.; Morini, S.; Vicente, E.; Concepcion, A.R.; et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 2013, 62, 899–910. [Google Scholar] [CrossRef]
- Berasain, C.; García-Trevijano, E.R.; Castillo, J.; Erroba, E.; Santamaría, M.; Lee, D.C.; Prieto, J.; Avila, M.A. Novel role for amphiregulin in protection from liver injury. J. Biol. Chem. 2005, 280, 19012–19020. [Google Scholar] [CrossRef]
- Berasain, C.; García-Trevijano, E.R.; Castillo, J.; Erroba, E.; Lee, D.C.; Prieto Avila, M.A. Amphiregulin: An early trigger of liver regeneration in mice. Gastroenterology 2005, 128, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Ingham, S.A.; Mohr, A.M.; Wehrkamp, C.J.; Ray, A.; Roy, S.; Cazanave, S.C.; Phillippi, M.A.; Mott, J.L. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology 2014, 60, 1942–1956. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirra, S.; Gavaldà-Navarro, A.; Manso, Y.; Higuera, M.; Serrat, R.; Salcedo, M.T.; Burgaya, F.; Balibrea, J.M.; Santamaría, E.; Uriarte, I.; et al. ARMCX3 Mediates Susceptibility to Hepatic Tumorigenesis Promoted by Dietary Lipotoxicity. Cancers 2021, 13, 1110. https://doi.org/10.3390/cancers13051110
Mirra S, Gavaldà-Navarro A, Manso Y, Higuera M, Serrat R, Salcedo MT, Burgaya F, Balibrea JM, Santamaría E, Uriarte I, et al. ARMCX3 Mediates Susceptibility to Hepatic Tumorigenesis Promoted by Dietary Lipotoxicity. Cancers. 2021; 13(5):1110. https://doi.org/10.3390/cancers13051110
Chicago/Turabian StyleMirra, Serena, Aleix Gavaldà-Navarro, Yasmina Manso, Mónica Higuera, Román Serrat, María Teresa Salcedo, Ferran Burgaya, José Maria Balibrea, Eva Santamaría, Iker Uriarte, and et al. 2021. "ARMCX3 Mediates Susceptibility to Hepatic Tumorigenesis Promoted by Dietary Lipotoxicity" Cancers 13, no. 5: 1110. https://doi.org/10.3390/cancers13051110
APA StyleMirra, S., Gavaldà-Navarro, A., Manso, Y., Higuera, M., Serrat, R., Salcedo, M. T., Burgaya, F., Balibrea, J. M., Santamaría, E., Uriarte, I., Berasain, C., Avila, M. A., Mínguez, B., Soriano, E., & Villarroya, F. (2021). ARMCX3 Mediates Susceptibility to Hepatic Tumorigenesis Promoted by Dietary Lipotoxicity. Cancers, 13(5), 1110. https://doi.org/10.3390/cancers13051110