
Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing


Simple Summary
Abstract
1. Introduction
2. Hypoxia-Inducible Factors: Their Regulation and Contribution to the Tumor Radioresistance
2.1. HIF-1-Mediated Radioprotective Mechanisms in Hypoxic Cancer Cells
2.2. Targeting HIF-1 to Sensitize Hypoxic Tumors to Radiation Exposure
3. Hypoxia-Induced Reprogramming of Energy Metabolism
3.1. Energy Metabolism in Hypoxic Tumor Cells and How It Is Linked to Their Radioresistance
3.2. Targeting Cellular Energy Metabolism to Radiosensitize Hypoxic Tumors
4. HSF1-Mediated Heat Stress Response and Heat Shock Proteins (HSPs)
4.1. Implication of HSF1 in the Cancer Cell Responses to Hypoxia and Radiation Exposure
4.2. HSP90 as a Potentially Druggable Target for Radiosensitizing Tumors
4.3. Roles of HSP70: A Radioprotector of Cancer Cells and Potential Target for Radiosensitizing Them
4.4. HSP27: Targeting the “Small” Chaperone to Radiosensitize Tumors
4.5. HSF1, HSPs and the Radiosensitizing Effects of Hyperthermia
5. Endoplasmic Reticulum Stress and Glucose-Regulated Proteins (GRPs)
5.1. ER Stress and Radioresistance of Hypoxic Tumors
5.2. GRPs as Potential Targets for Radiosensitizing Hypoxic Tumors
6. Hypoxia-Responsive Autophagy
6.1. Implication of Autophagy in Cellular Homeostasis and Stress Response
6.2. Autophagy and Radioresistance of Hypoxic Cancer Cells
7. Hypoxia-Induced Generation of Radioresistant CSC-Like Cells
7.1. Hypoxia-Induced Formation of the Radioresistant CSC Phenotype and “Runaway” of Migrating CSC-Like Cells from Therapeutic Radiation Exposure
7.2. Targeting CSCs and EMT to Overcome the Radioresistance of Hypoxic Tumors
8. How Do Epigenetic Regulators Affect the Radioresistance of Hypoxic Cancer Cells?
8.1. Hypoxia-Responsive MicroRNAs
8.2. Long Noncoding RNAs and Circular RNAs
8.3. Enzymes Participating in Epigenetic Regulation of Cancer Cell Responses to Hypoxia and Radiation
9. Hypoxia-Induced Exosome Generation by Tumor Cells
9.1. Hypoxia-Induced Exosomes Can Promote the Radioresistance of Hypoxic Tumors
9.2. Exosomes as Targets or Tools to Attenuate the Radioresistance of Hypoxic Tumors
10. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Valable, S.; Laurent, C.; Haghdoost, S.; Pérès, E.A.; Bernaudin, M.; Sichel, F.; Lesueur, P.; Césaire, M.; Petit, E.; et al. Hadrontherapy Interactions in Molecular and Cellular Biology. Int. J. Mol. Sci. 2019, 21, 133. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef]
- Forster, J.C.; Marcu, L.G.; Bezak, E. Approaches to combat hypoxia in cancer therapy and the potential for in silico models in their evaluation. Phys. Med. 2019, 64, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Bader, S.B.; Dewhirst, M.W.; Hammond, E.M. Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers 2020, 13, 23. [Google Scholar] [CrossRef]
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Kim, B.N.; Park, C.K.; Kim, J.W.; Kim, Y.K.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef]
- Carlson, D.J.; Stewart, R.D.; Semenenko, V.A. Effects of oxygen on intrinsic radiation sensitivity: A test of the relationship between aerobic and hypoxic linear-quadratic (LQ) model parameters. Med. Phys. 2006, 33, 3105–3115. [Google Scholar] [CrossRef]
- Grimes, D.R. Estimation of the oxygen enhancement ratio for charged particle radiation. Phys. Med. Biol. 2020, 65, 15NT01. [Google Scholar] [CrossRef]
- Hamming, L.C.; Slotman, B.J.; Verheul, H.M.V.; Thijssen, V.L. The clinical application of angiostatic therapy in combination with radiotherapy: Past, present, future. Angiogenesis 2017, 20, 217–232. [Google Scholar] [CrossRef]
- Levy, E.B.; Gacchina Johnson, C.; Jacobs, G.; Woods, D.L.; Sharma, K.V.; Bacher, J.D.; Lewis, A.L.; Dreher, M.R.; Wood, B.J. Direct Quantification and Comparison of Intratumoral Hypoxia following Transcatheter Arterial Embolization of VX2 Liver Tumors with Different Diameter Microspheres. J. Vasc. Interv. Radiol. 2015, 26, 1567–1573. [Google Scholar] [CrossRef]
- Lee, E.J.; Chung, H.W.; Jo, J.-H.; So, Y. Radioembolization for the Treatment of Primary and Metastatic Liver Cancers. Nucl. Med. Mol. Imaging 2019, 6, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Frost, J.; Frost, M.; Batie, M.; Jiang, H.; Rocha, S. Roles of HIF and 2-Oxoglutarate-Dependent Dioxygenases in Controlling Gene Expression in Hypoxia. Cancers 2021, 13, 350. [Google Scholar] [CrossRef]
- Harada, H. Hypoxia-inducible factor 1-mediated characteristic features of cancer cells for tumor radioresistance. J. Radiat. Res. 2016, 57 (Suppl. S1), i99–i105. [Google Scholar] [CrossRef]
- Chen, S.; Yin, C.; Lao, T.; Liang, D.; He, D.; Wang, C.; Sang, N. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1α and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle 2015, 14, 2520–2536. [Google Scholar] [CrossRef]
- Baek, J.H.; Liu, Y.V.; McDonald, K.R.; Wesley, J.B.; Hubbi, M.E.; Byun, H.; Gregg, L.; Semenza, G.L. Spermidine/spermine-N1-acetyltransferase 2 is an essential component of the ubiquitin ligase complex that regulates hypoxia-inducible factor 1alpha. J. Biol. Chem. 2007, 282, 23572–23580. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-Y.; Oh, S.H.; Woo, J.-K.; Hong, W.K.; Lee, H.-Y. Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1alpha. Cancer Res. 2009, 69, 1624–1632. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Costa, M.; Gao, J.; Huang, C. JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res. 2010, 70, 813–823. [Google Scholar] [CrossRef]
- Joo, H.-Y.; Yun, M.; Jeong, J.; Park, E.-R.; Shin, H.-J.; Woo, S.R.; Jung, J.K.; Kim, Y.-M.; Park, J.-J.; Kim, J.; et al. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1α (HIF-1α) via direct interactions during hypoxia. Biochem. Biophys. Res. Commun. 2015, 462, 294–300. [Google Scholar] [CrossRef]
- Filippopoulou, C.; Simos, G.; Chachami, G. The Role of Sumoylation in the Response to Hypoxia: An Overview. Cells 2020, 9, 2359. [Google Scholar] [CrossRef]
- Zhao, F.-L.; Qin, C.-F. EGF promotes HIF-1α expression in colorectal cancer cells and tumor metastasis by regulating phosphorylation of STAT3. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1055–1062. [Google Scholar] [PubMed]
- Wu, S.-L.; Li, Y.-J.; Liao, K.; Shi, L.; Zhang, N.; Liu, S.; Hu, Y.-Y.; Li, S.-L.; Wang, Y. 2-Methoxyestradiol inhibits the proliferation and migration and reduces the radioresistance of nasopharyngeal carcinoma CNE-2 stem cells via NF-κB/HIF-1 signaling pathway inactivation and EMT reversal. Oncol. Rep. 2017, 37, 793–802. [Google Scholar] [CrossRef]
- Li, B.; He, L.; Zuo, D.; He, W.; Wang, Y.; Zhang, Y.; Liu, W.; Yuan, Y. Mutual Regulation of MiR-199a-5p and HIF-1α Modulates the Warburg Effect in Hepatocellular Carcinoma. J. Cancer 2017, 8, 940–949. [Google Scholar] [CrossRef]
- Chen, X.; Wu, L.; Li, D.; Xu, Y.; Zhang, L.; Niu, K.; Kong, R.; Gu, J.; Xu, Z.; Chen, Z.; et al. Radiosensitizing effects of miR-18a-5p on lung cancer stem-like cells via downregulating both ATM and HIF-1α. Cancer Med. 2018, 7, 3834–3847. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, Y.; Sun, T.; Yang, W. LincRNA-p21 knockdown enhances radiosensitivity of hypoxic tumor cells by reducing autophagy through HIF-1/Akt/mTOR/P70S6K pathway. Exp. Cell Res. 2017, 358, 188–198. [Google Scholar] [CrossRef]
- Li, N.; Meng, D.-D.; Gao, L.; Xu, Y.; Liu, P.-J.; Tian, Y.-W.; Yi, Z.-Y.; Zhang, Y.; Tie, X.-J.; Xu, Z.-Q. Overexpression of HOTAIR leads to radioresistance of human cervical cancer via promoting HIF-1α expression. Radiat. Oncol. 2018, 13, 210. [Google Scholar] [CrossRef]
- Liu, A.-M.; Zhu, Y.; Huang, Z.-W.; Lei, L.; Fu, S.-Z.; Chen, Y. Long noncoding RNA FAM201A involves in radioresistance of non-small-cell lung cancer by enhancing EGFR expression via miR-370. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5802–5814. [Google Scholar]
- Wang, Y.; Chen, W.; Lian, J.; Zhang, H.; Yu, B.; Zhang, M.; Wei, F.; Wu, J.; Jiang, J.; Jia, Y.; et al. The lncRNA PVT1 regulates nasopharyngeal carcinoma cell proliferation via activating the KAT2A acetyltransferase and stabilizing HIF-1α. Cell Death Differ. 2020, 27, 695–710. [Google Scholar] [CrossRef]
- Weili, Z.; Zhikun, L.; Jianmin, W.; Qingbao, T. Knockdown of USP28 enhances the radiosensitivity of esophageal cancer cells via the c-Myc/hypoxia-inducible factor-1 alpha pathway. J. Cell Biochem. 2019, 120, 201–212. [Google Scholar] [CrossRef]
- Xie, G.; Liu, Y.; Yao, O.; Zheng, R.; Zhang, L.; Lin, J.; Guo, Z.; Du, S.; Ren, C.; Yuan, Q.; et al. Hypoxia-induced angiotensin II by the lactate-chymase-dependent mechanism mediates radioresistance of hypoxic tumor cells. Sci. Rep. 2017, 7, 42396. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Zani, B.M.; Popov, V.M.; Fratticci, A.; Cerasani, M.; Genova, D.D.; Mancini, M.; Ciccarelli, C.; Ficorella, C.; et al. Hypoxia sustains glioblastoma radioresistance through ERKs/DNA-PKcs/HIF-1α functional interplay. Int. J. Oncol. 2014, 44, 2121–2131. [Google Scholar] [CrossRef]
- Wang, G.; Li, Y.; Yang, Z.; Xu, W.; Yang, Y.; Tan, X. ROS mediated EGFR/MEK/ERK/HIF-1α Loop Regulates Glucose metabolism in pancreatic cancer. Biochem. Biophys. Res. Commun. 2018, 500, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Kuger, S.; Cörek, E.; Polat, B.; Kämmerer, U.; Flentje, M.; Djuzenova, C.S. Novel PI3K and mTOR Inhibitor NVP-BEZ235 Radiosensitizes Breast Cancer Cell Lines under Normoxic and Hypoxic Conditions. Breast Cancer 2014, 8, 39–49. [Google Scholar] [CrossRef]
- Miyasaka, A.; Oda, K.; Ikeda, Y.; Sone, K.; Fukuda, T.; Inaba, K.; Makii, C.; Enomoto, A.; Hosoya, N.; Tanikawa, M.; et al. PI3K/mTOR pathway inhibition overcomes radioresistance via suppression of the HIF1-α/VEGF pathway in endometrial cancer. Gynecol. Oncol. 2015, 138, 174–180. [Google Scholar] [CrossRef]
- Ushijima, H.; Suzuki, Y.; Oike, T.; Komachi, M.; Yoshimoto, Y.; Ando, K.; Okonogi, N.; Sato, H.; Noda, S.-E.; Saito, J.-I.; et al. Radio-sensitization effect of an mTOR inhibitor, temsirolimus, on lung adenocarcinoma A549 cells under normoxic and hypoxic conditions. J. Radiat. Res. 2015, 56, 663–668. [Google Scholar] [CrossRef][Green Version]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1α to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.; Wang, X.; Li, Y.; Chen, Y.; Li, K.; Zhang, J.; Yao, L.; Guo, G. HIF-1 and NDRG2 contribute to hypoxia-induced radioresistance of cervical cancer Hela cells. Exp. Cell Res. 2010, 316, 1985–1993. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, D.; Cheng, H.; Wang, F. Hypoxia-inducible factor-1α protects cervical carcinoma cells from apoptosis induced by radiation via modulation of vascular endothelial growth factor and p53 under hypoxia. Med. Sci. Monit. 2015, 21, 318–325. [Google Scholar] [PubMed]
- Hosokawa, Y.; Okumura, K.; Terashima, S.; Sakakura, Y. Radiation protective effect of hypoxia-inducible factor-1α (HIF-1α) on human oral squamous cell carcinoma cell lines. Radiat. Prot. Dosim. 2012, 152, 159–163. [Google Scholar] [CrossRef]
- Hennessey, D.; Martin, L.M.; Atzberger, A.; Lynch, T.H.; Hollywood, D.; Marignol, L. Exposure to hypoxia following irradiation increases radioresistance in prostate cancer cells. Urol. Oncol. 2013, 31, 1106–1116. [Google Scholar] [CrossRef]
- Luo, Y.; Li, M.; Zuo, X.; Basourakos, S.P.; Zhang, J.; Zhao, J.; Han, Y.; Lin, Y.; Wang, Y.; Jiang, Y.; et al. β-catenin nuclear translocation induced by HIF-1α overexpression leads to the radioresistance of prostate cancer. Int. J. Oncol. 2018, 52, 1827–1840. [Google Scholar] [CrossRef]
- Su, T.; Liu, P.; Ti, X.; Wu, S.; Xue, X.; Wang, Z.; Dioum, E.; Zhang, Q. HΙF1α, EGR1 and SP1 co-regulate the erythropoietin receptor expression under hypoxia: An essential role in the growth of non-small cell lung cancer cells. Cell Commun. Signal. 2019, 17, 152. [Google Scholar] [CrossRef] [PubMed]
- Hassouna, I.; Sperling, S.; Kim, E.; Schulz-Schaeffer, W.; Rave-Fränk, M.; Hasselblatt, M.; Jelkmann, W.; Giese, A.; Ehrenreich, H. Erythropoietin augments survival of glioma cells after radiation and temozolomide. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 927–934. [Google Scholar] [CrossRef]
- Pérès, E.A.; Gérault, A.N.; Valable, S.; Roussel, S.; Toutain, J.; Divoux, D.; Guillamo, J.-S.; Sanson, M.; Bernaudin, M.; Petit, E. Silencing erythropoietin receptor on glioma cells reinforces efficacy of temozolomide and X-rays through senescence and mitotic catastrophe. Oncotarget 2015, 6, 2101–2119. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Lin, Y.-J.; Wu, C.-P.; Lee, H.-T.; Shyu, W.-C.; Wang, C.C. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clin. Cancer Res. 2015, 21, 460–470. [Google Scholar] [CrossRef]
- Peng, C.; Liu, G.; Huang, K.; Zheng, Q.; Li, Y.; Yu, C. Hypoxia-Induced Upregulation of HE4 Is Responsible for Resistance to Radiation Therapy of Gastric Cancer. Mol. Ther. Oncolytics 2018, 12, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Xu, H.; Chen, G.; Zhao, G.; Gao, Y.; Liu, X.; Ma, S.; Dong, L. The role of hypoxia-inducible factor-1α in radiation-induced autophagic cell death in breast cancer cells. Tumour Biol. 2015, 36, 7077–7083. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.-M.; Hu, G.-Y.; Zhao, X.-Q.; Lu, T.; Zhu, F.; Yu, S.-Y.; Xiong, H. Hypoxia-induced autophagy contributes to radioresistance via c-Jun-mediated Beclin1 expression in lung cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 761–767. [Google Scholar] [CrossRef]
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xing, X.; Liu, Q.; Wang, Z.; Xin, Y.; Zhang, P.; Hu, C.; Liu, Y. Hypoxia-induced autophagy reduces radiosensitivity by the HIF-1α/miR-210/Bcl-2 pathway in colon cancer cells. Int. J. Oncol. 2015, 46, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Goto, Y.; Koyasu, S.; Kobayashi, M.; Morinibu, A.; Yoshimura, M.; Hiraoka, M.; Hammond, E.M.; Harada, H. UCHL1-HIF-1 axis-mediated antioxidant property of cancer cells as a therapeutic target for radiosensitization. Sci. Rep. 2017, 7, 6879. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Shen, H.; Cook, K.; Gee, H.E.; Hau, E. Hypoxia, metabolism, and the circadian clock: New links to overcome radiation resistance in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 129. [Google Scholar] [CrossRef] [PubMed]
- Grosso, S.; Doyen, J.; Parks, S.K.; Bertero, T.; Paye, A.; Cardinaud, B.; Gounon, P.; Lacas-Gervais, S.; Noël, A.; Pouysségur, J.; et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013, 4, e544. [Google Scholar] [CrossRef] [PubMed]
- Dang, K.; Myers, K.A. The role of hypoxia-induced miR-210 in cancer progression. Int. J. Mol. Sci. 2015, 16, 6353–6372. [Google Scholar] [CrossRef]
- Xing, Y.; Cui, D.; Wang, S.; Wang, P.; Xing, X.; Li, H. Oleuropein represses the radiation resistance of ovarian cancer by inhibiting hypoxia and microRNA-299-targetted heparanase expression. Food Funct. 2017, 8, 2857–2864. [Google Scholar] [CrossRef]
- Lehmann, S.; Boekhorst, V.T.; Odenthal, J.; Bianchi, R.; Helvert, S.V.; Ikenberg, K.; Ilina, O.; Stoma, S.; Xandry, J.; Jiang, L.; et al. Hypoxia Induces a HIF-1-Dependent Transition from Collective-to-Amoeboid Dissemination in Epithelial Cancer Cells. Curr. Biol. 2017, 27, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.-P.; Wong, C.C.-L.; Kai, A.K.-L.; Ho, D.W.-H.; Lau, E.Y.-T.; Tsui, Y.-M.; Chan, L.-K.; Cheung, T.-T.; Chok, K.S.-H.; Chan, A.C.Y.; et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 2017, 66, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Inoue, M.; Itasaka, S.; Hirota, K.; Morinibu, A.; Shinomiya, K.; Zeng, L.; Ou, G.; Zhu, Y.; Yoshimura, M.; et al. Cancer cells that survive radiation therapy acquire HIF-1 activity and translocate towards tumour blood vessels. Nat. Commun. 2012, 3, 783. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, X.; Qu, Y.; Xu, O.; Sun, Q. Hypoxia promotes radioresistance of CD133-positive Hep-2 human laryngeal squamous carcinoma cells in vitro. Int. J. Oncol. 2013, 43, 131–140. [Google Scholar] [CrossRef]
- Wozny, A.-S.; Lauret, A.; Battiston-Montagne, P.; Guy, J.-B.; Beuve, M.; Cunha, M.; Saintigny, Y.; Blond, E.; Magne, N.; Lalle, P.; et al. Differential pattern of HIF-1α expression in HNSCC cancer stem cells after carbon ion or photon irradiation: One molecular explanation of the oxygen effect. Br. J. Cancer 2017, 116, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Jiang, Z.; Bernhard, E.J.; Evans, S.M.; Koch, C.J.; Hahn, S.M.; Maity, A. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: Implications for radiotherapy. Cancer Res. 2006, 66, 9252–9259. [Google Scholar] [CrossRef] [PubMed]
- Palayoor, S.T.; Mitchell, J.B.; Cerna, D.; Degraff, W.; John-Aryankalayil, M.; Coleman, C.N. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radiosensitivity of prostate carcinoma cells. Int. J. Cancer 2008, 123, 2430–2437. [Google Scholar] [CrossRef]
- Yu, J.; Mi, J.; Wang, Y.; Wang, A.; Tian, X. Regulation of radiosensitivity by HDAC inhibitor trichostatin A in the human cervical carcinoma cell line Hela. Eur. J. Gynaecol. Oncol. 2012, 33, 285–290. [Google Scholar]
- Wang, B.-F.; Wang, X.-J.; Kang, H.-F.; Bai, M.-H.; Guan, H.-T.; Wang, Z.-W.; Zan, Y.; Song, L.-Q.; Min, W.-L.; Lin, S.; et al. Saikosaponin-D enhances radiosensitivity of hepatoma cells under hypoxic conditions by inhibiting hypoxia-inducible factor-1α. Cell Physiol. Biochem. 2014, 33, 37–51. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, M.; Xing, D.; Feng, Y. Atorvastatin enhances radiosensitivity in hypoxia-induced prostate cancer cells related with HIF-1α inhibition. Biosci. Rep. 2017, 37, BSR20170340. [Google Scholar] [CrossRef]
- Kim, E.-H.; Ko, H.Y.; Yu, A.R.; Kim, H.; Zaheer, J.; Kang, H.J.; Lim, Y.-C.; Cho, K.D.; Joo, H.-Y.; Kang, M.K.; et al. Inhibition of HIF-1α by Atorvastatin During 131I-RTX Therapy in Burkitt’s Lymphoma Model. Cancers 2020, 12, 1203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, X.; Zhang, Q.; Yang, B.; Xu, L.; Qin, Q.; Zhu, H.; Liu, J.; Cai, J.; Tao, G.; et al. Berberine radiosensitizes human nasopharyngeal carcinoma by suppressing hypoxia-inducible factor-1α expression. Acta Otolaryngol. 2014, 134, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, C.; Yang, X.; Yang, B.; Wang, J.; Kang, Y.; Wang, Z.; Li, D.; Huang, G.; Ma, Z.; et al. Berberine inhibits the expression of hypoxia induction factor-1alpha and increases the radiosensitivity of prostate cancer. Berberine inhibits the expression of hypoxia induction factor-1alpha and increases the radiosensitivity of prostate cancer. Diagn. Pathol. 2014, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhu, H.; Ge, Y.; Liu, J.; Cai, J.; Qin, Q.; Zhan, L.; Zhang, C.; Xu, L.; Liu, Z.; et al. Melittin enhances radiosensitivity of hypoxic head and neck squamous cell carcinoma by suppressing HIF-1α. Tumour Biol. 2014, 35, 10443–10448. [Google Scholar] [CrossRef] [PubMed]
- Helbig, L.; Koi, L.; Brüchner, K.; Gurtner, K.; Hess-Stumpp, H.; Unterschemmann, K.; Pruschy, M.; Baumann, M.; Yaromina, A.; Zips, D. Hypoxia-inducible factor pathway inhibition resolves tumor hypoxia and improves local tumor control after single-dose irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 159–166. [Google Scholar] [CrossRef]
- Moon, S.Y.; Chang, H.W.; Roh, J.-L.; Kim, G.C.; Choi, S.-H.; Lee, S.-W.; Cho, K.-J.; Nam, S.Y.; Kim, S.Y. Using YC-1 to overcome the radioresistance of hypoxic cancer cells. Oral Oncol. 2009, 45, 915–919. [Google Scholar] [CrossRef]
- Harada, H.; Itasaka, S.; Zhu, Y.; Zeng, L.; Xie, X.; Morinibu, A.; Shinomiya, K.; Hiraoka, M. Treatment regimen determines whether an HIF-1 inhibitor enhances or inhibits the effect of radiation therapy. Br. J. Cancer 2009, 100, 747–757. [Google Scholar] [CrossRef]
- Harada, T.; Hirose, K.; Wada, Y.; Sato, M.; Ichise, K.; Aoki, M.; Kato, T.; Takeda, K.; Takai, Y. YC-1 sensitizes the antitumor effects of boron neutron capture therapy in hypoxic tumor cells. J. Radiat. Res. 2020, 61, 524–534. [Google Scholar] [CrossRef]
- Yang, M.; Yang, Y.; Cui, H.; Guan, Z.; Yang, Y.; Zhang, H.; Chen, X.; Zhu, H.; Yang, X.; Cai, J.; et al. The natural compound gambogic acid radiosensitizes nasopharyngeal carcinoma cells under hypoxic conditions. Tumori 2016, 102, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Cai, D.; Ma, J. Pachymic Acid Sensitizes Gastric Cancer Cells to Radiation Therapy by Upregulating Bax through Hypoxia. Am. J. Chin. Med. 2018, 46, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, X.; Zhang, Q.; Guo, Q.; He, J.; Qin, Q.; Zhu, H.; Liu, J.; Zhan, L.; Lu, J.; et al. STAT3 inhibitor NSC74859 radiosensitizes esophageal cancer via the downregulation of HIF-1α. Tumour Biol. 2014, 35, 9793–9799. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Zhang, Q.; Yu, M.; Qi, X.; Wang, G.; Xiao, L.; Yi, Q.; Jin, W. Ursolic acid sensitizes radioresistant NSCLC cells expressing HIF-1α through reducing endogenous GSH and inhibiting HIF-1α. Oncol. Lett. 2017, 13, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Singh-Gupta, V.; Zhang, H.; Banerjee, S.; Kong, D.; Raffoul, J.J.; Sarkar, F.H.; Hillman, G.G. Radiation-induced HIF-1alpha cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int. J. Cancer 2009, 124, 1675–1684. [Google Scholar] [CrossRef]
- Singh-Gupta, V.; Joiner, M.C.; Runyan, L.; Yunker, C.K.; Sarkar, F.H.; Miller, S.; Gadgeel, S.M.; Konski, A.A.; Hillman, G.G. Soy isoflavones augment radiation effect by inhibiting APE1/Ref-1 DNA repair activity in non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 688–698. [Google Scholar] [CrossRef]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. HIF-1α inhibition by siRNA or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via CA9 expression. BMC Cancer 2010, 10, 605. [Google Scholar] [CrossRef]
- Staab, A.; Loeffler, J.; Said, H.M.; Diehlmann, D.; Katzer, A.; Beyer, M.; Fleischer, M.; Schwab, F.; Baier, K.; Einsele, H.; et al. Effects of HIF-1 inhibition by chetomin on hypoxia-related transcription and radiosensitivity in HT 1080 human fibrosarcoma cells. BMC Cancer 2007, 7, 213. [Google Scholar] [CrossRef]
- Jin, Z.; Aixi, Y.; Baiwen, Q.; Zonghuan, L.; Xiang, H. Inhibition of hypoxia-inducible factor-1 alpha radiosensitized MG-63 human osteosarcoma cells in vitro. Tumori 2015, 101, 578–584. [Google Scholar] [CrossRef]
- Lu, Y.-R.; Song, J.; Zhabihula, B.-X.; Zhang, J.-R. 2-Methoxyestradiol promotes radiosensitivity of esophageal squamous cell carcinoma by suppressing hypoxia-inducible factor-1α expression. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10785–10795. [Google Scholar] [PubMed]
- Zhao, H.; Jiang, H.; Li, Z.; Zhuang, Y.; Liu, Y.; Zhou, S.; Xiao, Y.; Xie, C.; Zhou, F.; Zhou, Y. 2-Methoxyestradiol enhances radiosensitivity in radioresistant melanoma MDA-MB-435R cells by regulating glycolysis via HIF-1α/PDK1 axis. Int. J. Oncol. 2017, 50, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Heo, K.; Park, H.S.; Yang, K.M.; Jeong, M.H. The resveratrol analog HS-1793 enhances radiosensitivity of mouse-derived breast cancer cells under hypoxic conditions. Int. J. Oncol. 2016, 49, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, H.; Zheng, Y.; Han, Y.; Wang, T.; Zhang, H.; Sun, Q.; Li, Z. Overcoming Radioresistance in Tumor Therapy by Alleviating Hypoxia and Using the HIF-1 Inhibitor. ACS Appl. Mater. Interfaces 2020, 12, 4231–4240. [Google Scholar] [CrossRef] [PubMed]
- Oommen, D.; Prise, K.M. KNK437, abrogates hypoxia-induced radioresistance by dual targeting of the AKT and HIF-1α survival pathways. Biochem. Biophys. Res. Commun. 2012, 421, 538–543. [Google Scholar] [CrossRef]
- Lai, K.-G.; Lin, Y.-H.; Ho, C.-T.; Chen, C.-Y.; Peng, C.-Y.; Liu, T.-Z.; Chiou, J.-F. Paclitaxel pretreatment overcomes hypoxia inducible factor-1α-induced radioresistance acquisition of human hepatoma and lung adenocarcinoma cells. Life Sci. 2015, 136, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Qin, Q.; Yang, M.; Zhang, H.; Liu, Z.; Yang, Y.; Chen, X.; Zhu, H.; Wang, D.; Meng, C.; et al. Bortezomib enhances the radiosensitivity of hypoxic cervical cancer cells by inhibiting HIF-1α expression. Int. J. Clin. Exp. Pathol. 2015, 8, 9032–9041. [Google Scholar] [PubMed]
- Wang, D.; Qin, Q.; Jiang, Q.-J.; Wang, D.-F. Bortezomib sensitizes esophageal squamous cancer cells to radiotherapy by suppressing the expression of HIF-1α and apoptosis proteins. J. Xray Sci. Technol. 2016, 24, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Shim, J.W.; Choi, Y.J.; Heo, K.; Yang, K. The combination of sorafenib and radiation preferentially inhibits breast cancer stem cells by suppressing HIF-1α expression. Oncol. Rep. 2013, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.; Nguewa, P.A.; Redrado, M.; Manrique, I.; Calvo, A. Sunitinib reduces tumor hypoxia and angiogenesis, and radiosensitizes prostate cancer stem-like cells. Prostate 2015, 75, 1137–1149. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Yu, W.; Zhao, X.; Xue, Y.; Xu, H. Radiosensitizing effect of irisquinone on glioma through the downregulation of HIF-1α evaluated by 18F-FDG and 18F-FMISO PET/CT. Nucl. Med. Commun. 2016, 37, 705–714. [Google Scholar] [CrossRef]
- Xu, T.; Xiao, D. Oleuropein enhances radiation sensitivity of nasopharyngeal carcinoma by downregulating PDRG1 through HIF1α-repressed microRNA-519d. J. Exp. Clin. Cancer Res. 2017, 36, 3. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Iijima, M.; Gombodorj, N.; Tachibana, Y.; Tachibana, K.; Yokobori, T.; Honma, K.; Nakano, T.; Takayuki Asao, T.; Kuwahara, R.; Aoyama, K.; et al. Development of single nanometer-sized ultrafine oxygen bubbles to overcome the hypoxia-induced resistance to radiation therapy via the suppression of hypoxia-inducible factor-1α. Int. J. Oncol. 2018, 52, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Hwang, J.; Lee, K.; Choi, Y.; Seo, Y.; Jeon, H.; Hong, J.W.; Choi, J. Anti-Tumor Drug-Loaded Oxygen Nanobubbles for the Degradation of HIF-1α and the Upregulation of Reactive Oxygen Species in Tumor Cells. Cancers 2019, 11, 1464. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef]
- Moeller, B.J.; Dreher, M.R.; Rabbani, Z.N.; Schroeder, T.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell 2005, 8, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.-Y. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Wang, W.-J.; Sui, H.; Qi, C.; Li, Q.; Zhang, J.; Wu, S.-F.; Mei, M.-Z.; Lu, Y.-Y.; Wan, Y.-T.; Chang, H.; et al. Ursolic acid inhibits proliferation and reverses drug resistance of ovarian cancer stem cells by downregulating ABCG2 through suppressing the expression of hypoxia-inducible factor-1α in vitro. Oncol. Rep. 2016, 36, 428–440. [Google Scholar] [CrossRef]
- Zhao, D.; Zhai, B.; He, C.; Tan, G.; Jiang, X.; Pan, S.; Dong, X.; Wei, Z.; Ma, L.; Qiao, H.; et al. Upregulation of HIF-2α induced by sorafenib contributes to the resistance by activating the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014, 26, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Tameemi, W.A.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Li, Y.; Gruber, J.J.; Litzenburger, U.M.; Zhou, Y.; Miao, Y.R.; LaGory, E.L.; Li, A.M.; Hu, Z.; Yip, M.; Hart, L.S.; et al. Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis. 2020, 11, 102. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chafe, S.C.; Brown, W.S.; Saberi, S.; Swayampakula, M.; Venkateswaran, G.; Nemirovsky, O.; Gillespie, J.A.; Karasinska, J.M.; Kalloger, S.E.; et al. Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells With Activated KRAS in Response to Hypoxia. Gastroenterology 2019, 157, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Cairns, R.A.; Chaudary, N.; Vellanki, R.N.; Kalliomaki, T.; Moriyama, E.H.; Mujcic, H.; Wilson, B.C.; Wouters, B.G.; Hill, R.; et al. Metabolic targeting of HIF-dependent glycolysis reduces lactate, increases oxygen consumption and enhances response to high-dose single-fraction radiotherapy in hypoxic solid tumors. BMC Cancer 2017, 17, 418. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Fatal Alliance of Hypoxia-/HIF-1α-Driven Microenvironmental Traits Promoting Cancer Progression. Adv. Exp. Med. Biol. 2020, 1232, 169–176. [Google Scholar] [PubMed]
- Riemann, A.; Rauschner, M.; Gießelmann, M.; Reime, S.; Haupt, V.; Thews, O. Extracellular Acidosis Modulates the Expression of Epithelial-Mesenchymal Transition (EMT) Markers and Adhesion of Epithelial and Tumor Cells. Neoplasia 2019, 21, 450–458. [Google Scholar] [CrossRef]
- Lim, S.-H.; Li, C.-H.; Jeong, Y.-I.; Jang, W.-Y.; Choi, J.-M.; Jung, S. Enhancing Radiotherapeutic Effect with Nanoparticle-Mediated Radiosensitizer Delivery Guided By Focused Gamma Rays In Lewis Lung Carcinoma-Bearing Mouse Brain Tumor Models. Int. J. Nanomed. 2019, 14, 8861–8874. [Google Scholar] [CrossRef]
- Kaplan, A.R.; Pham, H.; Liu, Y.; Oyaghire, S.; Bahal, R.; Engelman, D.M.; Glazer, P.M. Ku80-Targeted pH-Sensitive Peptide-PNA Conjugates Are Tumor Selective and Sensitize Cancer Cells to Ionizing Radiation. Mol. Cancer Res. 2020, 18, 873–882. [Google Scholar] [CrossRef]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef]
- Bamodu, O.A.; Chang, H.-L.; Ong, J.-R.; Lee, W.-H.; Yeh, C.-T.; Tsai, J.-T. Elevated PDK1 Expression Drives PI3K/AKT/MTOR Signaling Promotes Radiation-Resistant and Dedifferentiated Phenotype of Hepatocellular Carcinoma. Cells 2020, 9, 746. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Uemura, M.; Munakata, K.; Takahashi, H.; Haraguchi, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Ikenaga, M.; Murata, K.; et al. Fructose-bisphosphate aldolase A is a key regulator of hypoxic adaptation in colorectal cancer cells and involved in treatment resistance and poor prognosis. Int. J. Oncol. 2017, 50, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.; Maurer, G.D.; Wanka, C.; Hofmann, U.; Luger, A.-L.; Bruns, I.; Steinbach, J.P.; Rieger, J. Gene Suppression of Transketolase-Like Protein 1 (TKTL1) Sensitizes Glioma Cells to Hypoxia and Ionizing Radiation. Int. J. Mol. Sci. 2018, 19, 2168. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wang, R.; Yan, H.; Jin, L.; Dou, X.; Chen, D. MicroRNA-21 modulates radiation resistance through upregulation of hypoxia-inducible factor-1α-promoted glycolysis in non-small cell lung cancer cells. Mol. Med. Rep. 2016, 13, 4101–4107. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Wang, Y.; Zhu, Y.; Li, S.; Liu, Y.; Chen, C.; Wang, X.; Zen, K.; Li, L. The miR-125a/HK2 axis regulates cancer cell energy metabolism reprogramming in hepatocellular carcinoma. Sci. Rep. 2017, 7, 3089. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wang, M.; Dong, Y.; Xu, B.; Chen, J.; Ding, Y.; Qiu, S.; Li, L.; Karamfilova Zaharieva, E.; Zhou, X.; et al. Circular RNA circRNF20 promotes breast cancer tumorigenesis and Warburg effect through miR-487a/HIF-1α/HK2. Cell Death Dis. 2020, 11, 145. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhong, R.; Deng, C.; Zhou, Z. Circle RNA circABCB10 Modulates PFN2 to Promote Breast Cancer Progression, as Well as Aggravate Radioresistance Through Facilitating Glycolytic Metabolism Via miR-223-3p. Cancer Biother. Radiopharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Li, J.; Chen, J.; Qian, L.; Wang, A.; Chen, X.; Xiong, W.; Tang, J.; Tang, S.; Chen, Y.; et al. microRNA-33a-5p increases radiosensitivity by inhibiting glycolysis in melanoma. Oncotarget 2017, 8, 83660–83672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, Y.; Zhou, M.; Zhang, Y.; Wang, P.; Li, X.; Yang, J.; Wang, H.; Ding, Z. HOXA9 inhibits HIF-1α-mediated glycolysis through interacting with CRIP2 to repress cutaneous squamous cell carcinoma development. Nat. Commun. 2018, 9, 1480. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef]
- Murata, Y.; Hashimoto, T.; Urushihara, Y.; Shiga, S.; Takeda, K.; Jingu, K.; Hosoi, Y. Knockdown of AMPKα decreases ATM expression and increases radiosensitivity under hypoxia and nutrient starvation in an SV40-transformed human fibroblast cell line, LM217. Biochem. Biophys. Res. Commun. 2018, 495, 2566–2572. [Google Scholar] [CrossRef] [PubMed]
- Shiga, S.; Murata, Y.; Hashimoto, T.; Urushihara, Y.; Fujishima, Y.; Kudo, K.; Sonohara, Y.; Kurusu, M.; Takeda, K.; Jingu, K.; et al. DNA-PKcs is activated under nutrient starvation and activates Akt, MST1, FoxO3a, and NDR1. Biochem. Biophys. Res. Commun. 2020, 521, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M.; Khuchua, Z.; et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef]
- Saxena, M.; Balaji, S.A.; Deshpande, N.; Ranganathan, S.; Pillai, D.M.; Hindupur, S.K.; Rangarajan, A. AMP-activated protein kinase promotes epithelial-mesenchymal transition in cancer cells through Twist1 upregulation. J. Cell Sci. 2018, 131, jcs208314. [Google Scholar] [CrossRef]
- Guo, B.; Han, X.; Tkach, D.; Huang, S.-G.; Zhang, D. AMPK promotes the survival of colorectal cancer stem cells. Anim. Model. Exp. Med. 2018, 1, 134–142. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Chien, M.-H.; Liu, H.-Y.; Chang, Y.-C.; Chen, C.-K.; Lee, W.-J.; Kuo, T.-C.; Hsiao, M.; Hua, K.-T.; Cheng, T.-Y. Nuclear translocation of PKM2/AMPK complex sustains cancer stem cell populations under glucose restriction stress. Cancer Lett. 2018, 421, 28–40. [Google Scholar] [CrossRef]
- Avolio, R.; Matassa, D.S.; Criscuolo, D.; Landriscina, M.; Esposito, F. Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules 2020, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 regulation of cancer metabolism: Dual role as oncogene or tumor suppressor. Genes 2018, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Powers, J.F.; Tischler, A.S.; Pacak, K.; Fliedner, S.; et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat. Commun. 2013, 4, 2139. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef]
- Singh, D.; Banerji, A.K.; Dwarakanath, B.S.; Tripathi, R.P.; Gupta, J.P.; Mathew, T.L.; Ravindranath, T.; Jain, V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef]
- Dwarakanath, B.S.; Singh, D.; Banerji, A.K.; Sarin, R.; Venkataramana, N.K.; Jalali, R.; Vishwanath, P.N.; Mohanti, B.K.; Tripathi, R.P.; Kalia, V.K.; et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: Present status and future prospects. J. Cancer Res. Ther. 2009, 5 (Suppl. S1), S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.-Y.; Zhou, S.-H.; Lu, Z.-J.; Fan, J.; Huang, Y.-P. Inhibiting GLUT-1 expression and PI3K/Akt signaling using apigenin improves the radiosensitivity of laryngeal carcinoma in vivo. Oncol. Rep. 2015, 34, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Ming, J.; Zhou, Y.; Fan, L. Inhibition of Glut1 by WZB117 sensitizes radioresistant breast cancer cells to irradiation. Cancer Chemother. Pharmacol. 2016, 77, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Hlouschek, J.; Ritter, V.; Wirsdörfer, F.; Klein, D.; Jendrossek, V.; Matschke, J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018, 439, 24–38. [Google Scholar] [CrossRef]
- De Mey, S.; Jiang, H.; Corbet, C.; Wang, H.; Dufait, I.; Law, K.; Bastien, E.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Antidiabetic Biguanides Radiosensitize Hypoxic Colorectal Cancer Cells Through a Decrease in Oxygen Consumption. Front. Pharmacol. 2018, 9, 1073. [Google Scholar] [CrossRef]
- Mudassar, F.; Shen, H.; O’Neill, G.; Hau, E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 208. [Google Scholar] [CrossRef] [PubMed]
- De Bruycker, S.; Vangestel, C.; van den Wyngaert, T.; Pauwels, P.; Wyffels, L.; Staelens, S.; Stroobants, S. 18F-Flortanidazole Hypoxia PET Holds Promise as a Prognostic and Predictive Imaging Biomarker in a Lung Cancer Xenograft Model Treated with Metformin and Radiotherapy. J. Nucl. Med. 2019, 60, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Desai, J.; Palackdharry, S.M.; Morris, J.C.; Zhu, Z.; Jandarov, R.; Riaz, M.K.; Takiar, V.; Mierzwa, M.; Gutkind, J.S.; et al. Phase 1 dose-finding study of metformin in combination with concurrent cisplatin and radiotherapy in patients with locally advanced head and neck squamous cell cancer. Cancer 2020, 126, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K.; Shabeeb, D.; Musa, A.E.; Najafi, M.; Farhood, B. Metformin as a Radiation Modifier; Implications to Normal Tissue Protection and Tumor Sensitization. Curr. Clin. Pharmacol. 2019, 14, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Maurya, R.; Saran, S. Introducing a simple model system for binding studies of known and novel inhibitors of AMPK: A therapeutic target for prostate cancer. J. Biomol. Struct. Dyn. 2019, 37, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-T.; Li, Y.-S.; Shi, Z.-Y.; Guo, X.-L. New insights into molecular chaperone TRAP1 as a feasible target for future cancer treatments. Life Sci. 2020, 254, 117737. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, V.; Buchner, J. Functional principles and regulation of molecular chaperones. Adv. Protein Chem. Struct. Biol. 2019, 114, 1–60. [Google Scholar]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. Regulation of the mammalian heat shock factor 1. FEBS J. 2017, 284, 1606–1627. [Google Scholar] [CrossRef] [PubMed]
- Dai, C. The heat-shock, or HSF1-mediated proteotoxic stress, response in cancer: From proteomic stability to oncogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160525. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2019, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Mivechi, N.F.; Koong, A.C.; Giaccia, A.J.; Hahn, G.M. Analysis of HSF-1 phosphorylation in A549 cells treated with a variety of stresses. Int. J. Hypertherm. 1994, 10, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Jung, S.M.; Kim, M.J.; Bae, J.H.; Kim, H.B.; Kim, J.Y.; Park, S.J.; Song, H.S.; Kim, D.W.; Kang, C.D.; et al. DNA-dependent protein kinase is involved in heat shock protein-mediated accumulation of hypoxia-inducible factor-1alpha in hypoxic preconditioned HepG2 cells. FEBS J. 2008, 275, 5969–5981. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Meng, L.; Kim, G.; Mills, T.A.; Benjamin, I.J.; Sherman, M.Y. Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol. Cell Biol. 2012, 32, 929–940. [Google Scholar] [CrossRef]
- Jiang, S.; Tu, K.; Fu, Q.; Schmitt, D.C.; Zhou, L.; Lu, N.; Zhao, Y. Multifaceted roles of HSF1 in cancer. Tumour Biol. 2015, 36, 4923–4931. [Google Scholar] [CrossRef]
- Kovács, D.; Sigmond, T.; Hotzi, B.; Bohár, B.; Fazekas, D.; Deák, V.; Vellai, T.; Barna, J. HSF1Base: A Comprehensive Database of HSF1 (Heat Shock Factor 1) Target Genes. Int. J. Mol. Sci. 2019, 20, 5815. [Google Scholar] [CrossRef]
- Kabakov, A.E.; Malyutina, Y.V.; Latchman, D.S. Hsf1-mediated stress response can transiently enhance cellular radioresistance. Radiat. Res. 2006, 165, 410–423. [Google Scholar] [CrossRef]
- Kabakov, A.E.; Kudryavtsev, V.A. Heat shock proteins as molecular targets for anticancer therapy: Approaches, agents, and trends. In Heat Shock Proteins. Classifications, Functions, and Applications; Usmani, S., Ed.; Nova Science Publishers: New York, NY, USA, 2013; pp. 25–56. [Google Scholar]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef]
- Li, Q.; Martinez, J.D. Loss of HSF1 results in defective radiation-induced G(2) arrest and DNA repair. Radiat. Res. 2011, 176, 17–24. [Google Scholar] [CrossRef]
- Vilaboa, N.E.; Galán, A.; Troyano, A.; de Blas, E.; Aller, P. Regulation of multidrug resistance 1 (MDR1)/P-glycoprotein gene expression and activity by heat-shock transcription factor 1 (HSF1). J. Biol. Chem. 2000, 275, 24970–24976. [Google Scholar] [CrossRef]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Stuart, J.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypoxia-Activated Prodrugs in Combination with Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1183–1196. [Google Scholar] [CrossRef]
- Jackson, R.K.; Liew, L.P.; Hay, M.P. Overcoming Radioresistance: Small Molecule Radiosensitisers and Hypoxia-activated Prodrugs. Clin. Oncol. R Coll. Radiol. 2019, 31, 290–302. [Google Scholar] [CrossRef]
- Kim, M.Y.; Park, S.-J.; Shim, J.W.; Yang, K.; Kang, H.S.; Heo, K. Naphthazarin enhances ionizing radiation-induced cell cycle arrest and apoptosis in human breast cancer cells. Int. J. Oncol. 2015, 46, 1659–1666. [Google Scholar] [CrossRef]
- Park, E.M.; Lee, I.J.; Kim, S.H.; Song, G.Y.; Park, Y.M. Inhibitory effect of a naphthazarin derivative, S64, on heat shock factor (Hsf) activation and glutathione status following hypoxia. Cell Biol. Toxicol. 2003, 19, 273–284. [Google Scholar] [CrossRef]
- Yoon, T.; Kang, G.-Y.; Han, A.-R.; Seo, E.-K.; Lee, Y.-S. 2,4-Bis(4-hydroxybenzyl)phenol inhibits heat shock transcription factor 1 and sensitizes lung cancer cells to conventional anticancer modalities. J. Nat. Prod. 2014, 77, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Schilling, D.; Kühnel, A.; Konrad, S.; Tetzlaff, F.; Bayer, C.; Yaglom, J.; Multhoff, G. Sensitizing tumor cells to radiation by targeting the heat shock response. Cancer Lett. 2015, 360, 294–301. [Google Scholar] [CrossRef]
- Dong, B.; Jaeger, A.M.; Thiele, D.J. Inhibiting Heat Shock Factor 1 in Cancer: A Unique Therapeutic Opportunity. Trends Pharmacol. Sci. 2019, 40, 986–1005. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Gökmen-Polar, Y. HSF1 as a Cancer Biomarker and Therapeutic Target. Curr. Cancer Drug Targets 2019, 19, 515–524. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; You, Q.D.; Xu, X.L. Heat shock protein 90 inhibitors: An update on achievements, challenges, and future directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef]
- Schilling, D.; Bayer, C.; Li, W.; Molls, M.; Vaupel, P.; Multhoff, G. Radiosensitization of normoxic and hypoxic h1339 lung tumor cells by heat shock protein 90 inhibition is independent of hypoxia inducible factor-1α. PLoS ONE 2012, 7, e31110. [Google Scholar] [CrossRef]
- Djuzenova, C.S.; Blassl, C.; Roloff, K.; Kuger, S.; Katzer, A.; Niewidok, N.; Günther, N.; Polat, B.; Sukhorukov, V.L.; Flentje, M. Hsp90 inhibitor NVP-AUY922 enhances radiation sensitivity of tumor cell lines under hypoxia. Cancer Biol. Ther. 2012, 13, 425–434. [Google Scholar] [CrossRef][Green Version]
- Hartmann, S.; Günther, N.; Biehl, M.; Katzer, A.; Kuger, S.; Worschech, E.; Sukhorukov, V.L.; Krohne, G.; Zimmermann, H.; Flentje, M.; et al. Hsp90 inhibition by NVP-AUY922 and NVP-BEP800 decreases migration and invasion of irradiated normoxic and hypoxic tumor cell lines. Cancer Lett. 2013, 331, 200–210. [Google Scholar] [CrossRef]
- Li, H.K.; Matsumoto, Y.; Furusawa, Y.; Kamada, T. PU-H71, a novel Hsp90 inhibitor, as a potential cancer-specific sensitizer to carbon-ion beam therapy. J. Radiat. Res. 2016, 57, 572–575. [Google Scholar] [CrossRef]
- Lee, Y.; Li, H.K.; Masaoka, A.; Sunada, S.; Hirakawa, H.; Fujimori, A.; Nickoloff, J.A.; Okayasu, R. The purine scaffold Hsp90 inhibitor PU-H71 sensitizes cancer cells to heavy ion radiation by inhibiting DNA repair by homologous recombination and non-homologous end joining. Radiother. Oncol. 2016, 121, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.A.; No, M.; Lee, J.M.; Shin, J.H.; Oh, J.S.; Choi, E.J.; Kim, I.H.; Atadja, P.; Bernhard, E.J. Epigenetic modulation of radiation response in human cancer cells with activated EGFR or HER-2 signaling: Potential role of histone deacetylase 6. Radiother. Oncol. 2009, 92, 125–132. [Google Scholar] [CrossRef]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperon. 2016, 21, 379–404. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Choi, H.G.; Kim, J.-S.; Kim, K.H.; Kim, K.H.; Sung, M.-W.; Choe, J.-Y.; Kim, J.E.; Jung, Y.H. Expression of hypoxic signaling markers in head and neck squamous cell carcinoma and its clinical significance. Eur. Arch. Otorhinolaryngol. 2015, 272, 219–228. [Google Scholar] [CrossRef]
- Gabai, V.L.; Kabakov, A.E. Induction of heat-shock protein synthesis and thermotolerance in EL-4 ascites tumor cells by transient ATP depletion after ischemic stress. Exp. Mol. Pathol. 1994, 60, 88–99. [Google Scholar] [CrossRef]
- Patel, B.; Khaliq, A.; Jarvis-Evans, J.; Boulton, M.; Arrol, S.; Mackness, M.; McLeod, D. Hypoxia induces HSP 70 gene expression in human hepatoma (HEP G2) cells. Biochem. Mol. Biol. Int. 1995, 36, 907–912. [Google Scholar]
- Huang, W.-J.; Xia, L.-M.; Zhu, F.; Huang, B.; Zhou, C.; Zhu, H.-F.; Wang, B.; Chen, B.; Lei, P.; Shen, G.-X.; et al. Transcriptional upregulation of HSP70-2 by HIF-1 in cancer cells in response to hypoxia. Int. J. Cancer 2009, 124, 298–305. [Google Scholar] [CrossRef]
- Duan, X.; Zhou, G.; Zheng, C.; Liang, H.; Liang, B.; Song, S.; Feng, G. Heat shock protein 70 expression and effect of combined transcatheter arterial embolization and radiofrequency ablation in the rabbit VX2 liver tumour model. Clin. Radiol. 2014, 69, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.S.; Naik, R.; Kim, H.M.; Kim, B.-K.; Lee, H.; Kim, I.; Ahn, H.; Jang, Y.; Jang, K.; Eo, Y.; et al. Identification of Targets of the HIF-1 Inhibitor IDF-11774 Using Alkyne-Conjugated Photoaffinity Probes. Bioconjug. Chem. 2016, 27, 1911–1920. [Google Scholar] [CrossRef]
- Luo, W.; Zhong, J.; Chang, R.; Hu, H.; Pandey, A.; Semenzam, G.L. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J. Biol. Chem. 2010, 285, 3651–3663. [Google Scholar] [CrossRef]
- Habryka, A.; Gogler-Pigłowska, A.; Sojka, D.; Kryj, M.; Krawczyk, Z.; Scieglinska, D. Cell type-dependent modulation of the gene encoding heat shock protein HSPA2 by hypoxia-inducible factor HIF-1: Down-regulation in keratinocytes and up-regulation in HeLa cells. Biochim. Biophys. Acta 2015, 1849, 1155–1169. [Google Scholar] [CrossRef]
- Da Rocha, A.B.; Regner, A.; Grivicich, I.; Schunemann, D.P.; Diel, C.; Kovaleski, G.; De Farias, C.B.; Mondadori, E.; Almeida, L.; Filho, A.B.; et al. Radioresistance is associated to increased Hsp70 content in human glioblastoma cell lines. Int. J. Oncol. 2004, 25, 777–785. [Google Scholar]
- Du, X.-L.; Jiang, T.; Wen, Z.-Q.; Gao, R.; Cui, M.; Wang, F. Silencing of heat shock protein 70 expression enhances radiotherapy efficacy and inhibits cell invasion in endometrial cancer cell line. Croat. Med. J. 2009, 50, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Budagova, K.R.; Sherman, M.Y. Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 2005, 24, 3328–3338. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Sherman, M.Y.; Yaglom, J.A. HSP72 depletion suppresses gammaH2AX activation by genotoxic stresses via p53/p21 signaling. Oncogene 2010, 29, 1952–1962. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.-O.; Hong, S.-E.; Kim, J.-Y.; Kim, M.-R.; Chang, Y.H.; Hong, Y.J.; Lee, J.K.; Park, I.-C. Induction of HSP27 and HSP70 by constitutive overexpression of Redd1 confers resistance of lung cancer cells to ionizing radiation. Oncol. Rep. 2019, 41, 3119–3126. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, B.; Xiao, H.; Dong, J.; Li, Y.; Zhu, C.; Jin, Y.; Li, H.; Cui, M.; Fan, S. LncRNA HOTAIR enhances breast cancer radioresistance through facilitating HSPA1A expression via sequestering miR-449b-5p. Thorac. Cancer 2020, 11, 1801–1816. [Google Scholar] [CrossRef]
- Seo, J.H.; Park, J.-H.; Lee, E.J.; Vo, T.T.L.; Choi, H.; Kim, J.Y.; Jang, J.K.; Wee, H.-J.; Lee, H.S.; Jang, S.H.; et al. ARD1-mediated Hsp70 acetylation balances stress-induced protein refolding and degradation. Nat. Commun. 2016, 7, 12882. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, I.; Skvortsov, S.; Popper, B.-A.; Haidenberger, A.; Saurer, M.; Gunkel, A.R.; Zwierzina, H.; Lukas, P. Rituximab enhances radiation-triggered apoptosis in non-Hodgkin’s lymphoma cells via caspase-dependent and—Independent mechanisms. J. Radiat. Res. 2006, 47, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Schilling, D.; Gehrmann, M.; Steinem, C.; De Maio, A.; Pockley, A.G.; Abend, M.; Molls, M.; Multhoff, G. Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J. 2009, 23, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Kühnel, A.; Schmid, T.E.; Ilicic, K.; Stangl, S.; Braun, I.S.; Gehrmann, M.; Molls, M.; Itami, J.; Multhoff, G. Role of membrane Hsp70 in radiation sensitivity of tumor cells. Radiat. Oncol. 2015, 10, 149. [Google Scholar] [CrossRef] [PubMed]
- Schilling, D.; Düwel, M.; Molls, M.; Multhoff, G. Radiosensitization of wildtype p53 cancer cells by the MDM2-inhibitor PXN727 is associated with altered heat shock protein 70 (Hsp70) levels. Cell Stress Chaperon. 2013, 18, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Gaca, S.; Reichert, S.; Multhoff, G.; Wacker, M.; Hehlgans, S.; Botzler, C.; Gehrmann, M.; Rödel, C.; Kreuter, J.; Rödel, F. Targeting by cmHsp70.1-antibody coated and survivin miRNA plasmid loaded nanoparticles to radiosensitize glioblastoma cells. J. Control. Release 2013, 172, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.A.; Nikolaev, B.P.; Ryzhov, V.A.; Yakovleva, L.Y.; Marchenko, Y.Y.; Parr, M.A.; Rolich, V.I.; Mikhrina, A.L.; Dobrodumov, A.V.; Pitkin, E.; et al. Ionizing radiation improves glioma-specific targeting of superparamagnetic iron oxide nanoparticles conjugated with cmHsp70.1 monoclonal antibodies (SPION-cmHsp70.1). Nanoscale 2015, 7, 20652–20664. [Google Scholar] [CrossRef]
- Arrigo, A.P. Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones 2017, 22, 517–529. [Google Scholar] [CrossRef]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting heat shock protein 27 in cancer: A druggable target for cancer treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef]
- Cheng, J.; Lv, Z.; Weng, X.; Ye, S.; Shen, K.; Li, M.; Qin, Y.; Hu, C.; Zhang, C.; Wu, J.; et al. Hsp27 Acts as a Master Molecular Chaperone and Plays an Essential Role in Hepatocellular Carcinoma Progression. Digestion 2015, 92, 192–202. [Google Scholar] [CrossRef]
- McCollum, A.K.; Teneyck, C.J.; Sauer, B.M.; Toft, D.O.; Erlichman, C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006, 66, 10967–10975. [Google Scholar] [CrossRef] [PubMed]
- Marotta, D.; Karar, J.; Jenkins, W.T.; Kumanova, M.; Jenkins, K.W.; Tobias, J.W.; Baldwin, D.; Hatzigeorgiou, A.; Alexiou, P.; Evans, S.M.; et al. In vivo profiling of hypoxic gene expression in gliomas using the hypoxia marker EF5 and laser-capture microdissection. Cancer Res. 2011, 71, 779–789. [Google Scholar] [CrossRef]
- Lin, S.-P.; Lee, Y.-T.; Wang, J.-Y.; Miller, S.A.; Chiou, S.-H.; Hung, M.-C.; Hung, S.-C. Survival of cancer stem cells under hypoxia and serum depletion via decrease in PP2A activity and activation of p38-MAPKAPK2-Hsp27. PLoS ONE 2012, 7, e49605. [Google Scholar] [CrossRef] [PubMed]
- Musiani, D.; Konda, J.D.; Pavan, S.; Torchiaro, E.; Sassi, F.; Noghero, A.; Erriquez, J.; Perera, T.; Olivero, M.; Di Renzo, M.F. Heat-shock protein 27 (HSP27, HSPB1) is up-regulated by MET kinase inhibitors and confers resistance to MET-targeted therapy. FASEB J. 2014, 28, 4055–4067. [Google Scholar] [CrossRef]
- Teimourian, S.; Jalal, R.; Sohrabpour, M.; Goliaei, B. Down-regulation of Hsp27 radiosensitizes human prostate cancer cells. Int. J. Urol. 2006, 13, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Aloy, M.-T.; Hadchity, E.; Bionda, C.; Diaz-Latoud, C.; Claude, L.; Rousson, R.; Arrigo, A.-P.; Rodriguez-Lafrasse, C. Protective role of Hsp27 protein against gamma radiation-induced apoptosis and radiosensitization effects of Hsp27 gene silencing in different human tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 543–553. [Google Scholar] [CrossRef]
- Hadchity, E.; Aloy, M.-T.; Paulin, C.; Armandy, E.; Watkin, E.; Rousson, R.; Gleave, M.; Chapet, O.; Rodriguez-Lafrasse, C. Heat shock protein 27 as a new therapeutic target for radiation sensitization of head and neck squamous cell carcinoma. Mol. Ther. 2009, 17, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Qu, J.-Q.; Xiao, L.; Yi, H.; Zhang, P.-F.; Li, M.-Y.; Hu, R.; Wan, X.-X.; He, Q.-Y.; Li, J.-H.; et al. Identification of heat shock protein 27 as a radioresistance-related protein in nasopharyngeal carcinoma cells. J. Cancer Res. Clin. Oncol. 2012, 138, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lin, X.; Zheng, Y.; Zhou, H. Silencing of heat shock protein 27 increases the radiosensitivity of non-small cell lung carcinoma cells. Mol. Med. Rep. 2019, 20, 613–621. [Google Scholar] [CrossRef]
- Ernst, B.P.; Wiesmann, N.; Gieringer, R.; Eckrich, J.; Brieger, J. HSP27 regulates viability and migration of cancer cell lines following irradiation. J. Proteomics 2020, 226, 103886. [Google Scholar] [CrossRef]
- Guttmann, D.M.; Hart, L.; Du, K.; Seletsky, A.; Koumenis, C. Inhibition of Hsp27 radiosensitizes head-and-neck cancer by modulating deoxyribonucleic acid repair. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 168–175. [Google Scholar] [CrossRef]
- Chen, W.; Ren, X.; Wu, J.; Gao, X.; Cen, X.; Wang, S.; Sheng, S.; Chen, Q.; Tang, Y.-J.; Liang, X.-H.; et al. HSP27 associates with epithelial-mesenchymal transition, stemness and radioresistance of salivary adenoid cystic carcinoma. J. Cell Mol. Med. 2018, 22, 2283–2298. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, Y.; Biswas, A.; Banik, P.; Pal, I.; Das, S.; Borkar, S.A.; Sardana, H.; Saha, A.; Das, S.K.; Emdad, L.; et al. Transcriptional regulation of HSPB1 by Friend leukemia integration-1 factor modulates radiation and temozolomide resistance in glioblastoma. Oncotarget 2020, 11, 1097–1108. [Google Scholar] [CrossRef][Green Version]
- Kim, E.-H.; Lee, H.-J.; Lee, D.-H.; Bae, S.; Soh, J.-W.; Jeoung, D.; Kim, J.; Cho, C.-K.; Lee, Y.-J.; Lee, Y.-S. Inhibition of heat shock protein 27-mediated resistance to DNA damaging agents by a novel PKC delta-V5 heptapeptide. Cancer Res. 2007, 67, 6333–6341. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Kim, E.-H.; Seo, W.D.; Choi, T.H.; Cheon, G.-J.; Lee, Y.-J.; Lee, Y.-S. Heat shock protein 27-targeted heptapeptide of the PKCΔ catalytic V5 region sensitizes tumors with radio- and chemoresistance. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-H.; Lee, Y.-J.; Seo, W.D.; Lee, H.-J.; Nam, J.-W.; Lee, Y.-J.; Kim, J.; Seo, E.-K.; Lee, Y.-S. Altered cross-linking of HSP27 by zerumbone as a novel strategy for overcoming HSP27-mediated radioresistance. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Mikami, H.; Saito, Y.; Okamoto, N.; Kakihana, A.; Kuga, T.; Nakayama, Y. Requirement of Hsp105 in CoCl 2-induced HIF-1α accumulation and transcriptional activation. Exp. Cell Res. 2017, 352, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Datta, N.R.; Kok, H.P.; Crezee, H.; Gaipl, U.S.; Bodis, S. Integrating Loco-Regional Hyperthermia Into the Current Oncology Practice: SWOT and TOWS Analyses. Front. Oncol. 2020, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Vujaskovic, Z.; Poulson, J.M.; Gaskin, A.A.; Thrall, D.E.; Page, R.L.; Charles, H.C.; MacFall, J.R.; Brizel, D.M.; Meyer, R.E.; Prescott, D.M. Temperature-dependent changes in physiologic parameters of spontaneous canine soft tissue sarcomas after combined radiotherapy and hyperthermia treatment. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 179–185. [Google Scholar] [CrossRef]
- Jones, E.L.; Prosnitz, L.R.; Dewhirst, M.W.; Marcom, P.K.; Hardenbergh, P.H.; Marks, L.B.; Brizel, D.M.; Vujaskovic, Z. Thermochemoradiotherapy improves oxygenation in locally advanced breast cancer. Clin. Cancer Res. 2004, 10, 4287–4293. [Google Scholar] [CrossRef]
- Song, C.W.; Shakil, A.; Osborn, J.L.; Iwata, K. Tumour oxygenation is increased by hyperthermia at mild temperatures. Int. J. Hypertherm. 2009, 25, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H. Cell biological effects of hyperthermia alone or combined with radiation or drugs: A short introduction to newcomers in the field. Int. J. Hypertherm. 2006, 22, 191–196. [Google Scholar] [CrossRef]
- Pandita, T.K.; Pandita, S.; Bhaumik, S.R. Molecular parameters of hyperthermia for radiosensitization. Crit. Rev. Eukaryot. Gene Expr. 2009, 19, 235–251. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, K.; Braakman, I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016, 60, 227–235. [Google Scholar]
- Chipurupalli, S.; Kannan, E.; Tergaonkar, V.; D’Andrea, R.; Robinson, N. Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer. Int. J. Mol. Sci. 2019, 20, 749. [Google Scholar] [CrossRef] [PubMed]
- Isohashi, F.; Endo, H.; Mukai, M.; Inoue, T.; Inoue, M. Insulin-like growth factor stimulation increases radiosensitivity of a pancreatic cancer cell line through endoplasmic reticulum stress under hypoxic conditions. Cancer Sci. 2008, 99, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro Oncol. 2013, 15, 1186–1199. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; van der Kogel, A.J.; Sweep, F.C.G.J.; Span, P.N. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother. Oncol. 2013, 108, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; Nishihara, K.; Machleidt, T.; Hermanson, S.; Wang, J.; Sakamuru, S.; Huang, R.; Xia, M. Identification of known drugs targeting the endoplasmic reticulum stress response. Anal. Bioanal. Chem. 2015, 407, 5343–5351. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-I.; Chiou, S.-H.; Hueng, D.-Y.; Tai, L.-K.; Huang, P.-I.; Kao, C.-L.; Chen, Y.-W.; Sytwu, H.-K. Celecoxib and radioresistant glioblastoma-derived CD133+ cells: Improvement in radiotherapeutic effects. Laboratory investigation. J. Neurosurg. 2011, 114, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yue, J.-B.; Liu, J.; Sun, X.-D.; Hu, X.-D.; Sun, J.-J.; Li, Y.-H.; Yu, J.-M. Effect of celecoxib on inhibiting tumor repopulation during radiotherapy in human FaDu squamous cell carcinoma. Contemp. Oncol. 2014, 18, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; He, D.; Song, E.; Jiang, M.; Song, Y. Celecoxib enhances the sensitivity of non-small-cell lung cancer cells to radiation-induced apoptosis through downregulation of the Akt/mTOR signaling pathway and COX-2 expression. PLoS ONE 2019, 14, e0223760. [Google Scholar] [CrossRef]
- Prabhu, A.; Sarcar, B.; Kahali, S.; Shan, Y.; Chinnaiyan, P. Targeting the unfolded protein response in glioblastoma cells with the fusion protein EGF-SubA. PLoS ONE 2012, 7, e52265. [Google Scholar] [CrossRef]
- Bachelder, R.E. Chapter 6: Cell surface GRP78: A targetable marker of cancer stem-like cells. In Cell Surface GRP78, A New Paradigm in Signal Transduction Biology; Pizzo, S.V., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 99–109. [Google Scholar]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef]
- Ondrej, M.; Cechakova, L.; Durisova, K.; Pejchal, J.; Tichy, A. To live or let die: Unclear task of autophagy in the radiosensitization battle. Radiother. Oncol. 2016, 119, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Jiang, F.; Yang, C.; Yan, Q.; Guo, W.; Huang, Q.; Zhang, L.; Jiang, G. Role of autophagy in regulating the radiosensitivity of tumor cells. J. Cancer Res. Clin. Oncol. 2017, 143, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef]
- He, W.-S.; Dai, X.-F.; Jin, M.; Liu, C.-W.; Rent, J.-H. Hypoxia-induced autophagy confers resistance of breast cancer cells to ionizing radiation. Oncol. Res. 2012, 20, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Yao, Q.; Xie, G.; Du, S.; Ren, C.; Wang, Y.; Yuan, Y. TAT-ODD-p53 enhances the radiosensitivity of hypoxic breast cancer cells by inhibiting Parkin-mediated mitophagy. Oncotarget 2015, 6, 17417–17429. [Google Scholar] [CrossRef] [PubMed]
- Chaachouay, H.; Fehrenbacher, B.; Toulany, M.; Schaller, M.; Multhoff, G.; Rodemann, H.P. AMPK-independent autophagy promotes radioresistance of human tumor cells under clinical relevant hypoxia in vitro. Radiother. Oncol. 2015, 116, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, P.; Guo, F.; Wang, X.; Wang, J.; Xu, J.; Yuan, D.; Zhang, J.; Shao, C. Autophagy enhanced the radioresistance of non-small cell lung cancer by regulating ROS level under hypoxia condition. Int. J. Radiat. Biol. 2017, 93, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Liu, M.; Ding, C.; Wang, X.; Wang, R.; Wu, X.; Fan, R. Hypoxia-responsive miR-124 and miR-144 reduce hypoxia-induced autophagy and enhance radiosensitivity of prostate cancer cells via suppressing PIM1. Cancer Med. 2016, 5, 1174–1182. [Google Scholar] [CrossRef]
- Xu, C.-G.; Yang, M.-F.; Fan, J.-X.; Wang, W. MiR-30a and miR-205 are downregulated in hypoxia and modulate radiosensitivity of prostate cancer cells by inhibiting autophagy via TP53INP1. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1501–1508. [Google Scholar]
- Wang, W.; Liu, M.; Guan, Y.; Wu, Q. Hypoxia-Responsive Mir-301a and Mir-301b Promote Radioresistance of Prostate Cancer Cells via Downregulating NDRG2. Med. Sci. Monit. 2016, 22, 2126–2132. [Google Scholar] [CrossRef]
- Lin, T.; Zhang, Q.; Yuan, A.; Wang, B.; Zhang, F.; Ding, Y.; Cao, W.; Chen, W.; Guo, H. Synergy of Tumor Microenvironment Remodeling and Autophagy Inhibition to Sensitize Radiation for Bladder Cancer Treatment. Theranostics 2020, 10, 7683–7696. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tan, H.; Zhang, X.; Chen, F.; Zhou, Z.; Hu, X.; Chang, S.; Liu, P.; Zhang, H. Enhancement of radiotherapy efficacy by silver nanoparticles in hypoxic glioma cells. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. S3), S922–S930. [Google Scholar] [CrossRef]
- Jutten, B.; Keulers, T.G.; Peeters, H.J.M.; Schaaf, M.B.E.; Savelkouls, K.G.M.; Compter, I.; Clarijs, R.; Schijns, O.E.M.G.; Ackermans, L.; Teernstra, O.P.M.; et al. EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy 2018, 14, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Anbalagan, S.; Pires, I.M.; Blick, C.; Hill, M.A.; Ferguson, D.J.P.; Chan, D.A.; Hammond, E.M. Radiosensitization of renal cell carcinoma in vitro through the induction of autophagy. Radiother. Oncol. 2012, 103, 388–393. [Google Scholar] [CrossRef]
- Classen, F.; Kranz, P.; Riffkin, H.; Pompsch, M.; Wolf, A.; Göpelt, K.; Baumann, M.; Baumann, J.; Brockmeier, U.; Metzen, E. Autophagy induced by ionizing radiation promotes cell death over survival in human colorectal cancer cells. Exp. Cell Res. 2019, 374, 29–37. [Google Scholar] [CrossRef]
- Song, L.; Liu, S.; Zhang, L.; Yao, H.; Gao, F.; Xu, D.; Li, Q. MiR-21 modulates radiosensitivity of cervical cancer through inhibiting autophagy via the PTEN/Akt/HIF-1α feedback loop and the Akt-mTOR signaling pathway. Tumour Biol. 2016, 37, 12161–12168. [Google Scholar] [CrossRef]
- Liu, B.; Han, D.; Zhang, T.; Cheng, G.; Lu, Y.; Wang, J.; Zhao, H.; Zhao, Z. Hypoxia-induced autophagy promotes EGFR loss in specific cell contexts, which leads to cell death and enhanced radiosensitivity. Int. J. Biochem. Cell Biol. 2019, 111, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Compter, I.; Eekers, D.B.P.; Hoeben, A.; Rouschop, K.M.A.; Reymen, B.; Ackermans, L.; Beckervordersantforth, J.; Bauer, N.J.C.; Anten, M.M.; Wesseling, P.; et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: A phase IB trial. Autophagy 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer stem cells: The architects of the tumor ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef]
- Visan, K.S.; Lobb, R.J.; Moller, A. The role of exosomes in the promotion of epithelial-to-mesenchymal transition and metastasis. Front. Biosci. Landmark Ed. 2020, 25, 1022–1057. [Google Scholar]
- Van den Beucken, T.; Koch, E.; Chu, K.; Rupaimoole, R.; Prickaerts, P.; Adriaens, M.; Voncken, J.W.; Harris, A.L.; Buffa, F.M.; Haider, S.; et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 2014, 5, 5203. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, Y.; Deng, M.; Ye, R.; Zhao, S.; Wang, Y.; Li, J.; Zhao, Z. MicroRNA control of epithelial-mesenchymal transition in cancer stem cells. Int. J. Cancer 2014, 135, 1019–1027. [Google Scholar] [CrossRef]
- Mineo, M.; Ricklefs, F.; Rooj, A.K.; Lyons, S.M.; Ivanov, P.; Ansari, K.I.; Nakano, I.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. The Long Non-coding RNA HIF1A-AS2 Facilitates the Maintenance of Mesenchymal Glioblastoma Stem-like Cells in Hypoxic Niches. Cell Rep. 2016, 15, 2500–2509. [Google Scholar] [CrossRef]
- Jiao, X.; Qian, X.; Wu, L.; Li, B.; Wang, Y.; Kong, X.; Xiong, L. microRNA: The Impact on Cancer Stemness and Therapeutic Resistance. Cells 2019, 9, 8. [Google Scholar] [CrossRef]
- Theys, J.; Jutten, B.; Habets, R.; Paesmans, K.; Groot, A.J.; Lambin, P.; Wouters, B.G.; Lammering, G.; Vooijs, M. E-Cadherin loss associated with EMT promotes radioresistance in human tumor cells. Radiother. Oncol. 2011, 99, 392–397. [Google Scholar] [CrossRef]
- Marie-Egyptienne, D.T.; Lohse, I.; Peter Hill, R.P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013, 341, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Nie, X.; Zou, Y.; Gong, C.; Li, Y.; Wu, H.; Qiu, H.; Yang, L.; Zhuang, L.; Zhang, P.; et al. Twist1 Enhances Hypoxia Induced Radioresistance in Cervical Cancer Cells by Promoting Nuclear EGFR Localization. J. Cancer 2017, 8, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-G.; Lee, J.-H.; Kim, S.-Y.; Hwang, H.-M.; Kim, T.-R.; Cho, E.-W. Hypoxia-inducible transgelin 2 selects epithelial-to-mesenchymal transition and γ-radiation-resistant subtypes by focal adhesion kinase-associated insulin-like growth factor 1 receptor activation in non-small-cell lung cancer cells. Cancer Sci. 2018, 109, 3519–3531. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Weng, X.; Yao, Y.; Huang, C.; Li, J.; Peng, Y.; Lin, R.; Lin, Z. ALDH-1-positive cells exhibited a radioresistant phenotype that was enhanced with hypoxia in cervical cancer. BMC Cancer 2020, 20, 891. [Google Scholar] [CrossRef]
- Hannen, R.; Bartsch, J.W. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 2018, 592, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.H.; El-Zein, R.; Dave, B. Autophagy Inhibition to Increase Radiosensitization in Breast Cancer. J. Nucl. Med. Radiat. Ther. 2015, 6, 254. [Google Scholar] [CrossRef]
- Ke, Y.; Wu, C.; Zeng, Y.; Chen, M.; Li, Y.; Xie, C.; Zhou, Y.; Zhong, Y.; Yu, H. Radiosensitization of Clioquinol Combined with Zinc in the Nasopharyngeal Cancer Stem-like Cells by Inhibiting Autophagy in Vitro and in Vivo. Int. J. Biol. Sci. 2020, 16, 777–789. [Google Scholar] [CrossRef]
- Lagadec, C.; Dekmezian, C.; Bauché, L.; Pajonk, F. Oxygen levels do not determine radiation survival of breast cancer stem cells. PLoS ONE 2012, 7, e34545. [Google Scholar] [CrossRef]
- Barbato, L.; Bocchetti, M.; Biase, A.D.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Matchuk, O.N.; Zamulaeva, I.A.; Selivanova, E.I.; Lipunov, N.M.; Proniushkina, K.A.; Ul’ianenko, S.E.; Lychagin, A.A.; Smirnova, S.G.; Orlova, N.V.; Saenko, A.S. Sensitivity of melanoma B16 side population to low- and high-LET radiation. Radiat. Biol. Radioecol. 2012, 52, 261–267. [Google Scholar]
- Zhang, X.; Lin, S.H.; Fang, B.; Gillin, M.; Mohan, R.; Joe, Y.; Chang, J.Y. Therapy-resistant cancer stem cells have differing sensitivity to photon versus proton beam radiation. J. Thorac. Oncol. 2013, 8, 1484–1491. [Google Scholar] [CrossRef]
- Narang, H.; Kumar, A.; Bhat, N.; Pandey, B.N.; Ghosh, A. Effect of proton and gamma irradiation on human lung carcinoma cells: Gene expression, cell cycle, cell death, epithelial-mesenchymal transition and cancer-stem cell trait as biological end points. Mutat. Res. 2015, 780, 35–46. [Google Scholar] [CrossRef]
- Dini, V.; Belli, M.; Tabocchini, M.A. Targeting cancer stem cells: Protons versus photons. Br. J. Radiol. 2020, 93, 20190225. [Google Scholar] [CrossRef] [PubMed]
- Ceder, J.; Elgqvist, J. Targeting Prostate Cancer Stem Cells with Alpha-Particle Therapy. Front. Oncol. 2017, 6, 273. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Hikida, M.; Nakada, M.; Sakurai, Y.; Hirata, E.; Takeno, S.; Suzuki, M. Glioma Stem-Like Cells Can Be Targeted in Boron Neutron Capture Therapy with Boronophenylalanine. Cancers 2020, 12, 3040. [Google Scholar] [CrossRef] [PubMed]
- Matchuk, O.N.; Orlova, N.V.; Zamulaeva, I.A. Changes in the Relative Number of SP Cells of Melanoma Line B16 after Radiation Exposure in vivo. Radiat. Biol. Radioecol. 2016, 56, 487–493. [Google Scholar]
- Gameiro, S.R.; Malamas, A.S.; Bernstein, M.B.; Tsang, K.Y.; Vassantachart, A.; Sahoo, N.; Tailor, R.; Pidikiti, R.; Guha, C.P.; Hahn, S.M.; et al. Tumor cells surviving exposure to proton or photon radiation share a common immunogenic modulation signature, rendering them more sensitive to T cell-mediated killing. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 120–130. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef]
- Baek, S.-J.; Ishii, H.; Tamari, K.; Hayashi, K.; Nishida, N.; Konno, M.; Kawamoto, K.; Koseki, J.; Fukusumi, T.; Hasegawa, S.; et al. Cancer stem cells: The potential of carbon ion beam radiation and new radiosensitizers (Review). Oncol. Rep. 2015, 34, 2233–2237. [Google Scholar] [CrossRef]
- Moncharmont, C.; Guy, J.-B.; Wozny, A.-S.; Gilormini, M.; Battiston-Montagne, P.; Ardail, D.; Beuve, M.; Alphonse, G.; Simoëns, X.; Rancoule, C.; et al. Carbon ion irradiation withstands cancer stem cells’ migration/invasion process in Head and Neck Squamous Cell Carcinoma (HNSCC). Oncotarget 2016, 7, 47738–47749. [Google Scholar] [CrossRef] [PubMed]
- Wozny, A.-S.; Vares, G.; Alphonse, G.; Lauret, A.; Monini, C.; Magné, N.; Cuerq, C.; Fujimori, A.; Monboisse, J.-C.; Beuve, M.; et al. ROS Production and Distribution: A New Paradigm to Explain the Differential Effects of X-ray and Carbon Ion Irradiation on Cancer Stem Cell Migration and Invasion. Cancers 2019, 11, 468. [Google Scholar] [CrossRef]
- Chiblak, S.; Tang, Z.; Lemke, D.; Knoll, M.; Dokic, I.; Warta, R.; Moustafa, M.; Mier, W.; Brons, S.; Rapp, C.; et al. Carbon irradiation overcomes glioma radioresistance by eradicating stem cells and forming an antiangiogenic and immunopermissive niche. JCI Insight 2019, 4, e123837. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, G.; Maalouf, M.; Boivin, A.; Battiston-Montagne, P.; Beuve, M.; Levy, A.; Jalade, P.; Fournier, C.; Ardail, D.; Magné, N.; et al. Targeting head and neck cancer stem cells to overcome resistance to photon and carbon ion radiation. Stem Cell Rev. Rep. 2014, 10, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; Vriend, L.E.M.; Krawczyk, P.M.; Horsman, M.R.; Franken, N.A.P.; Crezee, J. Targeting therapy-resistant cancer stem cells by hyperthermia. Int. J. Hypertherm. 2017, 33, 419–427. [Google Scholar] [CrossRef]
- Ammendola, M.; Currò, G.; Memeo, R.; Curto, L.S.; Luposella, M.; Zuccalà, V.; Pessaux, P.; Navarra, G.; Gadaleta, C.D.; Ranieri, G. Targeting Stem Cells with Hyperthermia: Translational Relevance in Cancer Patients. Oncology 2020, 98, 755–762. [Google Scholar] [CrossRef]
- Croker, A.K.; Allan, A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res. Treat. 2012, 133, 75–87. [Google Scholar] [CrossRef]
- Marcu, L.G.; Marcu, D. In silico modelling of a cancer stem cell-targeting agent and its effects on tumour control during radiotherapy. Sci. Rep. 2016, 6, 32332. [Google Scholar] [CrossRef]
- Naujokat, C.; Steinhart, R. Salinomycin as a drug for targeting human cancer stem cells. J. Biomed. Biotechnol. 2012, 2012, 950658. [Google Scholar] [CrossRef]
- Liu, L.; Wang, O.; Mao, J.; Qin, T.; Sun, Y.; Yang, J.; Han, Y.; Li, L.; Li, Q. Salinomycin suppresses cancer cell stemness and attenuates TGF-β-induced epithelial-mesenchymal transition of renal cell carcinoma cells. Chem. Biol. Interact. 2018, 296, 145–153. [Google Scholar] [CrossRef]
- Shen, Y.-A.; Lin, C.-H.; Chi, W.-H.; Wang, C.-Y.; Hsieh, Y.-T.; Wei, Y.-H.; Chen, Y.-J. Resveratrol Impedes the Stemness, Epithelial-Mesenchymal Transition, and Metabolic Reprogramming of Cancer Stem Cells in Nasopharyngeal Carcinoma through p53 Activation. Evid. Based Complement. Alternat. Med. 2013, 2013, 590393. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Sato, C.; Shiozawa, N.; Sato, A.; Sato, H.; Virgona, N.; Yano, T. Suppressive Effect of Delta-Tocotrienol on Hypoxia Adaptation of Prostate Cancer Stem-like Cells. Anticancer Res. 2018, 38, 1391–1399. [Google Scholar] [PubMed]
- Koh, S.Y.; Moon, J.Y.; Unno, T.; Cho, S.K. Baicalein Suppresses Stem Cell-Like Characteristics in Radio- and Chemoresistant MDA-MB-231 Human Breast Cancer Cells through Up-Regulation of IFIT2. Nutrients 2019, 11, 624. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wei, J.; Guo, T.; Shen, Y.; Liu, F. Knockdown of miR-210 decreases hypoxic glioma stem cells stemness and radioresistance. Exp. Cell Res. 2014, 326, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bai, J.; Yin, H.; Long, L.; Zheng, Z.; Wang, Q.; Chen, F.; Yu, X.; Zhou, Y. Exosomal miR-1255b-5p targets human telomerase reverse transcriptase in colorectal cancer cells to suppress epithelial-to-mesenchymal transition. Mol. Oncol. 2020, 14, 2589–2608. [Google Scholar] [CrossRef]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Calin, G.A.; Lopez-Berestein, G.; Sood, A.K. miRNA Deregulation in Cancer Cells and the Tumor Microenvironment. Cancer Discov. 2016, 6, 235–246. [Google Scholar] [CrossRef]
- Ullmann, P.; Nurmik, M.; Begaj, R.; Haan, S.; Letellier, E. Hypoxia- and MicroRNA-Induced Metabolic Reprogramming of Tumor-Initiating Cells. Cells 2019, 8, 528. [Google Scholar] [CrossRef]
- Manem, V.S.; Dhawan, A. RadiationGeneSigDB: A database of oxic and hypoxic radiation response gene signatures and their utility in pre-clinical research. Br. J. Radiol. 2019, 92, 20190198. [Google Scholar] [CrossRef] [PubMed]
- Macedo-Silva, C.; Miranda-Gonçalves, V.; Henrique, R.; Jerónimo, C.; Bravo, I. The Critical Role of Hypoxic Microenvironment and Epigenetic Deregulation in Esophageal Cancer Radioresistance. Genes 2019, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- Rezaeian, A.-H.; Khanbabaei, H.; Calin, G.A. Therapeutic Potential of the miRNA-ATM Axis in the Management of Tumor Radioresistance. Cancer Res. 2020, 80, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Babar, I.A.; Czochor, J.; Steinmetz, A.; Weidhaas, J.B.; Glazer, P.M.; Slack, F.J. Inhibition of hypoxia-induced miR-155 radiosensitizes hypoxic lung cancer cells. Cancer Biol. Ther. 2011, 12, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Sun, T.; Cao, J.; Liu, F.; Tian, Y.; Zhu, W. Downregulation of miR-210 expression inhibits proliferation, induces apoptosis and enhances radiosensitivity in hypoxic human hepatoma cells in vitro. Exp. Cell Res. 2012, 318, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lan, F.; Xia, T. Hypoxic Glioma Cell-Secreted Exosomal miR-301a Activates Wnt/β-catenin Signaling and Promotes Radiation Resistance by Targeting TCEAL7. Mol. Ther. 2019, 27, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gao, C.-E.; Chen, W.-L.; Tang, Y.-Y.; Nie, J.-Y.; Shen, L.-D.; Ma, X.; Chen, D.-D. Opposite response to hypoxia by breast cancer cells between cell proliferation and cell migration: A clue from microRNA expression profile. Oncol. Lett. 2018, 15, 2771–2780. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.-H.; Wu, C.-Y.; Kuo, P.-L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef]
- Yu, Y.; Min, Z.; Zhihang, Z.; Linhong, M.; Tao, R.; Yan, L.; Song, H. Hypoxia-induced exosomes promote hepatocellular carcinoma proliferation and metastasis via miR-1273f transfer. Exp. Cell Res. 2019, 385, 111649. [Google Scholar] [CrossRef]
- Kuo, T.-C.; Kung, H.-J.; Shih, J.-W. Signaling in and out: Long-noncoding RNAs in tumor hypoxia. J. Biomed. Sci. 2020, 27, 59. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Ma, X.; Chen, B.; Lu, X.; Hu, Y.; Lin, Y.; Huang, R.; Qiu, Y. Upregulated AHIF-mediated radioresistance in glioblastoma. Biochem. Biophys. Res. Commun. 2019, 509, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Liao, K.; Zhuang, Z.; Chen, B.; Zhou, Z.; Zhou, S.; Lin, G.; Zhang, F.; Lin, Y.; Miao, Y.; et al. AHIF promotes glioblastoma progression and radioresistance via exosomes. Int. J. Oncol. 2019, 54, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Gao, R.; Xiu, Z.; Sun, T. Knockdown of cZNF292 suppressed hypoxic human hepatoma SMMC7721 cell proliferation, vasculogenic mimicry, and radioresistance. Cell Signal. 2019, 60, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, L.; Chen, T.; Shi, Y.; Yao, B.; Liu, Z.; Wang, Y.; Li, Q.; Liu, R.; Niu, Y.; et al. LncRNA RUNX1-IT1 which is downregulated by hypoxia-driven histone deacetylase 3 represses proliferation and cancer stem-like properties in hepatocellular carcinoma cells. Cell Death Dis. 2020, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Piao, H.-Y.; Guo, S.; Wang, Y.; Zhang, J. Exosome-transmitted lncRNA PCGEM1 promotes invasive and metastasis in gastric cancer by maintaining the stability of SNAI1. Clin. Transl. Oncol. 2020. [Google Scholar] [CrossRef]
- Joseph, N.A.; Chiou, S.-H.; Lung, Z.; Yang, C.-L.; Lin, T.-Y.; Chang, H.-W.; Sun, H.S.; Gupta, S.K.; Yen, L.; Wang, S.-D. The role of HGF-MET pathway and CCDC66 cirRNA expression in EGFR resistance and epithelial-to-mesenchymal transition of lung adenocarcinoma cells. J. Hematol. Oncol. 2018, 11, 74. [Google Scholar] [CrossRef]
- Tang, J.; Zhu, H.; Lin, J.; Wang, H. Knockdown of Circ_0081143 Mitigates Hypoxia-Induced Migration, Invasion, and EMT in Gastric Cancer Cells Through the miR-497-5p/EGFR Axis. Cancer Biother. Radiopharm. 2020. [Google Scholar] [CrossRef]
- Kim, J.-G.; Yi, J.M.; Park, S.-J.; Kim, J.-S.; Son, T.G.; Yang, K.; Yoo, M.-A.; Heo, K. Histone demethylase JMJD2B-mediated cell proliferation regulated by hypoxia and radiation in gastric cancer cell. Biochim. Biophys. Acta 2012, 1819, 1200–1207. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Heo, K.; Kim, J.S.; Im, J.W.; Lee, S.M.; Cho, M.; Kang, D.H.; Heo, J.; Lee, J.W.; Choi, C.W.; et al. Emodin attenuates radioresistance induced by hypoxia in HepG2 cells via the enhancement of PARP1 cleavage and inhibition of JMJD2B. Oncol. Rep. 2015, 33, 1691–1698. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Zhao, C.; Dong, H.; Xu, Q.; Zhang, Y. Histone deacetylase (HDAC) inhibitors in cancer: A patent review (2017-present). Expert Opin. Ther. Pat. 2020, 30, 263–274. [Google Scholar] [CrossRef]
- Saelen, M.G.; Ree, A.H.; Kristian, A.; Fleten, K.G.; Furre, T.; Hektoen, H.H.; Flatmark, K. Radiosensitization by the histone deacetylase inhibitor vorinostat under hypoxia and with capecitabine in experimental colorectal carcinoma. Radiat. Oncol. 2012, 7, 165. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.; Ragnum, H.B.; Julin, C.H.; Yeramian, A.; Clancy, T.; Frikstad, K.-A.M.; Seierstad, T.; Stokke, T.; Matias-Guiu, X.; Ree, A.H.; et al. Hypoxia-independent gene expression signature associated with radiosensitisation of prostate cancer cell lines by histone deacetylase inhibition. Br. J. Cancer 2016, 115, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Chow, J.-M.; Hsieh, Y.-Y.; Lin, C.-Y.; Hsu, K.-W.; Hsieh, W.-S.; Chi, W.-M.; Shabangu, B.M.; Lee, C.-H. HDAC1,2 Knock-Out and HDACi Induced Cell Apoptosis in Imatinib-Resistant K562 Cells. Int. J. Mol. Sci. 2019, 20, 2271. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, J.; Ye, S.; He, M.; Ren, R.; Yuan, D.; Shao, C. SirT1 regulates radiosensitivity of hepatoma cells differently under normoxic and hypoxic conditions. Cancer Sci. 2012, 103, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, J.; Xu, Y.; Shao, C. SirT1 confers hypoxia-induced radioresistance via the modulation of c-Myc stabilization on hepatoma cells. J. Radiat. Res. 2012, 53, 44–50. [Google Scholar] [CrossRef]
- Shao, C.; Yang, F.; Miao, S.; Liu, W.; Wang, C.; Shu, Y.; Shen, H. Role of hypoxia-induced exosomes in tumor biology. Mol. Cancer 2018, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Zonneveld, M.I.; Keulers, T.G.H.; Rouschop, K.M.A. Extracellular Vesicles as Transmitters of Hypoxia Tolerance in Solid Cancers. Cancers 2019, 11, 154. [Google Scholar] [CrossRef]
- Thomas, S.N.; Liao, Z.; Clark, D.; Chen, Y.; Samadani, R.; Mao, L.; Ann, D.K.; Baulch, J.E.; Shapiro, P.; Yang, A.J. Exosomal Proteome Profiling: A Potential Multi-Marker Cellular Phenotyping Tool to Characterize Hypoxia-Induced Radiation Resistance in Breast Cancer. Proteomes 2013, 1, 87–108. [Google Scholar] [CrossRef]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Yang, M.; Li, Y.; Yang, F.; Feng, Y. Exosomes Derived from Hypoxic Colorectal Cancer Cells Transfer Wnt4 to Normoxic Cells to Elicit a Prometastatic Phenotype. Int. J. Biol. Sci. 2018, 14, 2094–2102. [Google Scholar] [CrossRef]
- Mo, F.; Xu, Y.; Zhang, J.; Zhu, L.; Wang, C.; Chu, X.; Pan, Y.; Bai, Y.; Shao, C.; Zhang, J. Effects of Hypoxia and Radiation-Induced Exosomes on Migration of Lung Cancer Cells and Angiogenesis of Umbilical Vein Endothelial Cells. Radiat. Res. 2020, 194, 71–80. [Google Scholar] [CrossRef]
- Jung, K.O.; Jo, H.; Yu, J.H.; Gambhir, S.S.; Pratx, G. Development and MPI tracking of novel hypoxia-targeted theranostic exosomes. Biomaterials 2018, 177, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, K.E.; Dwyer, R.M. Engineering Exosomes for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1122. [Google Scholar] [CrossRef] [PubMed]
- Eckert, F.; Zwirner, K.; Boeke, S.; Thorwarth, D.; Zips, D.; Huber, S.M. Rationale for Combining Radiotherapy and Immune Checkpoint Inhibition for Patients with Hypoxic Tumors. Front. Immunol. 2019, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.-G.; Yuan, H.; Deng, W.; Li, J.; Huang, Y.; Kim, B.Y.S.; Story, M.D.; Jiang, W. The Reciprocity between Radiotherapy and Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 1709–1717. [Google Scholar] [CrossRef]






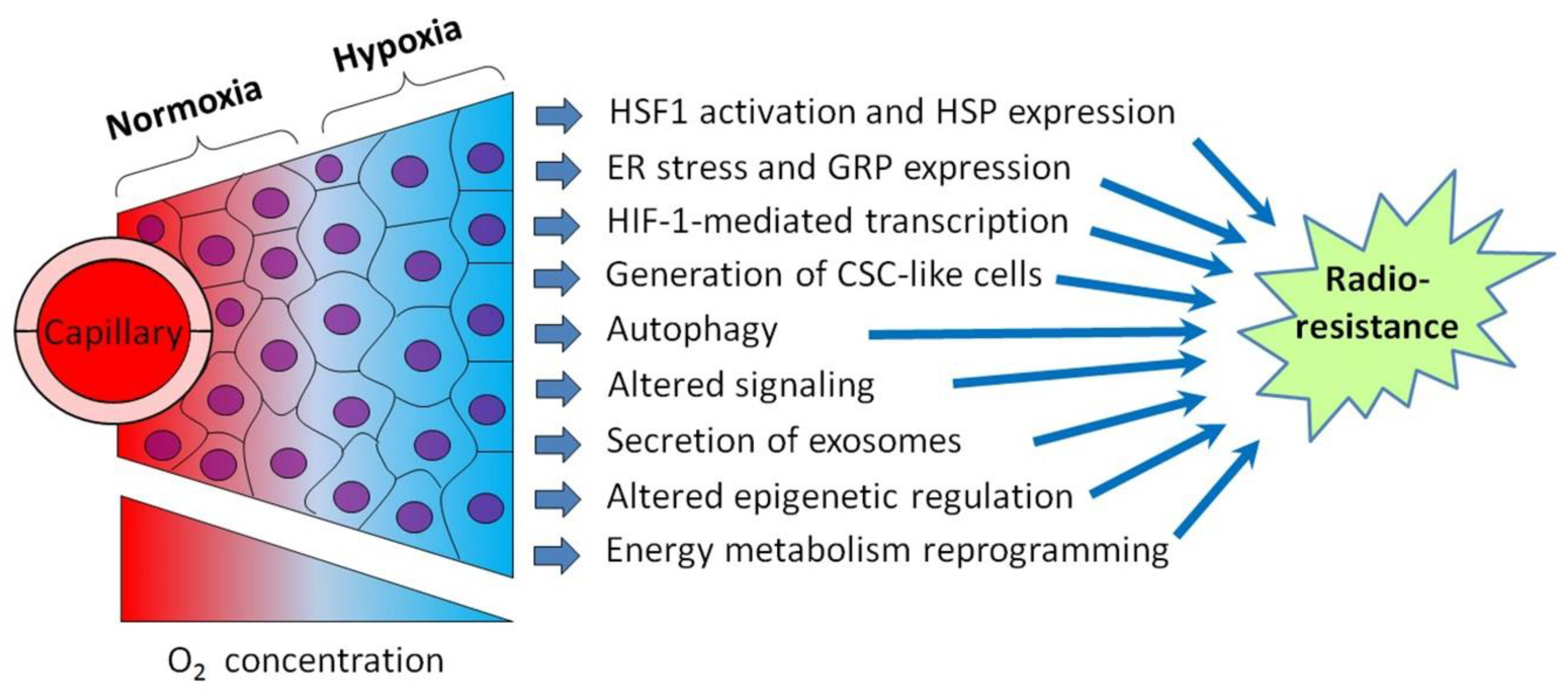{kind=link}
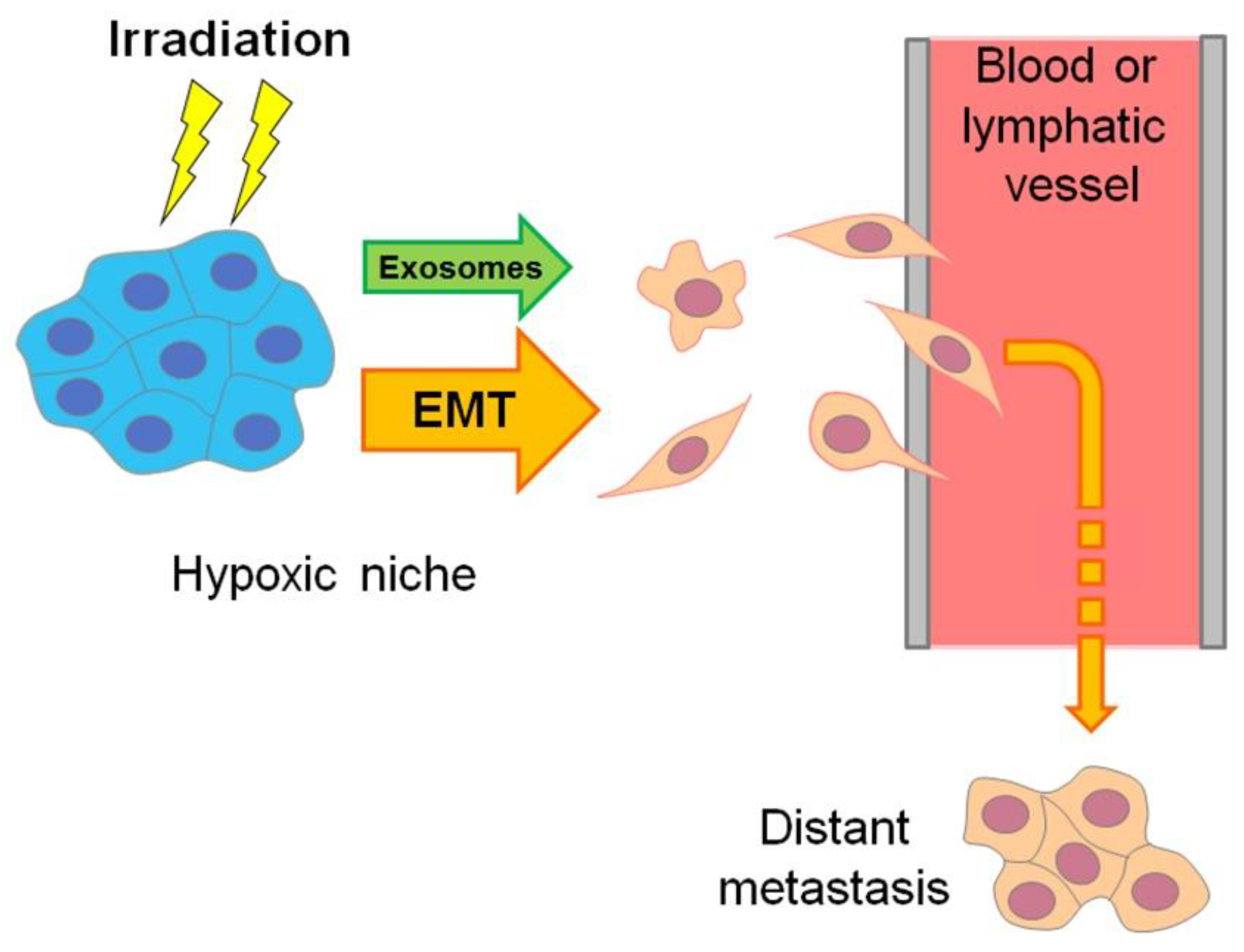{kind=link}
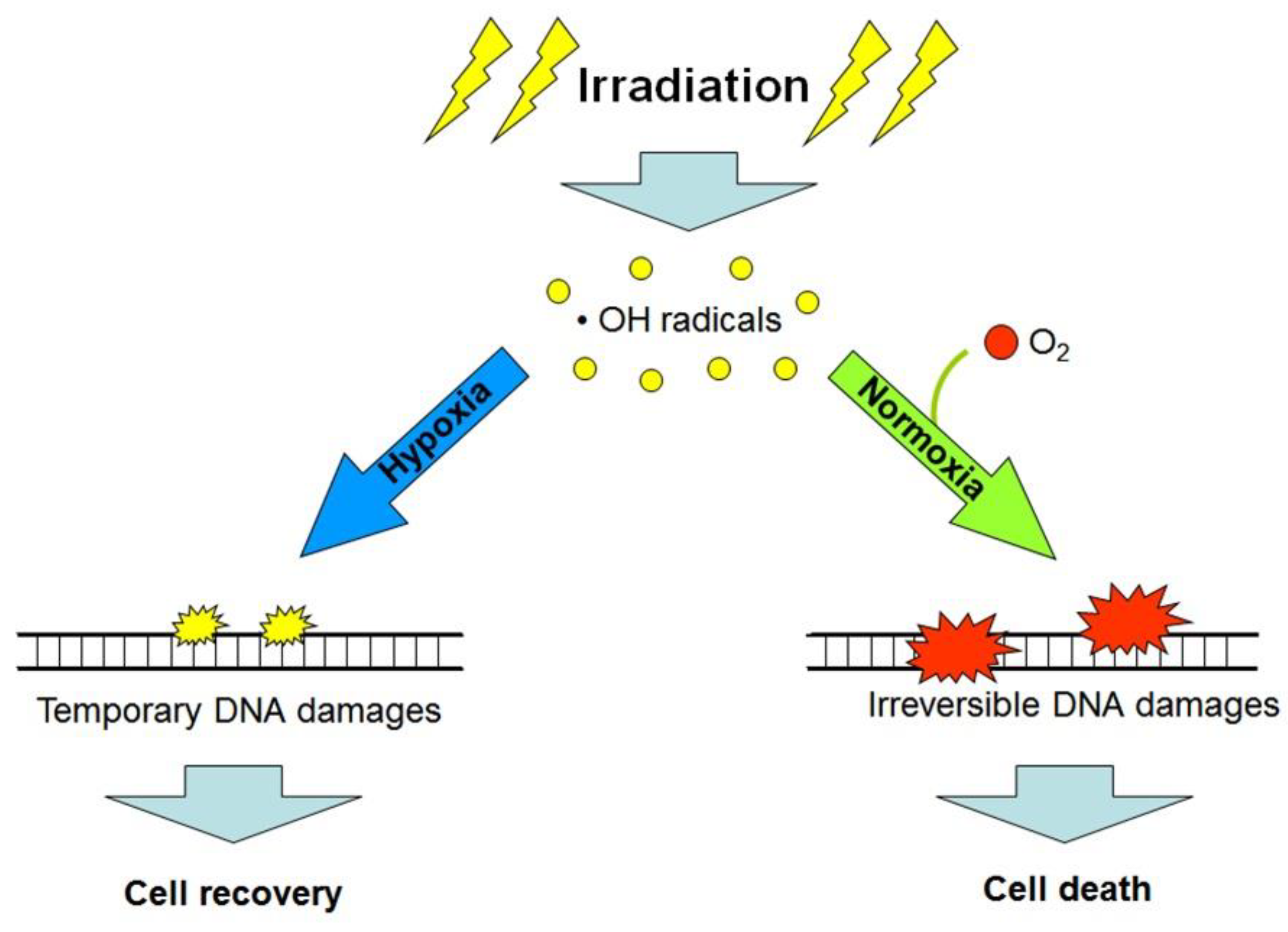{kind=link}
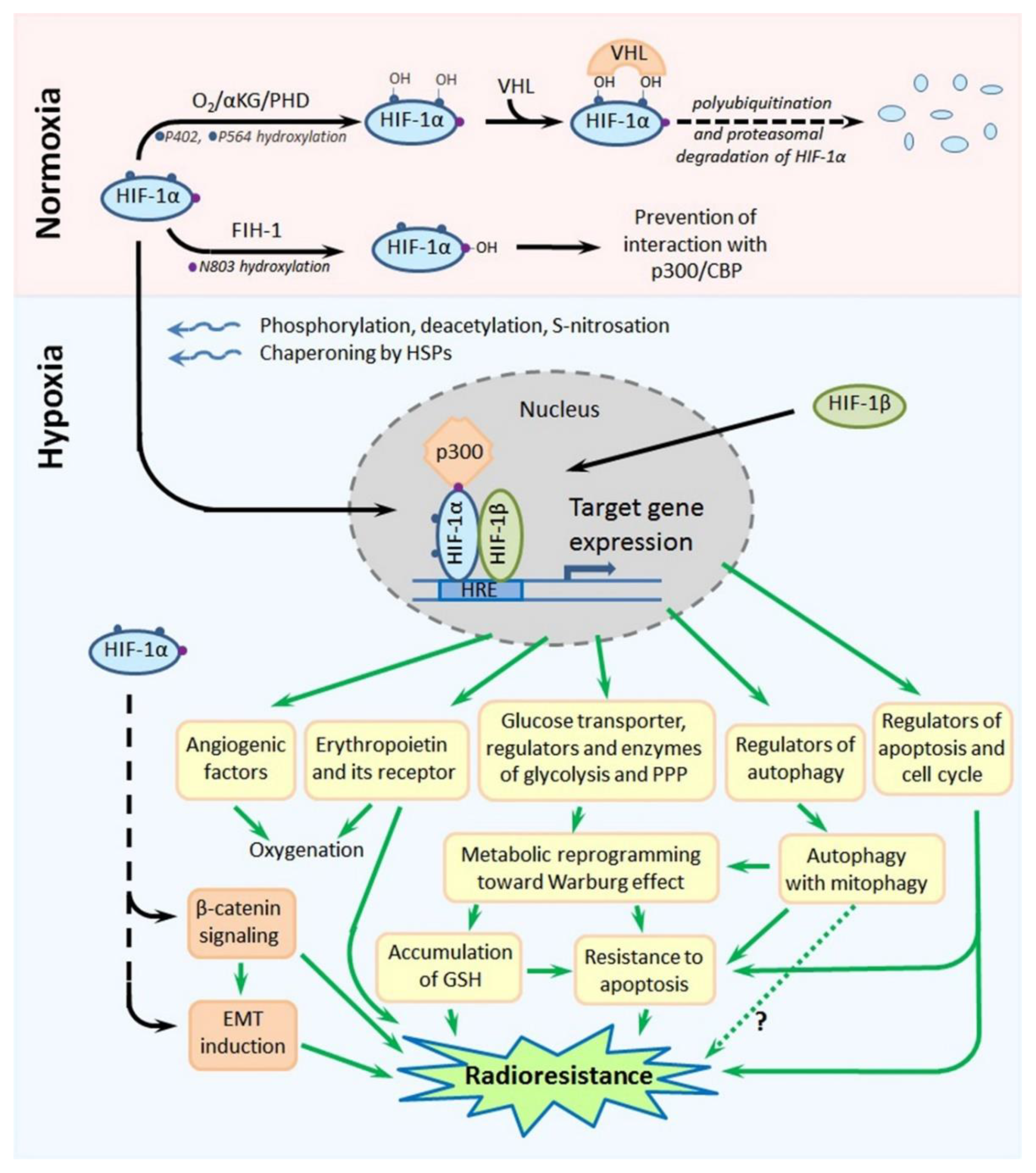{kind=link}
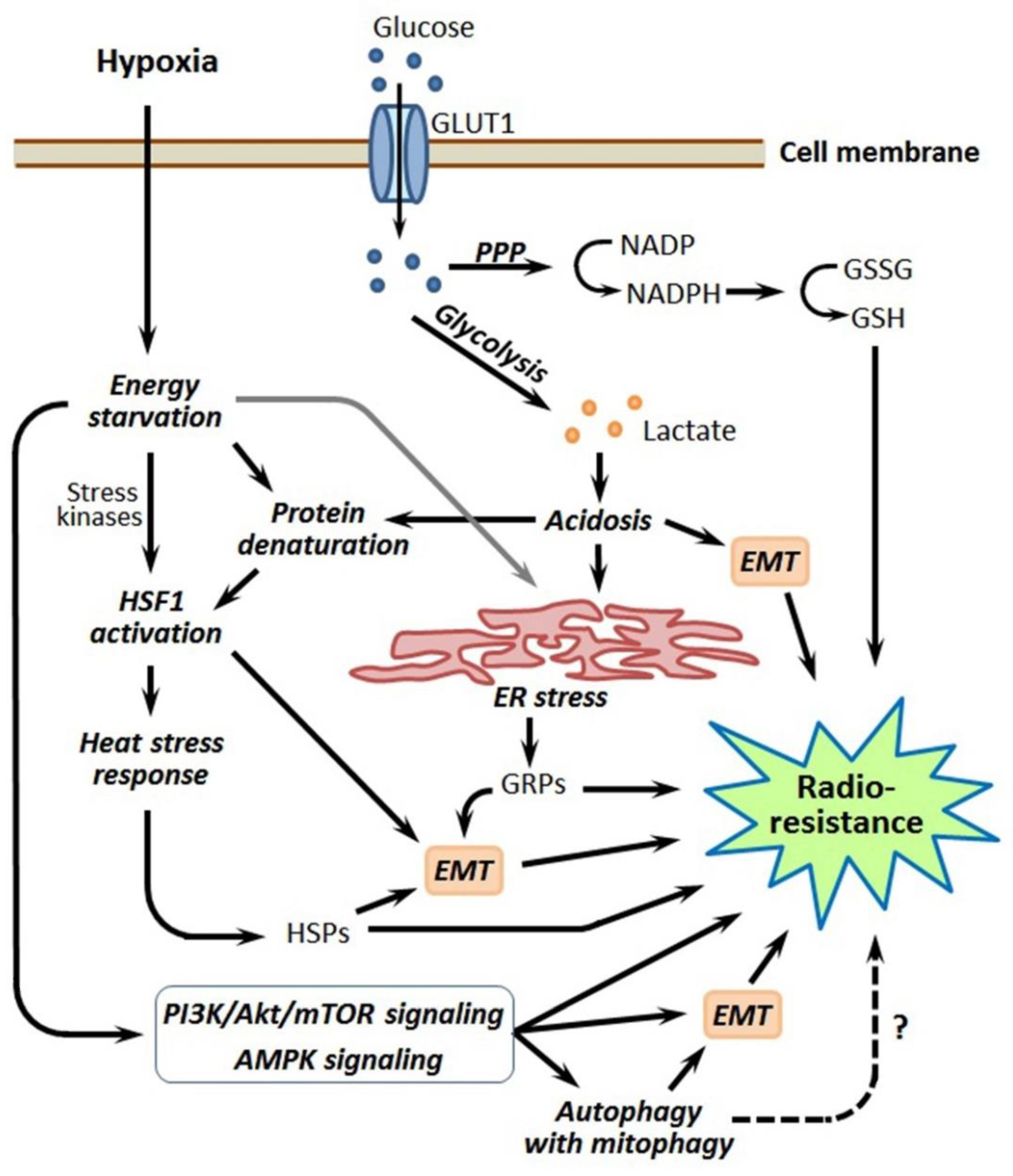{kind=link}
| Agent | Tumor | Molecular Targets | Achieved Effects |
|---|---|---|---|
| Nelfinavir | Head and neck squamous cell carcinoma, lung cancer [66] | HIF-1α and VEGF 1 expression | Decreased HIF-1α and VEGF levels, suppressed angiogenesis, radiosensitization |
| PX-478 | Prostate cancer [67] | HIF-1α expression | Decreased HIF-1α level, protracted DNA repair, radiosensitization |
| Trichostatin A | Cervical carcinoma [68] | HDAC 2 activity, HIF-1α and VEGF expression | Decreased HIF-1α and VEGF levels, radiosensitization |
| Saikosaponin-D (or with PX-478) | Hepatoma [69] | HIF-1α expression | Decreased HIF-1α level, upregulated p53 and Bax, downregulated Bcl-2, increased radiosensitivity |
| NVP-BEZ235 | Breast cancer [37], endometrial cancer [38] | PI3K/mTOR signaling pathway | Suppressed HIF-1α, protracted DNA repair, enhanced apoptosis |
| Atorvastatin | Prostate cancer [70], Burkitt’s lymphoma [71] | HIF-1α expression miR-346/HIF-1α/VEGF | Downregulated HIF-1α, increased radiosensitivity, anti-angiogenesis |
| Berberine | Nasopharyngeal [72] and prostate [73] carcinomas | HIF-1α and VEGF | Inhibition of upregulation of HIF-1α and VEGF, radiosensitization |
| Melittin | Squamous cell carcinoma [74] | HIF-1α and VEGF expression | Reduced expression of HIF-1α and VEGF, increased radiosensitivity |
| BAY-84-7296 | Squamous cell carcinoma [75] | HIF-1α activity | Decreased nuclear translocation of HIF-1α, reduced radioresistance |
| YC-1 | Squamous cell carcinoma [76], cervical carcinoma [77], breast cancer and others [78] | HIF-1α activity | Downregulation of HIF-1α, sensitization to γ-radiation [76,77] and neutron capture therapy [78] |
| Gambogic acid | Nasopharyngeal carcinoma [79] | HIF-1α expression | Decreased HIF-1α expression, cell cycle arrest, enhanced apoptosis |
| Pachymic acid | Gastric cancer [80] | HIF-1α expression, Bax expression | Decreased HIF-1α expression, increased Bax, radiosensitization |
| NSC74859 | Esophageal cancer [81] | STAT3, HIF-1α and VEGF expression | Reduced levels of HIF-1α and VEGF, increased radiosensitivity |
| Ursolic acid | Non-small cell lung cancer [82] | HIF-1α expression, endogenous GSH | Suppressed HIF-1α, decreased intracellular GSH levels |
| Soy isoflavones | Prostate cancer [83], lung cancer [84] | Src/STAT3/HIF-1α pathway, DNA repair | Inhibition of HIF-1-mediated transcription, radiosensitization |
| Chetomin | Glioma [85], fibrosarcoma [86], osteosarcoma [87] | HIF-1α–p300 interactions | Suppressed HIF-mediated gene transcription, radiosensitization |
| 2-ME2 3 | Nasopharyngeal [26] and esophageal [88] carcinomas, melanoma [89] | HIF-1α and VEGF expression, NF-κB, glycolysis | Downregulation of HIF-1α and VEGF, reversed EMT, increased radiosensitivity |
| Resveratrol analog | Murine breast cancer [90] | HIF-1α and VEGF expression | Decreased HIF-1α and VEGF levels, inhibited angiogenesis and enhanced apoptosis |
| Acriflavine | Murine breast cancer [91] | HIF-1α expression | Decreased HIF-1α expression, sensitization to radiotherapy |
| Temsirolimus | Lung cancer [39] | mTOR, HIF-1α | Decreased HIF-1α expression, radiosensitization |
| 17AAG 4, deguelin | Lung cancer [21] | HSP90, HIF-1α | Decreased HIF-1α expression, radiosensitization |
| KNK437 | Breast cancer and glioblastoma [92] | Akt signaling, HIF-1α expression | Decreased HIF-1α expression, radiosensitization |
| Paclitaxel | Hepatoma and lung adenocarcinoma [93] | HIF-1α-dependent bFGF 5/PI3K/Akt pathway | Overcoming HIF-1α-induced radioresistance |
| Bortezomib | Cervical [94] and esophageal [95] carcinomas | HIF-1α and VEGF expression | Downregulated HIF-1α and VEGF, apoptosis and delayed DNA repair, radiosensitization |
| Sorafenib | Breast cancer [96] | HIF-1α expression | Suppressing HIF-1α expression, elimination of irradiated CSCs |
| Sunitinib | Prostate cancer [97] | HIF-1α expression | Decreased HIF-1α expression, radiosensitization of CSCs |
| FM19G11 | Glioblastoma [35] | HIF-1α activity | Radiosensitization |
| Irisquinone | C6 rat glioma [98] | HIF-1α expression | Downregulation of HIF-1α, radiosensitization |
| Oleuropein | Nasopharyngeal carcinoma [99] | HIF-1α expression, HIF-1α/miR-519d/PDRG1 6 pathway | Decreased levels of HIF-1α and PDRG1, radiosensitization |
| Agent | Tumor | Molecular Targets | Achieved Effects |
|---|---|---|---|
| 2-deoxyglucose | Glioblastoma [143,144], breast cancer [126] | HK2 1, glycolysis CircABCB10 2, profilin-2 | Inhibition of glycolysis and radiosensitization |
| Apigenin | Laryngeal carcinoma [145] | PI3K/Akt signaling, GLUT1 3 expression | Downregulated GLUT1, increased radiosensitivity |
| WZB117 | Breast cancer [146] | GLUT1 expression, glucose metabolism | Downregulated GLUT1, radiosensitization |
| 2-ME2 | Melanoma [89] | HIF-1α, PDK1 4 and GLUT1 expression | Downregulated PDK1 and GLUT1, radiosensitization |
| BX795 | Hepatocellular carcinoma [120] | PDK1 activity | Increased Bax/Bcl-2 ratio and apoptosis, radiosensitization |
| Dichloroacetate | Melanoma [89] glioblastoma [119] | PDK1 activity | Increased DNA damage and apoptosis, radiosensitization |
| BAY-84-7296 | Squamous cell carcinoma [75] | Mitochondrial complex I, HIF-1α | Downregulated HIF-1α, impaired radioresistance |
| Butylmalonate | Lung and prostate cancer, glioblastoma [147] | SLC25A10 5 | Overcoming radioresistance induced by hypoxia |
| Metformin and phenformin | Colorectal cancer [148] | Mitochondrial complex I, AMPK | Overcoming radioresistance induced by hypoxia |
| Anti-parasitic drugs (atovaquone, others) | High-grade glioma [149] | Mitochondrial metabolism | Enhanced radiosensitivity (suggested) |
| Hypoxia-Altered MicroRNA Expression | Type of Tumor | Cellular Targets and Induced Effects Leading to the Tumor Radioresistance |
|---|---|---|
| Increased miR-155 | Lung cancer [318] | Decreased FOXO3A 1 expression |
| Increased miR-210 | Hepatoma [319] | Decreased AIFM3 2, protection from apoptosis |
| Lung cancer [58] | HIF-1-dependent DNA double-strand break repair | |
| Glioma stem cells [309] | Decreased MNT 3, protection from apoptosis | |
| Colon cancer [54] | HIF-1α/miR-210/Bcl-2 pathway, protection from apoptosis, enhanced autophagy | |
| Increased miR-21 | Lung cancer [123] | Upregulation of HIF-1α, activated glycolysis |
| Decreased miR-124/miR-144 | Prostate cancer [255] | Downregulated PIM1 4, induced autophagy |
| Decreased miR-30a/miR-205 | Prostate cancer [256] | Increased TP53INP1 5, induced autophagy |
| Increased miR-301a/miR-301b | Prostate cancer [257] | Decreased NDRG2 6, elevated autophagy |
| Decreased miR-519d | Nasopharyngeal cancer [99] | Decreased levels of HIF-1α and PDRG1 7 |
| Exosomal miR-301a | Glioma [320] | Suppressed TCEAL7 8 and activated Wnt/β-catenin pathway |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabakov, A.E.; Yakimova, A.O. Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing. Cancers 2021, 13, 1102. https://doi.org/10.3390/cancers13051102
Kabakov AE, Yakimova AO. Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing. Cancers. 2021; 13(5):1102. https://doi.org/10.3390/cancers13051102
Chicago/Turabian StyleKabakov, Alexander E., and Anna O. Yakimova. 2021. "Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing" Cancers 13, no. 5: 1102. https://doi.org/10.3390/cancers13051102
APA StyleKabakov, A. E., & Yakimova, A. O. (2021). Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing. Cancers, 13(5), 1102. https://doi.org/10.3390/cancers13051102