
THC Reduces Ki67-Immunoreactive Cells Derived from Human Primary Glioblastoma in a GPR55-Dependent Manner

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
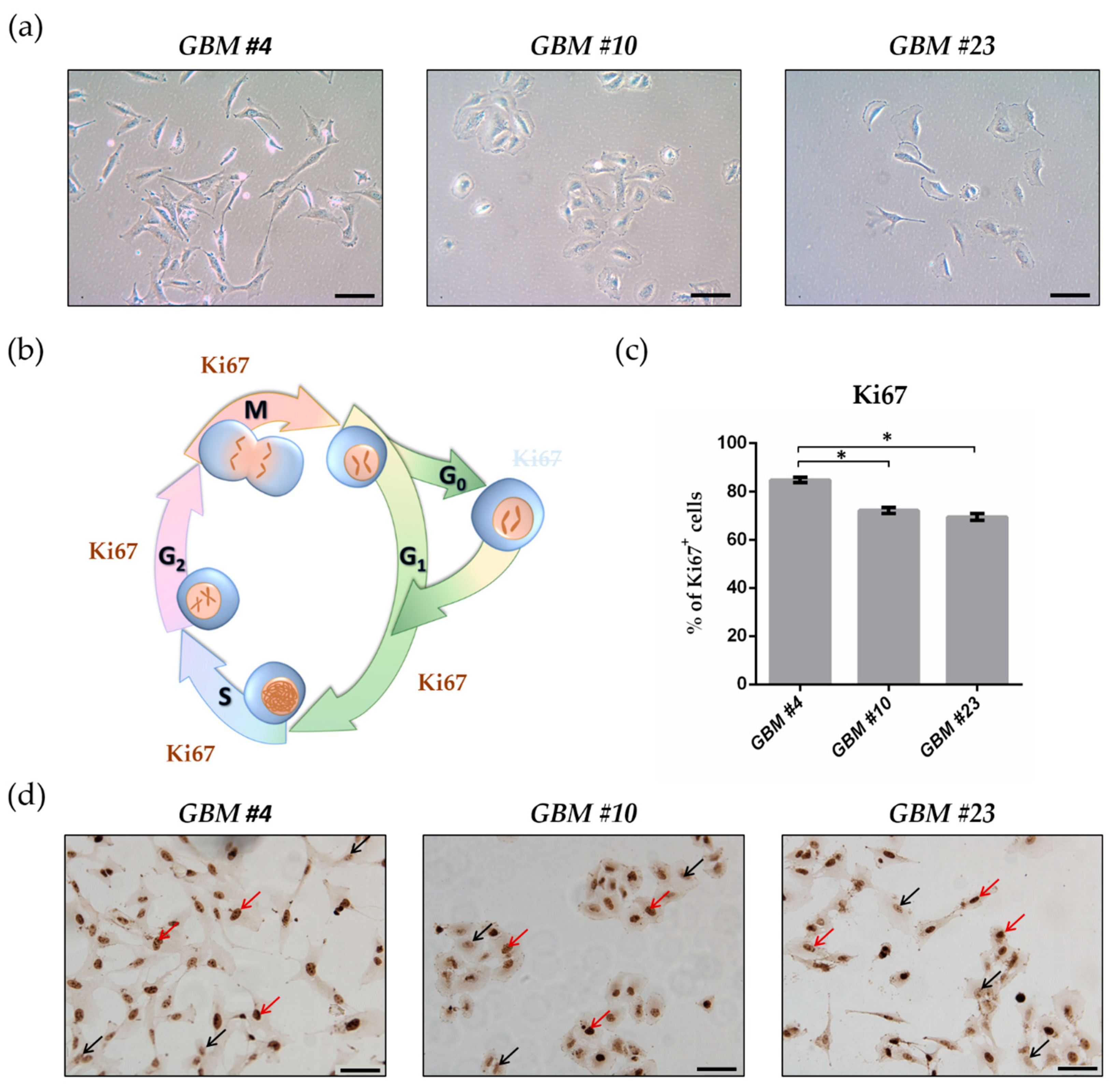
2.1. GBM Cells Differed in Their Morphology and Showed High Proliferation Capacity
2.2. THC Decreased Ki67+ Cells and Was Blocked by CBD
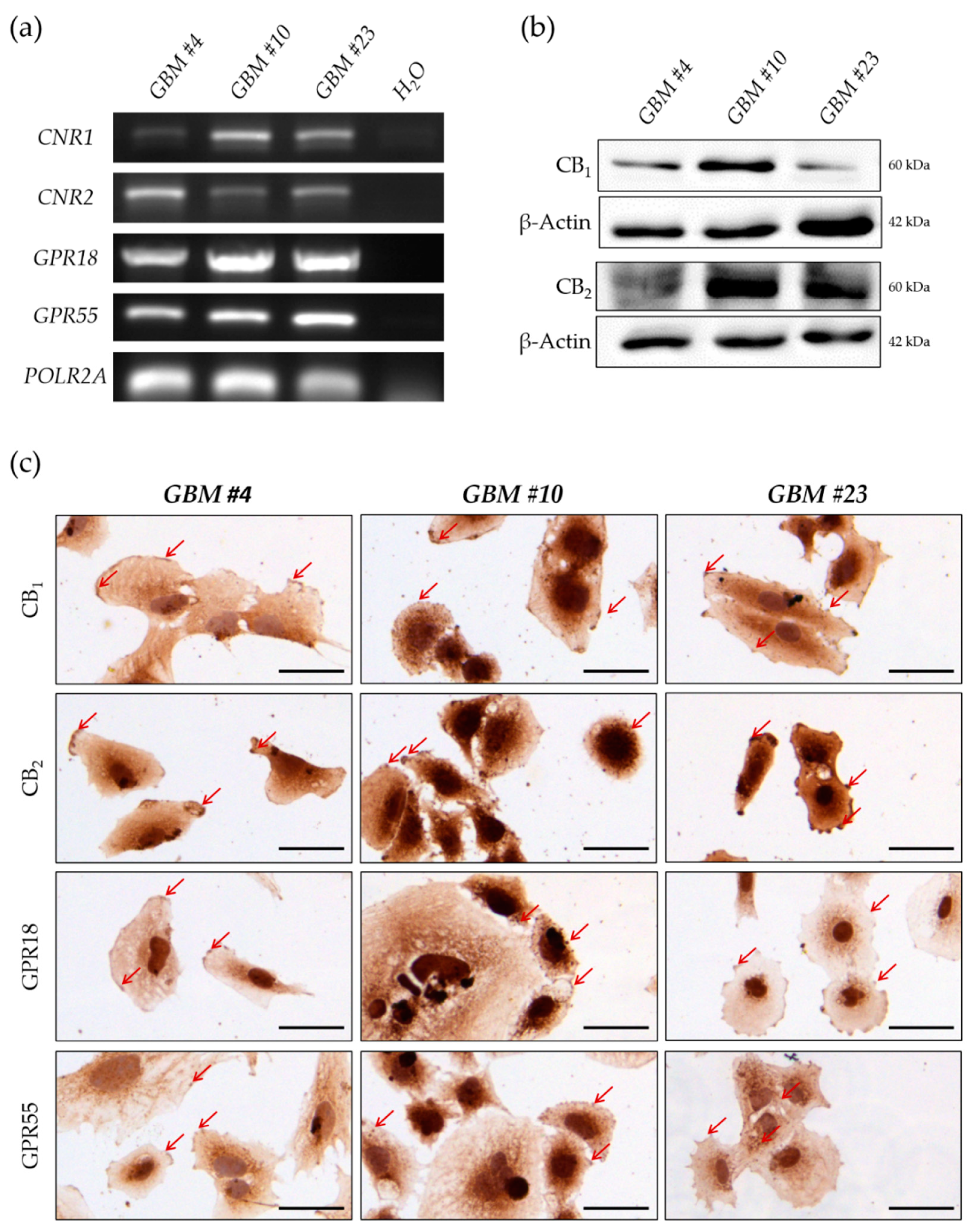
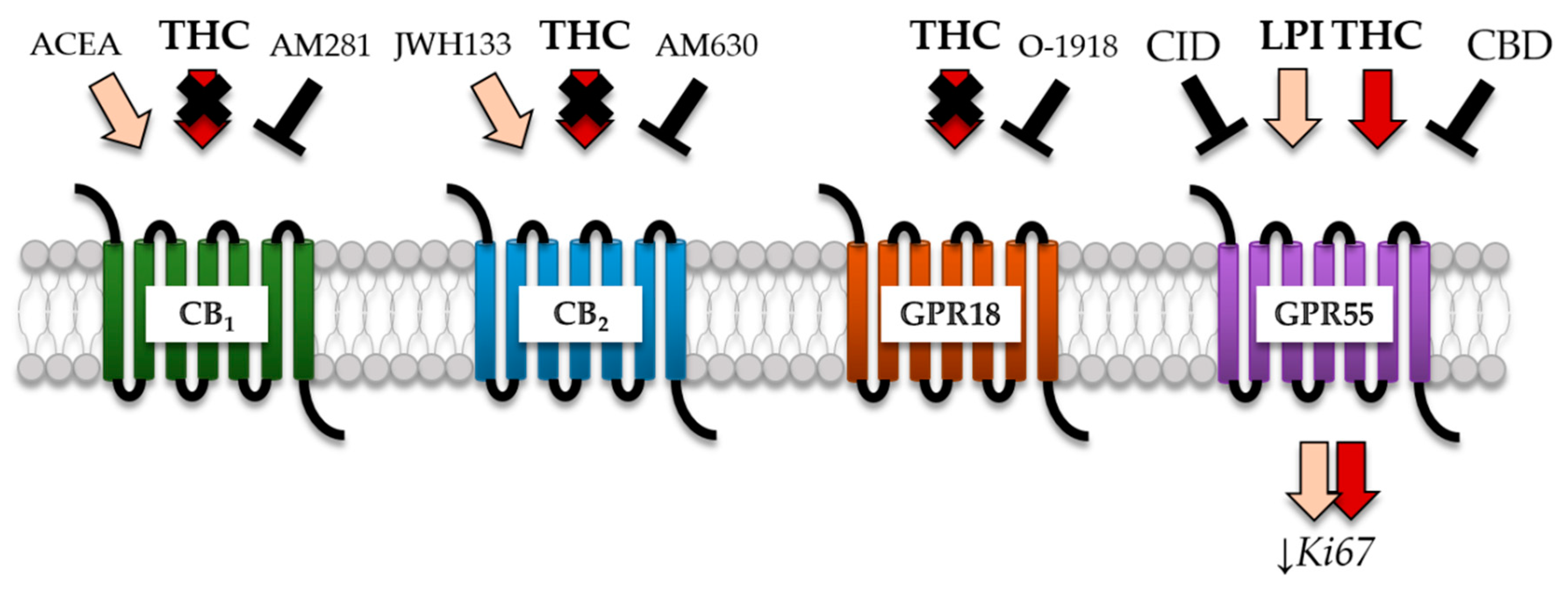
2.3. GBM Cells Expressed Classical and Orphan Cannabinoid Receptors
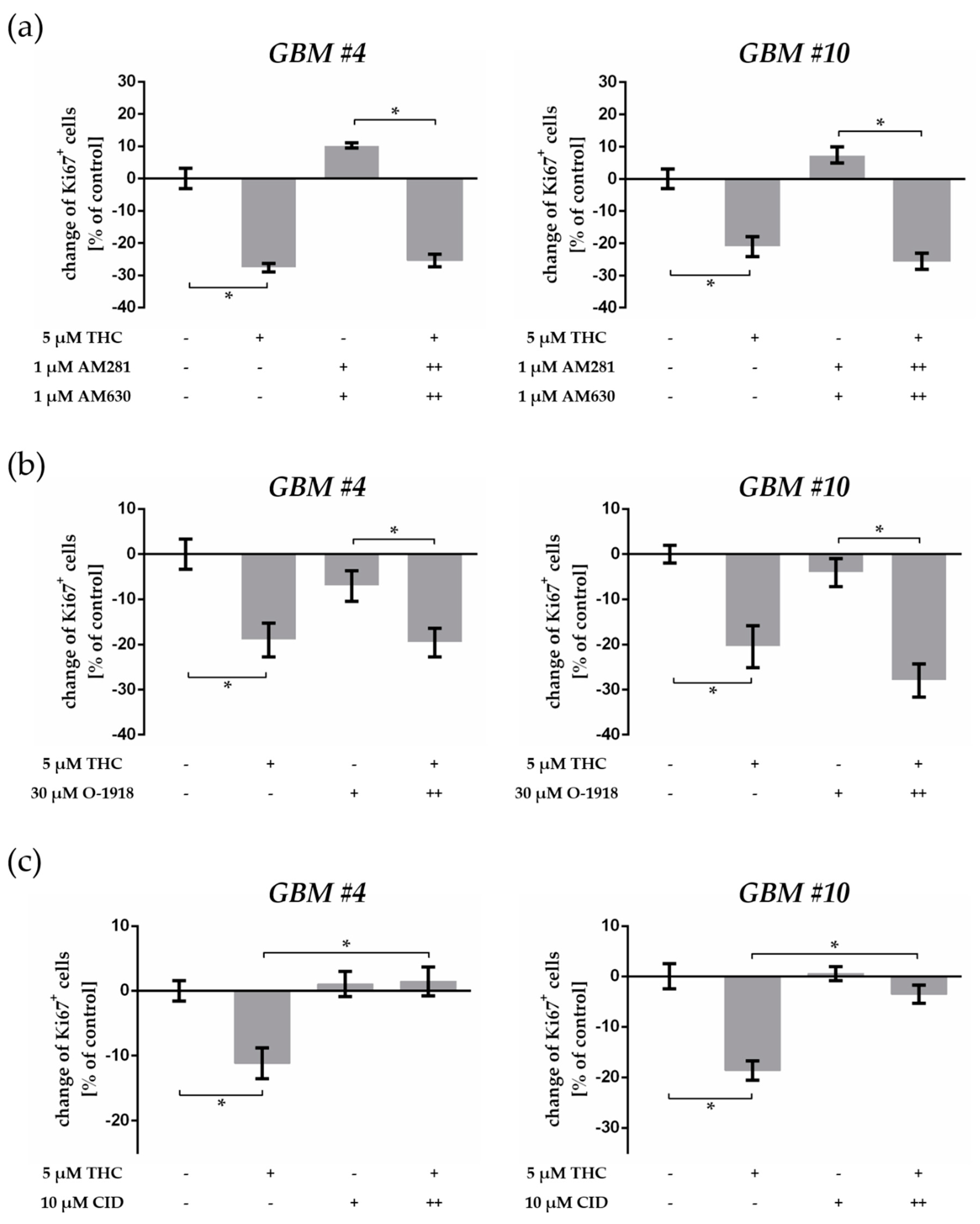
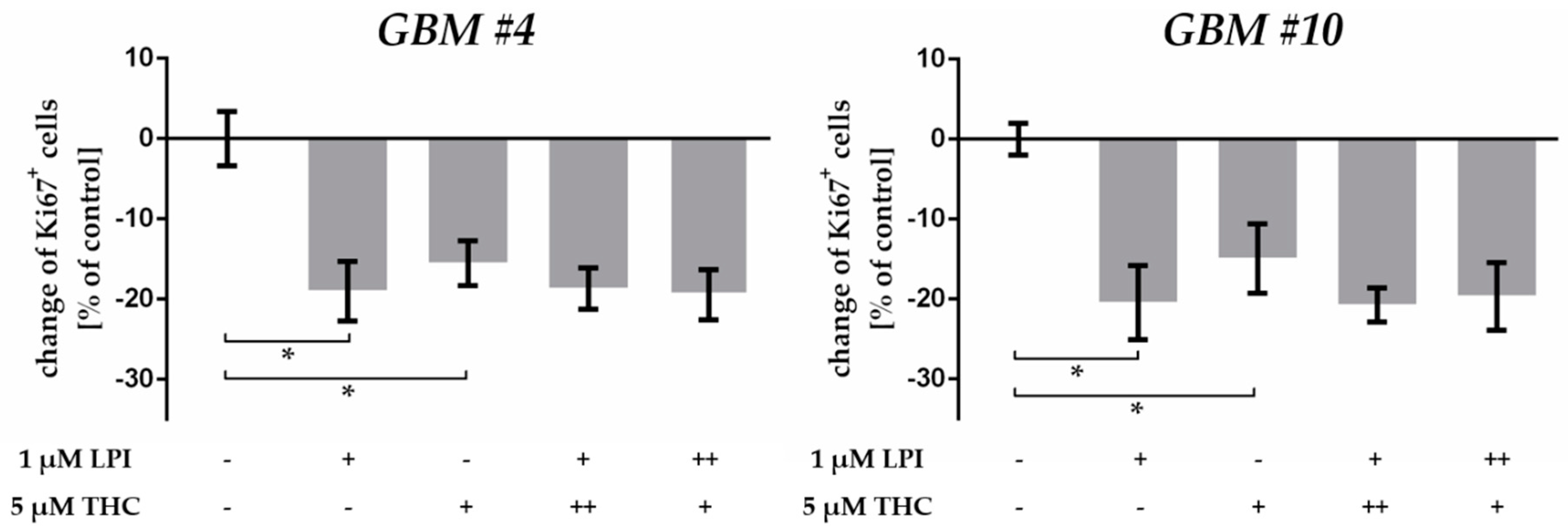
2.4. THC-Dependent Reduction of Ki67+ Cells Was Not Mediated by CB1, CB2, and GPR18
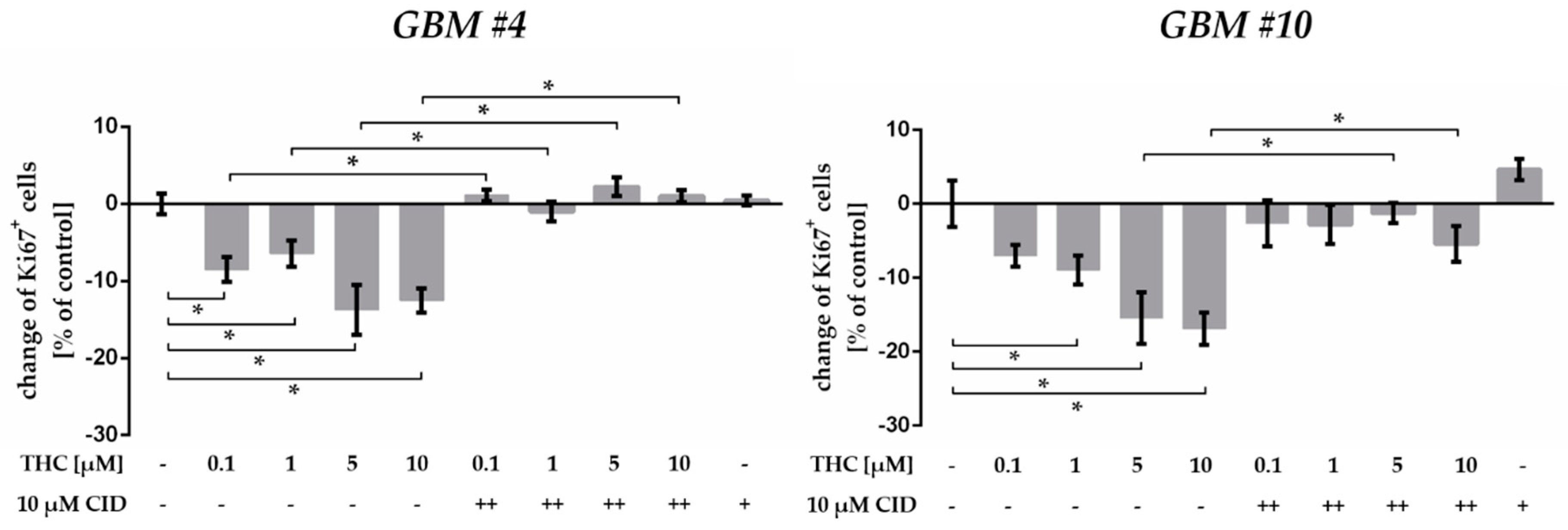
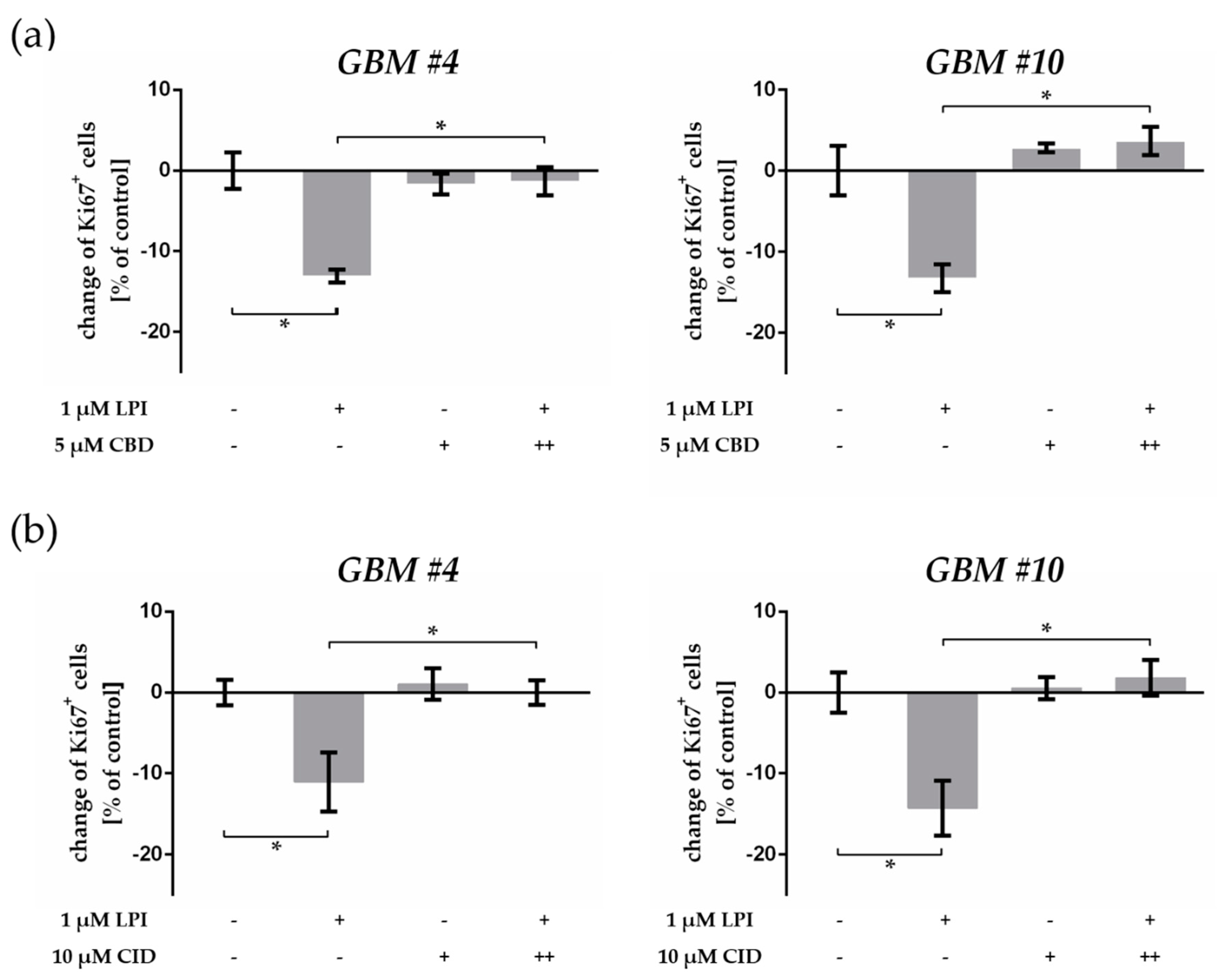
2.5. GPR55 Activation Was Involved in THC-Dependent Reduction of Ki67+ Cells
3. Discussion
3.1. THC Modified the Percentage of Ki67+ Patient-Derived GBM Cells
3.2. THC Effects Were Not Mediated via CB1 and CB2
3.3. THC Did Not Act via GPR18
3.4. THC and CBD Exhibited GPR55-Dependent Signaling in Patient-Derived GBM Cells
3.5. Cell Type-Specific Effects of THC and LPI
4. Materials and Methods
4.1. Isolation of Human Patient-Derived GBM Cells and Cell Culture
4.2. Treatment
4.3. PCR
4.4. Western Blot
4.5. Immunochemical Staining
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wen, P.Y.; Kesari, S. Malignant Gliomas in Adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Mooney, J.H.; Ilyas, A.; Chagoya, G.; Estevez-Ordonez, D.; Ibrahim, A.; Nakano, I. Molecular and cellular intratumoral heterogeneity in primary glioblastoma: Clinical and translational implications. J. Neurosurg. 2019, 1–9. [Google Scholar] [CrossRef]
- Stoyanov, G.S.; Dzhenkov, D.; Ghenev, P.; Iliev, B.; Enchev, Y.; Tonchev, A.B. Cell biology of glioblastoma multiforme: From basic science to diagnosis and treatment. Med. Oncol. 2018, 35, 27. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Javid, F.A.; Phillips, R.M.; Afshinjavid, S.; Verde, R.; Ligresti, A. Cannabinoid pharmacology in cancer research: A new hope for cancer patients? Eur. J. Pharmacol. 2016, 775, 1–14. [Google Scholar] [CrossRef]
- Benz, A.H.; Renné, C.; Maronde, E.; Koch, M.; Grabiec, U.; Kallendrusch, S.; Rengstl, B.; Newrzela, S.; Hartmann, S.; Hansmann, M.-L.; et al. Expression and Functional Relevance of Cannabinoid Receptor 1 in Hodgkin Lymphoma. PLoS ONE 2013, 8, e81675. [Google Scholar] [CrossRef]
- Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The Endocannabinoid System: A Target for Cancer Treatment. IJMS 2020, 21, 747. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Δ9-Tetrahydrocannabinol Inhibits Cell Cycle Progression in Human Breast Cancer Cells through Cdc2 Regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef]
- Hohmann, T.; Grabiec, U.; Ghadban, C.; Feese, K.; Dehghani, F. The influence of biomechanical properties and cannabinoids on tumor invasion. Cell Adh. Migr. 2017, 11, 54–67. [Google Scholar] [CrossRef]
- Torres, S.; Lorente, M.; Rodriguez-Fornes, F.; Hernandez-Tiedra, S.; Salazar, M.; Garcia-Taboada, E.; Barcia, J.; Guzman, M.; Velasco, G. A Combined Preclinical Therapy of Cannabinoids and Temozolomide against Glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef]
- Solinas, M.; Massi, P.; Cinquina, V.; Valenti, M.; Bolognini, D.; Gariboldi, M.; Monti, E.; Rubino, T.; Parolaro, D. Cannabidiol, a Non-Psychoactive Cannabinoid Compound, Inhibits Proliferation and Invasion in U87-MG and T98G Glioma Cells through a Multitarget Effect. PLoS ONE 2013, 8, e76918. [Google Scholar] [CrossRef] [PubMed]
- Marcu, J.P.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Horowitz, M.P.; Lee, J.; Pakdel, A.; Allison, J.; Limbad, C.; Moore, D.H.; et al. Cannabidiol enhances the inhibitory effects of delta9-tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Mol. Cancer 2010, 9, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, T.; Feese, K.; Greither, T.; Ghadban, C.; Jäger, V.; Dehghani, F.; Grabiec, U. Synthetic Cannabinoids Influence the Invasion of Glioblastoma Cell Lines in a Cell- and Receptor-Dependent Manner. Cancers 2019, 11, 161. [Google Scholar] [CrossRef]
- Hohmann, T.; Feese, K.; Ghadban, C.; Dehghani, F.; Grabiec, U. On the influence of cannabinoids on cell morphology and motility of glioblastoma cells. PLoS ONE 2019, 14, e0212037. [Google Scholar] [CrossRef]
- Pertwee, R.G. Ligands that target cannabinoid receptors in the brain: From THC to anandamide and beyond: Ligands that target cannabinoid receptors in the brain. Addict. Biol. 2008, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharm. 2007, 150, 613–623. [Google Scholar] [CrossRef]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. In Phytocannabinoids; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 103, pp. 103–131. ISBN 978-3-319-45539-6. [Google Scholar]
- Aizpurua-Olaizola, O.; Elezgarai, I.; Rico-Barrio, I.; Zarandona, I.; Etxebarria, N.; Usobiaga, A. Targeting the endocannabinoid system: Future therapeutic strategies. Drug Discov. Today 2017, 22, 105–110. [Google Scholar] [CrossRef]
- Soderstrom, K.; Soliman, E.; Van Dross, R. Cannabinoids Modulate Neuronal Activity and Cancer by CB1 and CB2 Receptor-Independent Mechanisms. Front. Pharm. 2017, 8, 720. [Google Scholar] [CrossRef]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef]
- Andradas, C.; Caffarel, M.M.; Pérez-Gómez, E.; Salazar, M.; Lorente, M.; Velasco, G.; Guzmán, M.; Sánchez, C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 2011, 30, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Ford, L.A.; Roelofs, A.J.; Anavi-Goffer, S.; Mowat, L.; Simpson, D.G.; Irving, A.J.; Rogers, M.J.; Rajnicek, A.M.; Ross, R.A. A role for L-α-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells: LPI and GPR55 modulate breast cancer cell migration. Br. J. Pharmacol. 2010, 160, 762–771. [Google Scholar] [CrossRef]
- Piñeiro, R.; Maffucci, T.; Falasca, M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2011, 30, 142–152. [Google Scholar] [CrossRef]
- Huang, L.; Ramirez, J.C.; Frampton, G.A.; Golden, L.E.; Quinn, M.A.; Pae, H.Y.; Horvat, D.; Liang, L.; DeMorrow, S. Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab. Investig. 2011, 91, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Pryce, G.; Davies, W.L.; Hiley, C.R. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol. Sci. 2006, 27, 1–4. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Page, J.; Dunn, E.; Bradshaw, H.B. Δ9-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells: Novel CB pharmacology at GPR18. Br. J. Pharmacol. 2012, 165, 2414–2424. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor: GPR55, a novel cannabinoid receptor. Br. J. Pharmacol. 2009, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Andradas, C.; Medrano, M.; Caffarel, M.M.; Pérez-Gómez, E.; Blasco-Benito, S.; Gómez-Cañas, M.; Pazos, M.R.; Irving, A.J.; Lluís, C.; et al. Targeting CB 2-GPR55 Receptor Heteromers Modulates Cancer Cell Signaling. J. Biol. Chem. 2014, 289, 21960–21972. [Google Scholar] [CrossRef]
- Likar, R.; Nahler, G. The use of cannabis in supportive care and treatment of brain tumor. Neuro-oncol. Pract. 2017, 4, 151–160. [Google Scholar] [CrossRef][Green Version]
- Blázquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-J.; He, D.-S.; Tang, R.-X.; Ren, F.-H.; Chen, G. Ki-67 is a valuable prognostic factor in gliomas: Evidence from a systematic review and meta-analysis. Asian Pac. J. Cancer Prev. 2015, 16, 411–420. [Google Scholar] [CrossRef]
- Ralte, A.M.; Sharma, M.C.; Karak, A.K.; Mehta, V.S.; Sarkar, C. Clinicopathological features, MIB-1 labeling index and apoptotic index in recurrent astrocytic tumors. Pathol. Oncol. Res. 2001, 7, 267–278. [Google Scholar] [CrossRef]
- Thotakura, M.; Tirumalasetti, N.; Krishna, R. Role of Ki-67 labeling index as an adjunct to the histopathological diagnosis and grading of astrocytomas. J. Cancer Res. 2014, 10, 641–645. [Google Scholar] [CrossRef]
- Moskowitz, S.I.; Jin, T.; Prayson, R.A. Role of MIB1 in Predicting Survival in Patients with Glioblastomas. J. Neurooncol. 2006, 76, 193–200. [Google Scholar] [CrossRef]
- Hillard, C.J.; Manna, S.; Greenberg, M.J.; DiCamelli, R.; Ross, R.A.; Stevenson, L.A.; Murphy, V.; Pertwee, R.G.; Campbell, W.B. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J. Pharm. Exp. 1999, 289, 1427–1433. [Google Scholar]
- Huffman, J.W.; Liddle, J.; Yu, S.; Aung, M.M.; Abood, M.E.; Wiley, J.L.; Martin, B.R. 3-(1’,1’-Dimethylbutyl)-1-deoxy-delta8-THC and related compounds: Synthesis of selective ligands for the CB2 receptor. Bioorg. Med. Chem. 1999, 7, 2905–2914. [Google Scholar] [CrossRef]
- Mallipeddi, S.; Janero, D.R.; Zvonok, N.; Makriyannis, A. Functional selectivity at G-protein coupled receptors: Advancing cannabinoid receptors as drug targets. Biochem. Pharmacol. 2017, 128, 1–11. [Google Scholar] [CrossRef]
- Glass, M.; Northup, J.K. Agonist Selective Regulation of G Proteins by Cannabinoid CB1 and CB2 Receptors. Mol. Pharm. 1999, 56, 1362–1369. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Biased Type 1 Cannabinoid Receptor Signaling Influences Neuronal Viability in a Cell Culture Model of Huntington Disease. Mol. Pharm. 2016, 89, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Diez-Alarcia, R.; Ibarra-Lecue, I.; Lopez-Cardona, Á.P.; Meana, J.; Gutierrez-Adán, A.; Callado, L.F.; Agirregoitia, E.; Urigüen, L. Biased Agonism of Three Different Cannabinoid Receptor Agonists in Mouse Brain Cortex. Front. Pharm. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Lauckner, J.E.; Hille, B.; Mackie, K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 19144–19149. [Google Scholar] [CrossRef]
- Finlay, D.B.; Cawston, E.E.; Grimsey, N.L.; Hunter, M.R.; Korde, A.; Vemuri, V.K.; Makriyannis, A.; Glass, M. Gαs signalling of the CB1 receptor and the influence of receptor number. Br. J. Pharm. 2017, 174, 2545–2562. [Google Scholar] [CrossRef]
- Al-Zoubi, R.; Morales, P.; Reggio, P.H. Structural Insights into CB1 Receptor Biased Signaling. IJMS 2019, 20, 1837. [Google Scholar] [CrossRef]
- Tomko, A.; O’Leary, L.; Trask, H.; Achenbach, J.C.; Hall, S.R.; Goralski, K.B.; Ellis, L.D.; Dupré, D.J. Antitumor Activity of Abnormal Cannabidiol and Its Analog O-1602 in Taxol-Resistant Preclinical Models of Breast Cancer. Front. Pharm. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Qin, Y.; Verdegaal, E.M.E.; Siderius, M.; Bebelman, J.P.; Smit, M.J.; Leurs, R.; Willemze, R.; Tensen, C.P.; Osanto, S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: The constitutively active orphan GPCR GPR18 as novel drug target: GPCR expression profiling in melanoma. Pigment Cell Melanoma Res. 2011, 24, 207–218. [Google Scholar] [CrossRef]
- Finlay, D.B.; Joseph, W.R.; Grimsey, N.L.; Glass, M. GPR18 undergoes a high degree of constitutive trafficking but is unresponsive to N-Arachidonoyl Glycine. PeerJ 2016, 4, e1835. [Google Scholar] [CrossRef]
- Brown, A.J. Novel cannabinoid receptors: Novel cannabinoid receptors. Br. J. Pharmacol. 2009, 152, 567–575. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.A.B.; Kargl, J.; Andradas, C.; Brown, A.J.; Irving, A.; Sanchez, C.; Waldhoer, M. Minireview: Recent Developments in the Physiology and Pathology of the Lysophosphatidylinositol-Sensitive Receptor GPR55. Mol. Endocrinol. 2011, 25, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Tudurí, E.; Imbernon, M.; Hernández-Bautista, R.J.; Tojo, M.; Fernø, J.; Diéguez, C.; Nogueiras, R. GPR55: A new promising target for metabolism? J. Mol. Endocrinol. 2017, 58, R191–R202. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, J.; Lehmann, C. GPR55—A putative “type 3” cannabinoid receptor in inflammation. J. Basic Clin. Physiol. Pharmacol. 2016, 27. [Google Scholar] [CrossRef]
- Kargl, J.; Andersen, L.; Hasenöhrl, C.; Feuersinger, D.; Stančić, A.; Fauland, A.; Magnes, C.; El-Heliebi, A.; Lax, S.; Uranitsch, S.; et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis: GPR55 in colon cancer. Br. J. Pharmacol. 2016, 173, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Abood, M.E. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharm. Ther. 2010, 126, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A. The enigmatic pharmacology of GPR55. Trends Pharmacol. Sci. 2009, 30, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.E.; Arifin, S.A.; Fyffe, C.A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 2018, 37, 6368–6382. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and cannabinoid CB 2 receptors modulates signalling: Heteromerization of GPR55 and CB2 receptors. Br. J. Pharm. 2014, 171, 5387–5406. [Google Scholar] [CrossRef]
- Kargl, J.; Balenga, N.; Parzmair, G.P.; Brown, A.J.; Heinemann, A.; Waldhoer, M. The Cannabinoid Receptor CB1 Modulates the Signaling Properties of the Lysophosphatidylinositol Receptor GPR55. J. Biol. Chem. 2012, 287, 44234–44248. [Google Scholar] [CrossRef]
- Pertwee, R.G. Pharmacology of cannabinoid receptor ligands. Curr. Med. Chem. 1999, 6, 635–664. [Google Scholar]
- Lan, R.; Gatley, J.; Lu, Q.; Fan, P.; Fernando, S.R.; Volkow, N.D.; Pertwee, R.; Makriyannis, A. Design and synthesis of the CB1 selective cannabinoid antagonist AM281: A potential human SPECT ligand. AAPS Pharmsci. 1999, 1, E4. [Google Scholar] [CrossRef]
- Hosohata, Y.; Quock, R.M.; Hosohata, K.; Makriyannis, A.; Consroe, P.; Roeske, W.R.; Yamamura, H.I. AM630 antagonism of cannabinoid-stimulated [35S]GTP gamma S binding in the mouse brain. Eur. J. Pharm. 1997, 321, R1–R3. [Google Scholar] [CrossRef]
- Salazar, M.; Carracedo, A.; Salanueva, Í.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef]
- Kargl, J.; Brown, A.J.; Andersen, L.; Dorn, G.; Schicho, R.; Waldhoer, M.; Heinemann, A. A Selective Antagonist Reveals a Potential Role of G Protein-Coupled Receptor 55 in Platelet and Endothelial Cell Function. J. Pharm. Exp. 2013, 346, 54–66. [Google Scholar] [CrossRef]
- Kallendrusch, S.; Kremzow, S.; Nowicki, M.; Grabiec, U.; Winkelmann, R.; Benz, A.; Kraft, R.; Bechmann, I.; Dehghani, F.; Koch, M. The G Protein-Coupled Receptor 55 Ligand l -α-Lysophosphatidylinositol Exerts Microglia-Dependent Neuroprotection After Excitotoxic Lesion: Microglia-Dependent GPR55-Driven Neuroprotection. Glia 2013, 61, 1822–1831. [Google Scholar] [CrossRef]
- McHugh, D.; Bradshaw, H.B. GPR18 and NAGly Signaling: New Members of the Endocannabinoid Family or Distant Cousins? In Endocannabinoids; Abood, M.E., Sorensen, R.G., Stella, N., Eds.; Springer: New York, NY, USA, 2013; pp. 135–142. ISBN 978-1-4614-4668-2. [Google Scholar]
- Grabiec, U.; Hohmann, T.; Ghadban, C.; Rothgänger, C.; Wong, D.; Antonietti, A.; Groth, T.; Mackie, K.; Dehghani, F. Protective Effect of N-Arachidonoyl Glycine-GPR18 Signaling after Excitotoxical Lesion in Murine Organotypic Hippocampal Slice Cultures. IJMS 2019, 20, 1266. [Google Scholar] [CrossRef]
- McHugh, D.; Hu, S.S.; Rimmerman, N.; Juknat, A.; Vogel, Z.; Walker, J.M.; Bradshaw, H.B. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010, 11, 44. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substances | Targets | Behavior | Solvent | Concentration | Company | Article Number |
|---|---|---|---|---|---|---|
| ACEA | CB1 | agonist [61] | ethanol | 10 µM [11,15,16] | Tocris, Bristol, UK | 1319 |
| SMILES: CCCCC/C=C\C/C=C\C/C=C\C/C=C\CCCC(=O)NCCCl | ||||||
| AM281 | CB1 | antagonist [62] | DMSO | 1 µM [15,16] | Tocris, Bristol, UK | 1115 |
| SMILES: CC1=C(N(N=C1C(=O)NN2CCOCC2)C3=C(C=C(C=C3)Cl)Cl)C4=CC=C(C=C4)I | ||||||
| AM630 | CB2 | antagonist [63] | DMSO | 1 µM [15,16] | Tocris, Bristol, UK | 1120 |
| SMILES: CC1=C(C2=C(N1CCN3CCOCC3)C=C(C=C2)I)C(=O)C4=CC=C(C=C4)OC | ||||||
| Cannabidiol (CBD) | CB1 CB2 | weak agonist [19] | DMSO | 5 µM [64] | Tocris, Bristol, UK | 1570 |
| GPR18 GPR55 | antagonist [19] | |||||
| SMILES: CCCCCC1=CC(=C(C(=C1)O)C2C=C(CCC2C(=C)C)C)O | ||||||
| CID16020046 | GPR55 | antagonist [65] | DMSO | 10 µM (Figure S2) | Tocris, Bristol, UK | 4959 |
| SMILES: CC1=CC=C(C=C1)C2=NNC3=C2C(N(C3=O)C4=CC=C(C=C4)C(=O)O)C5=CC(=CC=C5)O | ||||||
| Dronabinol (THC) | CB1 CB2 GPR18 GPR55 | agonist [19] | DMSO | 5 µM [64] | THC pharm GmbH, Frankfurt am Main, Germany | THC-1016 |
| SMILES: CCCCCC1=CC(=C2C3C=C(CCC3C(OC2=C1)(C)C)C)O | ||||||
| JWH133 | CB2 | agonist [61] | DMSO | 10 µM [11,15,16] | Tocris, Bristol, UK | 1343 |
| SMILES: CCCC(C)(C)C1=CC2=C(C=C1)C3CC(=CCC3C(O2)(C)C)C | ||||||
| Lysophosphatidylinositol (LPI) | GPR55 | agonist [22] | DMSO | 1 µM [66] | Sigma-Aldrich® Chemie GmbH, Steinheim, Germany | L7635 |
| SMILES: CC(=O)OCC(COP(=O)(O)OC1C(C(C(C(C1O)O)O)O)O)O | ||||||
| O-1918 | GPR18 | antagonist [67] | DMSO | 30 µM [68] | Tocris, Bristol, UK | 2288 |
| SMILES: CC1=CC(C(CC1)C(=C)C)C2=C(C=C(C=C2OC)C)OC | ||||||
| Gene | Accession Number | Forward Primer (5′-> 3′) | Reverse Primer (5′-> 3′) | Product Size |
|---|---|---|---|---|
| CD44 | NM_000610 | CTGGCGCAGATCGATTTGAA | TTGCTGCACAGATGGAGTTGG | 244 bp |
| CNR1 | NM_033181 | GCATCCAAGGAAGGGATGTA | CCGTTGTGTGTCTCATCCAC | 250 bp |
| CNR2 | NM_001841 | GCTCCTCATCTGTTGGTTCC | TGACCATGGAGTTGATGAGGC | 208 bp |
| GPR18 | NM_005292 | CCACCAAGAAGAGAACCAC | GAAGGGCATAAAGCAGACG | 596 bp |
| GPR55 | NM_005683 | GGTGCTCTCCCTCCCATT | GCTCACCAGTAGCGGGTAAC | 172 bp |
| MSI1 | NM_002442 | GATGGTCACTCGGACGAAGAA | CAAACCCTCTGTGCCTGTTG | 149 bp |
| NES | NM_006617 | CAGCGTTGGAACAGAGGTTGG | TGGCACAGGTGTCTCAAGGGTAG | 389 bp |
| POLR2A | NM_000937 | CTTGCCCCGTGCCATGCAGA | CTCGCACCCGGCCTTCCTTG | 83 bp |
| SOX2 | NM_003106 | ACTCCTACGTGGGCGACGAGG | CAGGTCCAGACGCAGGATGGC | 389 bp |
| Antibodies | Species | Concentration (Application) | Company | Article Number |
|---|---|---|---|---|
| Anti-β-actin | mouse | 1:5000 (WB 1) | Cell Signaling, Danvers, MA, USA | 3700 |
| Anti-CB1 | rabbit | 1:200 (ICC 2) 1:1200 (WB) | Cayman Chemicals, Ann Arbor, MI, USA | 101500 |
| Anti-CB2 | rabbit | 1:200 (ICC, WB) | Alomon Labs, Jerusalem, Israel | ACR-002 |
| Anti-CD133 | mouse | 1:40 (ICC) | Miltenyi Biotec, Bergisch, Gladbach, Germany | 130-092-395 |
| Anti-GPR18 | rabbit | 1:300 (ICC) | gifted from Ken Mackie | described in [69] |
| Anti-GPR55 | rabbit | 1:200 (ICC) | Cayman Chemicals, Ann Arbor, MI, USA | 10224 |
| Anti-IDH1 R132H | mouse | 1:500 (ICC) | Dianova GmbH | DIA-H09 |
| Anti-Ki67 | rabbit | 1:200 (ICC) | DSC innovative Diagnostic-System, Hamburg, Germany | KI681C002 |
| Anti-mouse IgG horseradish peroxidase (HRP)-conjugated | horse | 1:10,000 (WB) | Vector, Burlingame, CA, USA | PI-2000 |
| Anti-rabbit IgG, biotin conjugated | goat | 1:100 (ICC) | Sigma-Aldrich® Chemie GmbH, Steinheim, Germany | B7389 |
| Anti-rabbit IgG, horseradish peroxidase (HRP)-conjugated | goat | 1:20,000 (WB) | Vector, Burlingame, CA, USA | PI-1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolbe, M.R.; Hohmann, T.; Hohmann, U.; Ghadban, C.; Mackie, K.; Zöller, C.; Prell, J.; Illert, J.; Strauss, C.; Dehghani, F. THC Reduces Ki67-Immunoreactive Cells Derived from Human Primary Glioblastoma in a GPR55-Dependent Manner. Cancers 2021, 13, 1064. https://doi.org/10.3390/cancers13051064
Kolbe MR, Hohmann T, Hohmann U, Ghadban C, Mackie K, Zöller C, Prell J, Illert J, Strauss C, Dehghani F. THC Reduces Ki67-Immunoreactive Cells Derived from Human Primary Glioblastoma in a GPR55-Dependent Manner. Cancers. 2021; 13(5):1064. https://doi.org/10.3390/cancers13051064
Chicago/Turabian StyleKolbe, Marc Richard, Tim Hohmann, Urszula Hohmann, Chalid Ghadban, Ken Mackie, Christin Zöller, Julian Prell, Jörg Illert, Christian Strauss, and Faramarz Dehghani. 2021. "THC Reduces Ki67-Immunoreactive Cells Derived from Human Primary Glioblastoma in a GPR55-Dependent Manner" Cancers 13, no. 5: 1064. https://doi.org/10.3390/cancers13051064
APA StyleKolbe, M. R., Hohmann, T., Hohmann, U., Ghadban, C., Mackie, K., Zöller, C., Prell, J., Illert, J., Strauss, C., & Dehghani, F. (2021). THC Reduces Ki67-Immunoreactive Cells Derived from Human Primary Glioblastoma in a GPR55-Dependent Manner. Cancers, 13(5), 1064. https://doi.org/10.3390/cancers13051064