
A Palette of Cytokines to Measure Anti-Tumor Efficacy of T Cell-Based Therapeutics

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. T Cell-Derived Cytokines as Biomarkers of Anti-Tumor Activity
3. The Dual Face of IFN-γ and TGF-β
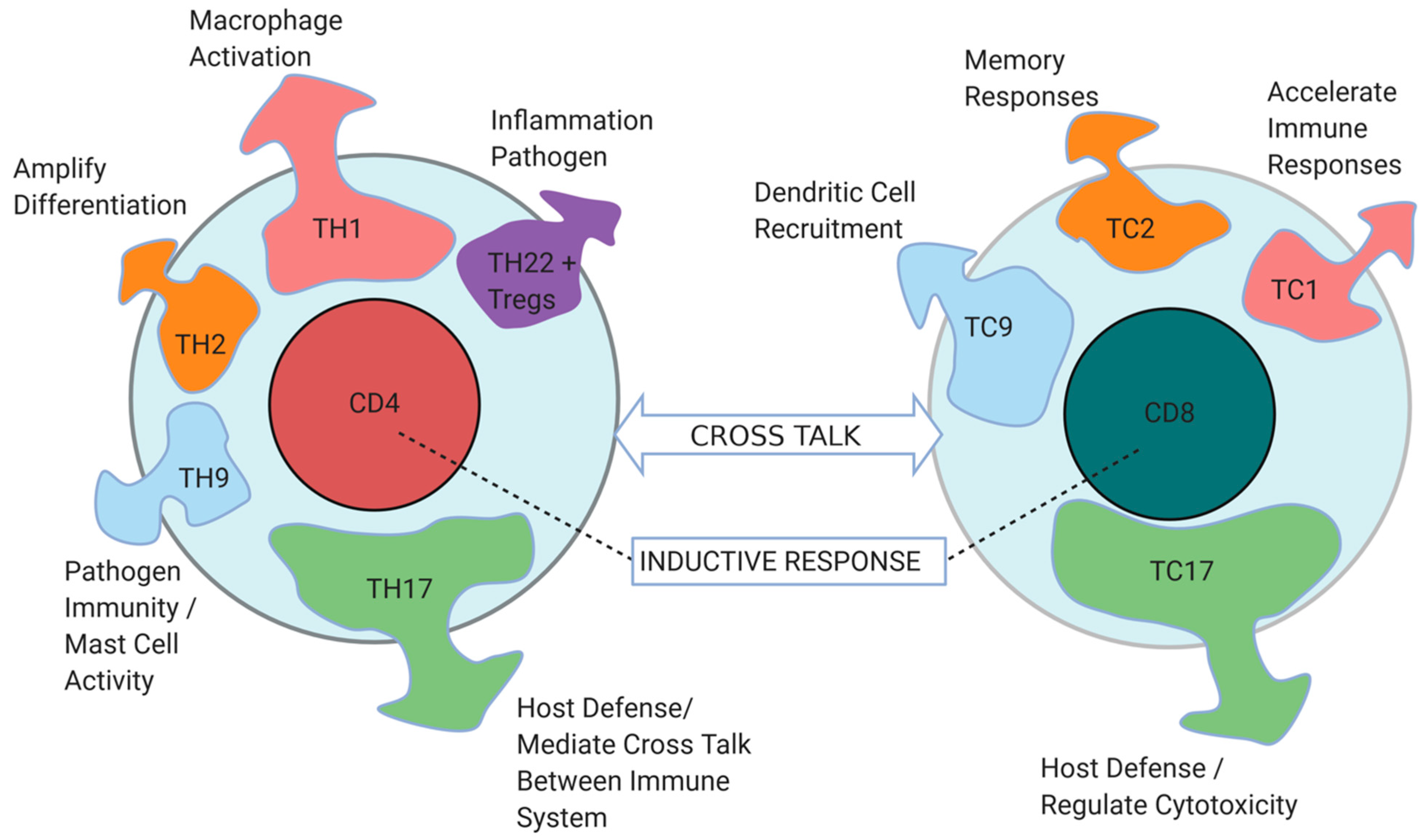
4. The Interplay of CD4+ and CD8+ T Cells Define Immune Responses in the TME
5. T Cell Polyfunctionality
6. Monitoring Cytokine Abundance
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Foster, J.R. The functions of cytokines and their uses in toxicology. Int. J. Exp. Pathol. 2001, 82, 171–192. [Google Scholar] [CrossRef]
- Bonini, C.; Mondino, A. Adoptive T-cell therapy for cancer: The era of engineered T cells. Eur. J. Immunol. 2015, 45, 2457–2469. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Allavena, P.; Mantovani, A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008, 43, 374–379. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Jennifer Gillary Segal, N.C.L.; Tsung, Y.L.; Jeffrey, A.; Norton, K.T. The Role of IFN-y in Rejection of Established Tumors by IL-12: Source of Productiona and Target. Cancer Res. 2002, 62, 4696–4703. [Google Scholar]
- Le Poole, I.C.; Riker, A.I.; Quevedo, M.E.; Stennett, L.S.; Wang, E.; Marincola, F.M.; Kast, W.M.; Robinson, J.K.; Nickoloff, B.J. Interferon-gamma reduces melanosomal antigen expression and recognition of melanoma cells by cytotoxic T cells. Am. J. Pathol. 2002, 160, 521–528. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naive to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Disis, M.L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Vegran, F.; Apetoh, L.; Ghiringhelli, F. Th9 cells: A novel CD4 T-cell subset in the immune war against cancer. Cancer Res. 2015, 75, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct Role of Antigen-specific T Helper Type 1 (Th1) and Th2 Cells in Tumor Eradication in Vivo. Rockefeller Univ. Press 1999, 190, 617–627. [Google Scholar] [CrossRef]
- van Belzen, I.; Kesmir, C. Immune biomarkers for predicting response to adoptive cell transfer as cancer treatment. Immunogenetics 2019, 71, 71–86. [Google Scholar] [CrossRef]
- Goswami, R.; Kaplan, M.H. A brief history of IL-9. J. Immunol. 2011, 186, 3283–3288. [Google Scholar] [CrossRef]
- Guery, L.; Hugues, S. Th17 Cell Plasticity and Functions in Cancer Immunity. Biomed. Res. Int 2015, 2015, 314620. [Google Scholar] [CrossRef]
- Badri, K.R.; Gao, L.; Hyjek, E.; Schuger, N.; Schuger, L.; Qin, W.; Chekaluk, Y.; Kwiatkowski, D.J.; Zhe, X. Exonic mutations of TSC2/TSC1 are common but not seen in all sporadic pulmonary lymphangioleiomyomatosis. Am. J. Respir. Crit. Care Med. 2013, 187, 663–665. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-M. Th1 cytokine-based immunotherapy for cancer. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 482–494. [Google Scholar] [CrossRef]
- Han, Q.; Bagheri, N.; Bradshaw, E.M.; Hafler, D.A.; Lauffenburger, D.A.; Love, J.C. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 1607–1612. [Google Scholar] [CrossRef]
- Attig, S.; Hennenlotter, J.; Pawelec, G.; Klein, G.; Koch, S.D.; Pircher, H.; Feyerabend, S.; Wernet, D.; Stenzl, A.; Rammensee, H.G.; et al. Simultaneous infiltration of polyfunctional effector and suppressor T cells into renal cell carcinomas. Cancer Res. 2009, 69, 8412–8419. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Kjeldsen, J.W.; Andersen, R.; Westergaard, M.C.W.; Bianchi, V.; Legut, M.; Attaf, M.; Szomolay, B.; Ott, S.; Dolton, G.; et al. PD-1(+) Polyfunctional T Cells Dominate the Periphery after Tumor-Infiltrating Lymphocyte Therapy for Cancer. Clin. Cancer Res. 2017, 23, 5779–5788. [Google Scholar] [CrossRef] [PubMed]
- Reap, E.A.; Suryadevara, C.M.; Batich, K.A.; Sanchez-Perez, L.; Archer, G.E.; Schmittling, R.J.; Norberg, P.K.; Herndon, J.E., 2nd; Healy, P.; Congdon, K.L.; et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res. 2018, 78, 256–264. [Google Scholar] [CrossRef]
- Tran, E.; Nielsen, J.S.; Wick, D.A.; Ng, A.V.; Johnson, L.D.; Nesslinger, N.J.; McMurtrie, E.; Webb, J.R.; Nelson, B.H. Polyfunctional T-cell responses are disrupted by the ovarian cancer ascites environment and only partially restored by clinically relevant cytokines. PLoS ONE 2010, 5, e15625. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.; Van Loenen, M.M.; Guislain, A.; Nicolet, B.P.; Freen-Van Heeren, J.J.; Verhagen, O.; Van Den Heuvel, M.M.; De Jong, J.; Burger, P.; Van Der Schoot, C.E.; et al. Polyfunctional tumor-reactive T cells are effectively expanded from non-small cell lung cancers, and correlate with an immune-engaged T cell profile. Oncoimmunology 2019, 8, e1648170. [Google Scholar] [CrossRef] [PubMed]
- Alshaker, H.A.; Matalka, K.Z. IFN-gamma, IL-17 and TGF-beta involvement in shaping the tumor microenvironment: The significance of modulating such cytokines in treating malignant solid tumors. Cancer Cell Int. 2011, 11, 33. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, W.; Pan, M.; Scully, E.; Girardi, M.; Augenlicht, L.H.; Craft, J.; Yin, Z. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J. Exp. Med. 2003, 198, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R. The Interferon-Gamma Paradox in Cancer. J. Interferon Cytokine Res. 2019, 39, 30–38. [Google Scholar] [CrossRef]
- Beatty, G.L.; Paterson, Y. Regulation of Tumor Growth by IFN-y in Cancer Immunotherapy. Immunol. Res. 2001, 24, 201–210. [Google Scholar] [CrossRef]
- Kursunel, M.A.; Esendagli, G. The untold story of IFN-gamma in cancer biology. Cytokine Growth Factor Rev. 2016, 31, 73–81. [Google Scholar] [CrossRef]
- Lu, Y.; Gu, X.; Chen, L.; Yao, Z.; Song, J.; Niu, X.; Xiang, R.; Cheng, T.; Qin, Z.; Deng, W.; et al. Interferon-γ produced by tumor-infiltrating NK cells and CD4+ T cells downregulates TNFSF15 expression in vascular endothelial cells. Angiogenesis 2014, 17, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-gamma: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19, 89. [Google Scholar] [CrossRef]
- Ni, L.; Lu, J. Interferon gamma in cancer immunotherapy. Cancer Med. 2018, 7, 4509–4516. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.J.; Zhang, H.R.; Tian, G.F.; Tian, J.; Mao, X.L.; Jia, Z.H.; Meng, Z.Y.; Zhao, L.Q.; Yin, Z.N.; et al. Tumor-derived transforming growth factor-beta is critical for tumor progression and evasion from immune surveillance. Asian Pac. J. Cancer Prev. 2014, 15, 5181–5186. [Google Scholar] [CrossRef][Green Version]
- Tu, E.; Chia, P.Z.; Chen, W. TGFbeta in T cell biology and tumor immunity: Angel or devil? Cytokine Growth Factor Rev. 2014, 25, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-beta Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef]
- Dahmani, A.; Delisle, J.S. TGF-beta in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kuhnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4(+)T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef]
- Takeuchi, A.; Saito, T. CD4 CTL, a Cytotoxic Subset of CD4(+) T Cells, Their Differentiation and Function. Front. Immunol. 2017, 8, 194. [Google Scholar] [CrossRef]
- Di Lullo, G.; Marcatti, M.; Heltai, S.; Brunetto, E.; Tresoldi, C.; Bondanza, A.; Bonini, C.; Ponzoni, M.; Tonon, G.; Ciceri, F.; et al. Th22 cells increase in poor prognosis multiple myeloma and promote tumor cell growth and survival. Oncoimmunology 2015, 4, e1005460. [Google Scholar] [CrossRef]
- Petrausch, U.; Poehlein, C.H.; Jensen, S.M.; Twitty, C.; Thompson, J.A.; Assmann, I.; Puri, S.; LaCelle, M.G.; Moudgil, T.; Maston, L.; et al. Cancer immunotherapy: The role regulatory T cells play and what can be done to overcome their inhibitory effects. Curr. Mol. Med. 2009, 9, 673–682. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perusina Lanfranca, M.; Lin, Y.; Fang, J.; Zou, W.; Frankel, T. Biological and pathological activities of interleukin-22. J. Mol. Med. 2016, 94, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef]
- Rivera Vargas, T.; Humblin, E.; Vegran, F.; Ghiringhelli, F.; Apetoh, L. TH9 cells in anti-tumor immunity. Semin. Immunopathol. 2017, 39, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Morales, J.M.G.; Puig, L.; Dauden, E.; Canete, J.D.; Pablos, J.L.; Martin, A.O.; Juanatey, C.G.; Adan, A.; Montalban, X.; Borruel, N.; et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun. Rev. 2020, 19, 102429. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. Tapping CD4 T cells for cancer immunotherapy: The choice of personalized genomics. J. Immunol. 2015, 194, 2049–2056. [Google Scholar] [CrossRef]
- Jackson, S.R.; Yuan, J.; Teague, R.M. Targeting CD8+ T-cell tolerance for cancer immunotherapy. Immunotherapy 2014, 6, 833–852. [Google Scholar] [CrossRef] [PubMed]
- Mittrucker, H.W.; Visekruna, A.; Huber, M. Heterogeneity in the differentiation and function of CD8(+) T cells. Arch. Immunol. Ther. Exp. 2014, 62, 449–458. [Google Scholar] [CrossRef]
- Fousek, K.; Ahmed, N. The Evolution of T-cell Therapies for Solid Malignancies. Clin. Cancer Res. 2015, 21, 3384–3392. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Takada, K.; Jameson, S.C. Self-class I MHC molecules support survival of naive CD8 T cells, but depress their functional sensitivity through regulation of CD8 expression levels. J. Exp. Med. 2009, 206, 2253–2269. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Young, H.A.; Rosenberg, A.S. An overview of cytokines and cytokine antagonists as therapeutic agents. Ann. N. Y. Acad. Sci. 2009, 1182, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.I.; Ishikawa, H. Transcriptional Regulation of Differentiation and Functions of Effector T Regulatory Cells. Cells 2019, 8, 939. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.C.; Huang, L.; Blazar, B.R.; Yagita, H.; Mellor, A.L.; Munn, D.H.; Zhou, G. Polyfunctional CD4(+) T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood 2012, 120, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Eby, J.M.; Smith, A.R.; Riley, T.P.; Cosgrove, C.; Ankney, C.M.; Henning, S.W.; Paulos, C.M.; Garrett-Mayer, E.; Luiten, R.M.; Nishimura, M.I.; et al. Molecular properties of gp100-reactive T-cell receptors drive the cytokine profile and antitumor efficacy of transgenic host T cells. Pigment. Cell Melanoma Res. 2019, 32, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Greenplate, A.R.; Johnson, D.B.; Ferrell, P.B., Jr.; Irish, J.M. Systems immune monitoring in cancer therapy. Eur. J. Cancer 2016, 61, 77–84. [Google Scholar] [CrossRef]
- Clay, T.M.; Hobeika, A.C.; Mosca, P.J.; Lyerly, H.K.; Morse, M.A. Assays for Monitoring Cellular Immune Responses to Active Immunotherapy of Cancer. Clin. Cancer Res. 2001, 7, 1127–1135. [Google Scholar]
- Diefenbach, C.; Lam, L.; Raphael, B.G.; Hymes, K.B.; Grossbard, M.L.; Moskovits, T.; Martin, P.; Ruan, J.; Leonard, J.P.; Chattopadhyay, P. Evaluation of the Functional Landscape of Systemic Immunity in Classical Hodgkin Using a Novel Single Cell Platform (Isolight). Blood 2019, 134, 3980. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstrahle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514. [Google Scholar] [CrossRef]
- Mocciaro, A.; Roth, T.L.; Bennett, H.M.; Soumillon, M.; Shah, A.; Hiatt, J.; Chapman, K.; Marson, A.; Lavieu, G. Light-activated cell identification and sorting (LACIS) for selection of edited clones on a nanofluidic device. Commun. Biol. 2018, 1, 41. [Google Scholar] [CrossRef] [PubMed]
- Siebert, J.C.; Walker, E.B. Monitoring cytokine profiles during immunotherapy. Immunotherapy 2010, 2, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, P.; Maecker, H.T. Multiparameter intracellular cytokine staining. Methods Mol. Biol. 2011, 699, 165–178. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef]
- Gadalla, R.; Noamani, B.; MacLeod, B.L.; Dickson, R.J.; Guo, M.; Xu, W.; Lukhele, S.; Elsaesser, H.J.; Razak, A.R.A.; Hirano, N.; et al. Validation of CyTOF Against Flow Cytometry for Immunological Studies and Monitoring of Human Cancer Clinical Trials. Front. Oncol. 2019, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Park, L.M.; Lannigan, J.; Jaimes, M.C. OMIP-069: Forty-Color Full Spectrum Flow Cytometry Panel for Deep Immunophenotyping of Major Cell Subsets in Human Peripheral Blood. Cytom. A 2020, 97, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef]
- Choi, J.R.; Yong, K.W.; Choi, J.Y.; Cowie, A.C. Single-Cell RNA Sequencing and Its Combination with Protein and DNA Analyses. Cells 2020, 9, 1130. [Google Scholar] [CrossRef]
- Mocellin, S.; Provenzano, M.; Rossi, C.R.; Pilati, P.; Nitti, D.; Lise, M. Use of quantitative real-time PCR to determine immune cell density and cytokine gene profile in the tumor microenvironment. J. Immunol. Methods 2003, 280, 1–11. [Google Scholar] [CrossRef]
- Chang, L.; Rissin, D.M.; Fournier, D.R.; Piech, T.; Patel, P.P.; Wilson, D.H.; Duffy, D.C. Single molecule enzyme-linked immunosorbent assays: Theoretical considerations. J. Immunol. Methods 2012, 378, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Chiswick, E.L.; Duffy, E.; Japp, B.; Remick, D. Detection and quantification of cytokines and other biomarkers. Methods Mol. Biol. 2012, 844, 15–30. [Google Scholar] [CrossRef]
- Van, T.M.; Blank, C.U. A user’s perspective on GeoMxTM digital spatial profiling. Immuno-Oncol. Technol. 2019, 1, 11–18. [Google Scholar] [CrossRef]
- Young, H.A. Cytokine multiplex analysis. Methods Mol. Biol. 2009, 511, 85–105. [Google Scholar] [CrossRef]
- Chowdhury, F.; Williams, A.; Johnson, P. Validation and comparison of two multiplex technologies, Luminex and Mesoscale Discovery, for human cytokine profiling. J. Immunol. Methods 2009, 340, 55–64. [Google Scholar] [CrossRef]
- Thurin, M.; Cesano, A.; Marincola, F.M. Biomarkers for Immunotherapy of Cancer: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2020; p. 731. [Google Scholar]
- Teves, J.M.; Won, K.J. Mapping Cellular Coordinates through Advances in Spatial Transcriptomics Technology. Mol. Cells 2020, 43, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Ho, J.Y.; Du, H.; Xuan, F.; Wu, X.; Wang, Q.; Wang, L.; Liu, Y.; Ba, M.; Wang, Y.; et al. Evidence of long-lasting anti-CD19 activity of engrafted CD19 chimeric antigen receptor–modified T cells in a phase I study targeting pediatrics with acute lymphoblastic leukemia. Hematol. Oncol. 2019, 37, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Romero, F.A.; Taur, Y.; Sadelain, M.; Brentjens, R.J.; Hohl, T.M.; Seo, S.K. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin. Infect. Dis. 2018, 67, 533–540. [Google Scholar] [CrossRef]
- Gofshteyn, J.S.; Shaw, P.A.; Teachey, D.T.; Grupp, S.A.; Maude, S.; Banwell, B.; Chen, F.; Lacey, S.F.; Melenhorst, J.J.; Edmonson, M.J.; et al. Neurotoxicity after CTL019 in a pediatric and young adult cohort. Ann. Neurol. 2018, 84, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef]
- Rossi, J.; Paczkowski, P.; Shen, Y.-W.; Morse, K.; Flynn, B.; Kaiser, A.; Ng, C.; Gallatin, K.; Cain, T.; Fan, R.; et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018, 132, 804–814. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Cytokines Probed and Monitoring Methods | Patients/Source of T Cells | Cancer Type and Treatment | T cell Treatment Outcome Response and Correlated Cytokine |
|---|---|---|---|---|
| #1 [27] | IFN-γ, IL-2, TNF-α FACS analysis, ICS, ELISpot | Healthy donors | CD3+ T cells isolated from peripheral blood | IL-2 (mostly among CD8− T cells) and IFN-γ secreting cells increased, TNF-α secreting cells decreased. IFN-γ and IL-2 secreting cytokines showed functional state persistence. |
| #2 [28] | TNF-α IFN-γ, IL-10, IL-17, IL-2 Intracellular cytokine staining of CD4+ and CD8+ T cells, in renal parenchyma tissues | Peripheral blood, fresh tumor, and autologous renal parenchyma | Renal cell carcinoma PBMC and TIL thawed and analyzed for cytokine release. | IL-10 increased among CD4+ and CD8+ subsets; TNF-α (CD4+ and CD8+), IFN-γ (CD8+) increased after activation. Some patients had increased IL-17 in CD4+ TIL. CD107a surface expression found in CD8+ and some CD4+ cells post- activation. Cytokine secretion pattern of responders: TNF-α, IFN-γ, IL-2 with little IL-5. |
| #3 [29] | TNF, IFN-γ, CD107a (cytotoxicity marker). FACS analysis, cytotoxicity assays, phenotype analysis, flow cytometry | Serial blood sample obtained from TIL treated patients | Melanoma IL-2 based TIL therapy | CD8+ T cells expressing CD107a were fewer than cytokine producing cells. Most CD107a + cells also produced one cytokine. |
| #4 [30] | IFN-γ, TNF-α, CCL3 IFNγ ELISPOT assay, FlowJo | 22 CMV seropositive patients | Glioblastoma In vitro generation of (CMV) pp65 T cells and CMV pp65- DCs from PBMCs | Patients receiving CMV pp65 T cells had more IFNγ+, TNFα+ CCL3+ pp65 specific CD8+ T cells. Survival in treated patients correlated with expression of IFNγ, TNFα and CCL3. |
| #5 [31]. | IFN-γ2,TNF-α1,2, IL-2, IL-12, IL-18, IL-21 CCL41,2, CD107a2(cytotoxicity marker) Flow cytometry, ELISA, Bio-Plex | Bulk ascites cell preparations from high-grade serous EOC patients | Epithelial ovarian cancer (EOC) Exogenous cytokine therapy and introduction of EOC ascites environment on T—cell polyfunctionality | IL-1+, IL-12+ IL-18 enhanced IFNγ (by CD8+ cells), TNF-α, and CCL4 expression Cytokine combination synergistically induced polyfunctional responses and decreased cytokine negative or monofunctional T cells. |
| #6 [32] | IFN-γ3, TNF-α3 IL-23 Flow cytometry, immunohistochemistry | 25 treatment- naïve NSCLC patients with clinical stage I-Iva tumors. | Non-small cell lung cancers (NSCLC) TIL therapy | CD4+/CD8+ cells producing either 2 or 3 of the cytokines were most informative. TNFα and IL-2 were crucial to T cell mediated immunity. |
| Study | Patient/Sample Measured | Cytokines Probed | Monitoring Techniques | Patient Outcomes | Correlation between Cytokines Detected and CAR T Cell Function |
|---|---|---|---|---|---|
| Study 1: NCT02963038 [87] | 10 patients Serum concentration | Il-1β, IFN-α2, IFN-γ, TNF-α, MCP-1, IL-6, IL-8, IL-10, IL-12p70, IL-17A, Il-18A, IL-18, IL-23, IL-33 | Flow cytometry, qPCR | 80% achieved MRD 30% have remained in remission state. 10% achieved complete remission. Long-term engrafted CAR-T cell clone CD19 activity observed in one patient for >2 years. 40% experienced grade 2 or higher CRS. | High concentrations of IFN-γ, IL-6, IL-8, IL-18, and MCP-1 correlate with CAR-T cell expansion. |
| Study 2: NCT01044069 [88] | 53 patients Serial serum samples | IFN-γ, IL-6, IL-10, IL-15, TNF-α | Luminex FlexMAP 3D system, 38-plex cytokine detection assays | 83% achieved complete remission 42% experienced infections. | Cytokine Release Syndrome (CRS) secretion of: IFN-γ: expressed by a greater # of patients w/o infection. IL-6: Patients with CRS grade 2 and 3 had more infections than without. IL-10: expressed by a greater # of patients without infection. Il-15: Patients with CRS grade 3 had slightly more infections than without. TNF-α: Patients with grades 4-5 CRS had more infections than without. |
| Study 3: NCT01626495 [89] | 50 patients Serum concentration levels | 43 cytokines tested (not individually listed) | Luminex bead array, FlexMap 3D system | 98% saw B-cell ALL with CD19 expression Neurotoxicity observed in 46% patients. | Serum IL-2, IL-15, IL-4, and HGF concentrations were notably higher in patients with neurotoxicity. TNFR-1 significantly higher in patients with encephalopathy. 22 cytokines accurately predict neurotoxicity. Predicting regression: IL-12, sgp130, sRAGE, sTNFR-1, sVEGFR, and sVEGFR2. |
| Study 4: NCT01865617 [90] | 47 patients Serum concentrations | IL-7, IL-15 | qPCR, Luminex Assay | Objective response observed in 51% of patients. 40% achieved complete remission CRS grade 1–3 observed in a subset of patients | High levels of IL-7 correlate with favorable outcomes. IL-7 concentration increases along with serum IL-15 levels. |
| Study 5: NCT00924326. [91] | 22 patients Coculture and Serum concentrations | 32 total cytokines: Granzyme B, IFN-γ, MIP-1α, perforin, TNF-α, TNF-β,IL-2, IL-5, IL-7, IL-8, IL-9, IL-12, IL-15, IL-21 IL-2, IL-10, IL-13, IL-22, TGF-β1, IL-1B, IL-6, IL-17A, IL-17F, MCP-1, MCP-4, CCL-11, IP-10, MIP-1β, sCD137, sCD40L, RANTES | PCR, MULTI-SPOT, EMD Millipore Luminex xMAP multiplex assays. | 70% objective response to CAR-T cell therapy. 65% observable CRS of grade 3 or higher. | Both polyfunctional CD4+ and CD8+ T cells secrete: IFN-γ, IL-8, IL-5, granzyme B, and/or MIP-1α. CD4+ population contained IL-17A+ polyfunctional T cells. Responders had higher levels of inflammatory, regulatory, chemoattractive, stimulatory, and effector cytokines). |
| Study 6: NCT00924326. [92] | 8 patients Serum concentration | IFN-γ, TNF, IL-2, CD107a (cytotoxicity marker) | ELISA, ICS followed by flow cytometry detection, CD107a degranulation assay. PCR | 75% attained remission. 50% had prominently elevated serum levels of IFN-γ and TNF | CAR T cells were the source of inflammatory cytokines IFN-γ and TNF found in some patient sera. |
| Study 7: NCT01865617 [93] | 37 patients: relapsed or refractory CD19+ NHL Serum concentrations | IFN-γ IL-6, IL-8, IL-10, IL-15, TGF-β | Luminex Assay | 89% receiving CAR T cell infusion saw objective response. Severe CRS observed in 4/32 patients post Cy/Flu conditioning. Severe neurotoxicity observed in 9/32 patients. | Cy/Flu conditioning induced higher response rates. Peak serum concentrations for IL-6, and IFN-γ observed in correlation with sCRS and Cy/Flu conditioning. Patients with grade ≥ 3 CRS saw increased serum concentrations of IL-6, IFN-γ, IL-15, IL-2, IL-18, and reduced TGF-β. High levels of IL-6, IFN-γ, IL-15, IL- 8, and IL-10 and low levels of TGF-β correlated with severe neurotoxicity. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramesh, P.; Shivde, R.; Jaishankar, D.; Saleiro, D.; Le Poole, I.C. A Palette of Cytokines to Measure Anti-Tumor Efficacy of T Cell-Based Therapeutics. Cancers 2021, 13, 821. https://doi.org/10.3390/cancers13040821
Ramesh P, Shivde R, Jaishankar D, Saleiro D, Le Poole IC. A Palette of Cytokines to Measure Anti-Tumor Efficacy of T Cell-Based Therapeutics. Cancers. 2021; 13(4):821. https://doi.org/10.3390/cancers13040821
Chicago/Turabian StyleRamesh, Prathyaya, Rohan Shivde, Dinesh Jaishankar, Diana Saleiro, and I. Caroline Le Poole. 2021. "A Palette of Cytokines to Measure Anti-Tumor Efficacy of T Cell-Based Therapeutics" Cancers 13, no. 4: 821. https://doi.org/10.3390/cancers13040821
APA StyleRamesh, P., Shivde, R., Jaishankar, D., Saleiro, D., & Le Poole, I. C. (2021). A Palette of Cytokines to Measure Anti-Tumor Efficacy of T Cell-Based Therapeutics. Cancers, 13(4), 821. https://doi.org/10.3390/cancers13040821