
Prognostic Effect of Inflammatory Genes on Stage I–III Colorectal Cancer—Integrative Analysis of TCGA Data

Abstract
Simple Summary
Abstract
1. Introduction
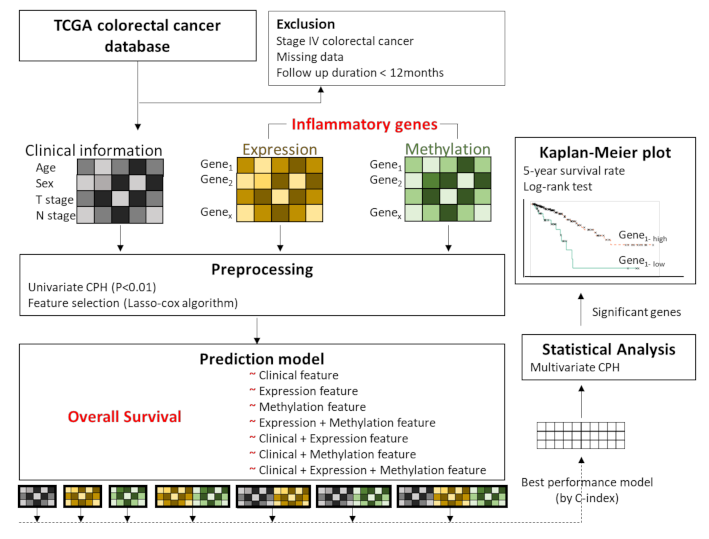
2. Methods
2.1. Data Acquisition and Preprocessing
2.2. Ethics Statement
2.3. Feature Selection
2.4. Overall Survival Prediction Based on Omic Features
3. Results
3.1. Patient Demographics
3.2. Feature Selection for Overall Survival with Lasso-Cox
3.3. Training Prediction Models for Overall Survival with Lasso-Cox
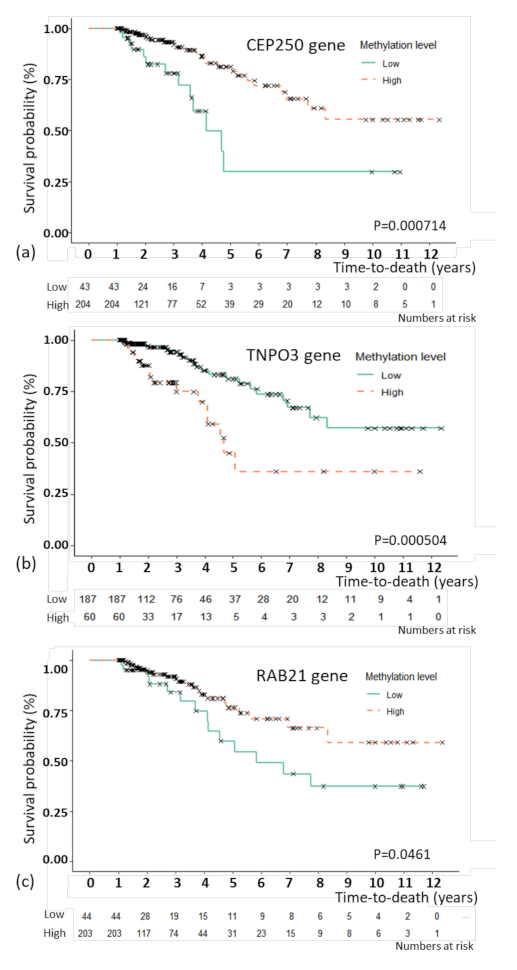
3.4. Association Analysis of Overall Survival with Multivariate Cox Proportional Hazard Regression
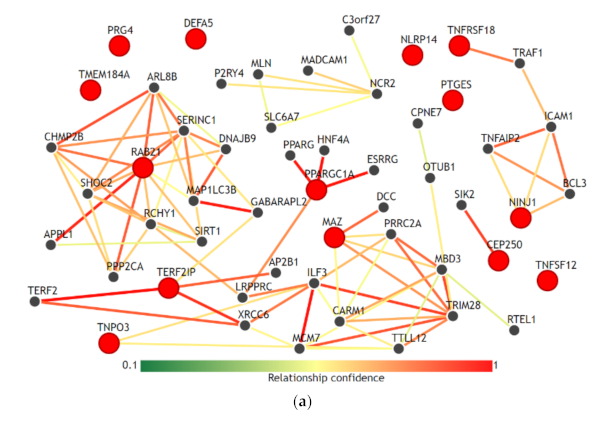
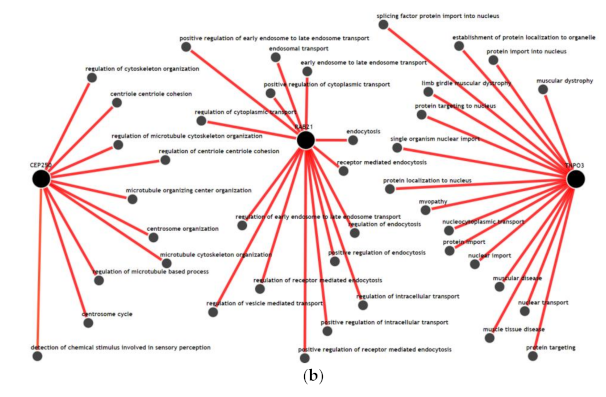
3.5. Gene-Gene Network and Biological Process Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CRC | Colorectal |
| TCGA-COREAD | The Cancer Genome Atlas-Colorectal Cancer |
| Lasso-Cox | Lasso-penalized Cox proportional hazards regression |
| CPH | Cox proportional hazard regression |
| IMP | Integrative multispecies prediction |
| LMR | Lymphocyte-to-monocyte ratio |
| NLR | Neutrophil-to-lymphocyte ratio |
| PLR | Platelet-to-lymphocyte ratio |
| C | Clinical features |
| E | Gene expression features |
| M | Gene methylation features |
| EM | Concatenated expression and methylation features |
References
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Sun, K.; Chen, S.; Xu, J.; Li, G.; He, Y. The prognostic significance of the prognostic nutritional index in cancer: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2014, 140, 1537–1549. [Google Scholar] [CrossRef]
- Templeton, A.J.; Ace, O.; McNamara, M.G.; Al-Mubarak, M.; Vera-Badillo, F.E.; Hermanns, T.; Seruga, B.; Ocana, A.; Tannock, I.F.; Amir, E. Prognostic role of platelet to lymphocyte ratio in solid tumors: A systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1204–1212. [Google Scholar] [CrossRef]
- Gu, L.; Li, H.; Chen, L.; Ma, X.; Li, X.; Gao, Y.; Zhang, Y.; Xie, Y.; Zhang, X. Prognostic role of lymphocyte to monocyte ratio for patients with cancer: Evidence from a systematic review and meta-analysis. Oncotarget 2016, 7, 31926–31942. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Rao, C.V. The role of inflammation in colon cancer. Adv. Exp. Med. Biol. 2014, 816, 25–52. [Google Scholar] [CrossRef] [PubMed]
- Axelrad, J.E.; Lichtiger, S.; Yajnik, V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J. Gastroenterol. 2016, 22, 4794–4801. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Chan, D.L.; Diakos, C.I.; Engel, A.; Pavlakis, N.; Gill, A.; Clarke, S.J. The Lymphocyte-to-Monocyte Ratio is a Superior Predictor of Overall Survival in Comparison to Established Biomarkers of Resectable Colorectal Cancer. Ann. Surg. 2017, 265, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Liu, X.M.; Zhang, X.F.; Zhang, J.F.; Wang, W.L.; Zhu, Y.; Dong, J.; Cheng, J.W.; Liu, Z.W.; Ma, L.; et al. Prognostic role of neutrophil-to-lymphocyte ratio in colorectal cancer: A systematic review and meta-analysis. Int. J. Cancer 2014, 134, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Zhu, G.Q.; Xie, L.; Liu, W.Y.; Shi, L.; Wang, O.C.; Huang, Z.H.; Braddock, M.; Guo, G.L.; Zheng, M.H. Preoperative platelet to lymphocyte ratio is a valuable prognostic biomarker in patients with colorectal cancer. Oncotarget 2016, 7, 25516–25527. [Google Scholar] [CrossRef]
- Song, Y.; Yang, Y.; Gao, P.; Chen, X.; Yu, D.; Xu, Y.; Zhao, J.; Wang, Z. The preoperative neutrophil to lymphocyte ratio is a superior indicator of prognosis compared with other inflammatory biomarkers in resectable colorectal cancer. BMC Cancer 2017, 17, 744. [Google Scholar] [CrossRef]
- Sipos, F.; Firneisz, G.; Muzes, G. Therapeutic aspects of c-MYC signaling in inflammatory and cancerous colonic diseases. World J. Gastroenterol. 2016, 22, 7938–7950. [Google Scholar] [CrossRef]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114 e2105. [Google Scholar] [CrossRef]
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.t.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Shahal, T.; Gabrieli, T.; Gilat, N.; Torchinsky, D.; Michaeli, Y.; Vogel, V.; Ebenstein, Y. Global modulation in DNA epigenetics during pro-inflammatory macrophage activation. Epigenetics 2019, 14, 1183–1193. [Google Scholar] [CrossRef]
- Hanash, S. Integrated global profiling of cancer. Nat. Rev. Cancer 2004, 4, 638–644. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Kim, D.; Joung, J.G.; Sohn, K.A.; Shin, H.; Park, Y.R.; Ritchie, M.D.; Kim, J.H. Knowledge boosting: A graph-based integration approach with multi-omics data and genomic knowledge for cancer clinical outcome prediction. J. Am. Med. Inform. Assoc. 2015, 22, 109–120. [Google Scholar] [CrossRef]
- Kristensen, V.N.; Lingjaerde, O.C.; Russnes, H.G.; Vollan, H.K.; Frigessi, A.; Borresen-Dale, A.L. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 2014, 14, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Shin, H.; Song, Y.S.; Kim, J.H. Synergistic effect of different levels of genomic data for cancer clinical outcome prediction. J. Biomed. Inform. 2012, 45, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Li, R.; Lucas, A.; Verma, S.S.; Dudek, S.M.; Ritchie, M.D. Using knowledge-driven genomic interactions for multi-omics data analysis: Metadimensional models for predicting clinical outcomes in ovarian carcinoma. J. Am. Med. Inform. Assoc. 2017, 24, 577–587. [Google Scholar] [CrossRef]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.D.; Maglott, D.R.; et al. Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 2015, 43, D36–D42. [Google Scholar] [CrossRef]
- Harrell, F.E., Jr.; Lee, K.L.; Mark, D.B. Multivariable prognostic models: Issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat. Med. 1996, 15, 361–387. [Google Scholar] [CrossRef]
- Hothorn, T.; Lausen, B. On the exact distribution of maximally selected rank statistics. J. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
- Wong, A.K.; Park, C.Y.; Greene, C.S.; Bongo, L.A.; Guan, Y.; Troyanskaya, O.G. IMP: A multi-species functional genomics portal for integration, visualization and prediction of protein functions and networks. Nucleic Acids Res. 2012, 40, W484–W490. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef]
- Oliveira, T.; Silva, A.; Satoh, K.; Julian, V.; Leao, P.; Novais, P. Survivability Prediction of Colorectal Cancer Patients: A System with Evolving Features for Continuous Improvement. Sensors 2018, 18, 2983. [Google Scholar] [CrossRef]
- Gene [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. 2004. Available online: https://www.ncbi.nlm.nih.gov/gene/ (accessed on 8 August 2019).
- Li, J.; Lan, C.N.; Kong, Y.; Feng, S.S.; Huang, T. Identification and Analysis of Blood Gene Expression Signature for Osteoarthritis With Advanced Feature Selection Methods. Front. Genet. 2018, 9, 246. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Feng, Y.; Bowers, R.; Becker-Hapak, M.; Gardner, J.; Council, L.; Linette, G.; Zhao, H.; Cornelius, L.A. Ras-associated protein-1 regulates extracellular signal-regulated kinase activation and migration in melanoma cells: Two processes important to melanoma tumorigenesis and metastasis. Cancer Res. 2006, 66, 7880–7888. [Google Scholar] [CrossRef]
- Zhang, L.; Chenwei, L.; Mahmood, R.; van Golen, K.; Greenson, J.; Li, G.; D’Silva, N.J.; Li, X.; Burant, C.F.; Logsdon, C.D.; et al. Identification of a putative tumor suppressor gene Rap1GAP in pancreatic cancer. Cancer Res. 2006, 66, 898–906. [Google Scholar] [CrossRef]
- Li, P.; Wu, Y.H.; Zhu, Y.T.; Li, M.X.; Pei, H.H. Requirement of Rab21 in LPS-induced TLR4 signaling and pro-inflammatory responses in macrophages and monocytes. Biochem. Biophys. Res. Commun. 2019, 508, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L.; Ng, E.L. Rabs and cancer cell motility. Cell Motil. Cytoskeleton 2009, 66, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.; Gaggioli, C.; Sahai, E. A chemical biology screen reveals a role for Rab21-mediated control of actomyosin contractility in fibroblast-driven cancer invasion. Br. J. Cancer 2010, 102, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Milbrandt, J. Ninjurin2, a novel homophilic adhesion molecule, is expressed in mature sensory and enteric neurons and promotes neurite outgrowth. J. Neurosci. 2000, 20, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Ifergan, I.; Kebir, H.; Terouz, S.; Alvarez, J.I.; Lecuyer, M.A.; Gendron, S.; Bourbonniere, L.; Dunay, I.R.; Bouthillier, A.; Moumdjian, R.; et al. Role of Ninjurin-1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann. Neurol. 2011, 70, 751–763. [Google Scholar] [CrossRef]
- Cho, S.J.; Rossi, A.; Jung, Y.S.; Yan, W.; Liu, G.; Zhang, J.; Zhang, M.; Chen, X. Ninjurin1, a target of p53, regulates p53 expression and p53-dependent cell survival, senescence, and radiation-induced mortality. Proc. Natl. Acad. Sci. USA 2013, 110, 9362–9367. [Google Scholar] [CrossRef]
- Langefeld, C.D.; Ainsworth, H.C.; Cunninghame Graham, D.S.; Kelly, J.A.; Comeau, M.E.; Marion, M.C.; Howard, T.D.; Ramos, P.S.; Croker, J.A.; Morris, D.L.; et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun. 2017, 8, 16021. [Google Scholar] [CrossRef]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Komatsu, M.; Li, H.O.; Tsutsui, H.; Itakura, K.; Matsumura, M.; Yokoyama, K.K. MAZ, a Myc-associated zinc finger protein, is essential for the ME1a1-mediated expression of the c-myc gene during neuroectodermal differentiation of P19 cells. Oncogene 1997, 15, 1123–1131. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, D.R.; Goh, H.S. Overexpression of the c-myc proto-oncogene in colorectal carcinoma is associated with a reduced mortality that is abrogated by point mutation of the p53 tumor suppressor gene. Clin. Cancer Res. 1996, 2, 1049–1053. [Google Scholar]
- Haberman, Y.; Karns, R.; Dexheimer, P.J.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef]
- Ahechu, P.; Zozaya, G.; Marti, P.; Hernandez-Lizoain, J.L.; Baixauli, J.; Unamuno, X.; Fruhbeck, G.; Catalan, V. NLRP3 Inflammasome: A Possible Link Between Obesity-Associated Low-Grade Chronic Inflammation and Colorectal Cancer Development. Front. Immunol. 2018, 9, 2918. [Google Scholar] [CrossRef]
- Seo, T.; Tatsuguchi, A.; Shinji, S.; Yonezawa, M.; Mitsui, K.; Tanaka, S.; Fujimori, S.; Gudis, K.; Fukuda, Y.; Sakamoto, C. Microsomal prostaglandin E synthase protein levels correlate with prognosis in colorectal cancer patients. Virchows Arch. 2009, 454, 667–676. [Google Scholar] [CrossRef]
- Yoshimatsu, K.; Golijanin, D.; Paty, P.B.; Soslow, R.A.; Jakobsson, P.J.; DeLellis, R.A.; Subbaramaiah, K.; Dannenberg, A.J. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 2001, 7, 3971–3976. [Google Scholar] [PubMed]
- Williams, A.D.; Korolkova, O.Y.; Sakwe, A.M.; Geiger, T.M.; James, S.D.; Muldoon, R.L.; Herline, A.J.; Goodwin, J.S.; Izban, M.G.; Washington, M.K.; et al. Human alpha defensin 5 is a candidate biomarker to delineate inflammatory bowel disease. PLoS ONE 2017, 12, e0179710. [Google Scholar] [CrossRef]
- Alquraini, A.; Jamal, M.; Zhang, L.; Schmidt, T.; Jay, G.D.; Elsaid, K.A. The autocrine role of proteoglycan-4 (PRG4) in modulating osteoarthritic synoviocyte proliferation and expression of matrix degrading enzymes. Arthritis Res. Ther. 2017, 19, 89. [Google Scholar] [CrossRef]
- Iqbal, S.M.; Leonard, C.; Regmi, S.C.; De Rantere, D.; Tailor, P.; Ren, G.; Ishida, H.; Hsu, C.; Abubacker, S.; Pang, D.S.; et al. Lubricin/Proteoglycan 4 binds to and regulates the activity of Toll-Like Receptors In Vitro. Sci. Rep. 2016, 6, 18910. [Google Scholar] [CrossRef]
- Sarkar, A.; Chanda, A.; Regmi, S.C.; Karve, K.; Deng, L.; Jay, G.D.; Jirik, F.R.; Schmidt, T.A.; Bonni, S. Recombinant human PRG4 (rhPRG4) suppresses breast cancer cell invasion by inhibiting TGFbeta-Hyaluronan-CD44 signalling pathway. PLoS ONE 2019, 14, e0219697. [Google Scholar] [CrossRef]
- Farwell, S.L.; Kanyi, D.; Hamel, M.; Slee, J.B.; Miller, E.A.; Cipolle, M.D.; Lowe-Krentz, L.J. Heparin Decreases in Tumor Necrosis Factor alpha (TNFalpha)-induced Endothelial Stress Responses Require Transmembrane Protein 184A and Induction of Dual Specificity Phosphatase 1. J. Biol. Chem. 2016, 291, 5342–5354. [Google Scholar] [CrossRef] [PubMed]
- Surveillance, Epidemiology, and End Results (SEER) Program SEER*Stat Database: Incidence—SEER 18 Regs Public-Use. October 2010 Sub (1973–2008 Varying); National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch, US, 2011. Available online: https://seer.cancer.gov (accessed on 14 December 2020).
- Hari, D.M.; Leung, A.M.; Lee, J.H.; Sim, M.S.; Vuong, B.; Chiu, C.G.; Bilchik, A.J. AJCC Cancer Staging Manual 7th edition criteria for colon cancer: Do the complex modifications improve prognostic assessment? J. Am. Coll. Surg. 2013, 217, 181–190. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Alive | Dead | p Value |
|---|---|---|---|
| Age | 62.9 ± 12.5 | 71.7 ± 12.6 | <0.001 |
| <65 | 111 (53.1%) | 7 (18.4%) | |
| ≥65 | 98 (46.9%) | 31 (81.6%) | |
| Continuous | 62.9 ± 12.5 | 71.7 ± 12.6 | |
| Gender | 0.407 | ||
| Male | 108 (51.7%) | 23 (60.5%) | |
| Female | 101 (48.3%) | 15 (39.5%) | |
| Overall stage | 0.201 | ||
| Stage 1, 2 | 136 (65.1%) | 20 (52.6%) | |
| Stage 3 | 73 (34.9%) | 18 (47.4%) | |
| Stage 1 | 46 (22.0%) | 2 (5.3%) | |
| Stage 2 | 90 (43.1%) | 18 (47.4%) | |
| Stage 3 | 73 (34.9%) | 18 (47.4%) | |
| T stage | 0.046 | ||
| T1, T2 | 50 (23.9%) | 3 (7.9%) | |
| T3, T4 | 159 (76.1%) | 35 (92.1%) | |
| T1 | 8 (3.8%) | 1 (2.6%) | |
| T2 | 42 (20.1%) | 2 (5.3%) | |
| T3 | 146 (69.9%) | 32 (84.2%) | |
| T4 | 13 (6.2%) | 3 (7.9%) | |
| N stage | 0.113 | ||
| LN negative | 136 (65.1%) | 19 (50.0%) | |
| LN positive | 73 (34.9%) | 19 (50.0%) | |
| N0 | 136 (65.1%) | 19 (50.0%) | |
| N1 | 49 (23.4%) | 9 (23.7%) | |
| N2 | 24 (11.5%) | 10 (26.3%) | |
| Tumor location | 1 | ||
| Right colon | 102 (50.2%) | 17 (48.6%) | |
| Left colon | 101 (49.8%) | 18 (51.4%) | |
| Venous invasion | 0.916 | ||
| Negative | 147 (80.8%) | 25 (78.1%) | |
| Positive | 35 (19.2%) | 7 (21.9%) | |
| Lymphatic invasion | 0.887 | ||
| Negative | 140 (75.7%) | 24 (72.7%) | |
| Positive | 45 (24.3%) | 9 (27.3%) | |
| Follow up duration (months) | 38.54 ± 31.26 | 39.97 ± 22.79 | 0.739 |
| Features Used | Min. | 1st Qu. | Median | Mean | 3rd Qu. | Max. |
|---|---|---|---|---|---|---|
| Clinical features | 0.318 | 0.648 | 0.726 | 0.706 | 0.791 | 0.921 |
| Expression features | 0.449 | 0.614 | 0.691 | 0.688 | 0.774 | 0.899 |
| Methylation features | 0.447 | 0.580 | 0.686 | 0.683 | 0.772 | 0.899 |
| Expression features + Methylation features | 0.337 | 0.647 | 0.727 | 0.715 | 0.826 | 0.884 |
| Clinical features + Expression features | 0.438 | 0.609 | 0.667 | 0.673 | 0.761 | 0.832 |
| Clinical features + Methylation features | 0.333 | 0.628 | 0.704 | 0.682 | 0.757 | 0.866 |
| Clinical features + Expression features + Methylation features | 0.326 | 0.655 | 0.756 | 0.708 | 0.818 | 0.883 |
| Features | Hazard Ratio (HR) | 95% CI, Lower | 95% CI, Upper | Z Value | Adjusted p Value |
|---|---|---|---|---|---|
| Age | 1.697 | 0.609 | 4.733 | 1.011 | 0.312 |
| N stage | 2.942 | 1.253 | 6.912 | 2.477 | 0.013 |
| T stage | 0.919 | 0.237 | 3.557 | −0.122 | 0.903 |
| Gender | 0.910 | 0.390 | 2.121 | −0.219 | 0.827 |
| CEP250 (methylation) | 0.592 | 0.364 | 0.963 | −2.110 | 0.035 |
| DEFA5 (expression) | 0.786 | 0.462 | 1.337 | −0.888 | 0.374 |
| MAZ (methylation) | 0.967 | 0.809 | 1.156 | −0.369 | 0.712 |
| NINJ1 (methylation) | 1.339 | 0.910 | 1.968 | 1.482 | 0.138 |
| NLRP14 (expression) | 0.797 | 0.525 | 1.211 | −1.063 | 0.288 |
| PPARGC1A (expression) | 0.808 | 0.636 | 1.027 | −1.744 | 0.081 |
| PRG4 (expression) | 1.256 | 0.871 | 1.811 | 1.219 | 0.223 |
| PTGES (expression) | 1.399 | 0.937 | 2.087 | 1.644 | 0.100 |
| RAB21 (methylation) | 1.556 | 1.172 | 2.065 | 3.060 | 0.002 |
| TERF2IP (expression) | 1.452 | 0.893 | 2.360 | 1.502 | 0.133 |
| TMEM184A (expression) | 1.251 | 0.819 | 1.911 | 1.038 | 0.299 |
| TNFRSF18 (methylation) | 1.489 | 0.783 | 2.829 | 1.214 | 0.225 |
| TNFSF12 (methylation) | 1.132 | 0.801 | 1.600 | 0.704 | 0.481 |
| TNPO3 (methylation) | 1.465 | 1.092 | 1.967 | 2.543 | 0.011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choe, E.K.; Lee, S.; Kim, S.Y.; Shivakumar, M.; Park, K.J.; Chai, Y.J.; Kim, D. Prognostic Effect of Inflammatory Genes on Stage I–III Colorectal Cancer—Integrative Analysis of TCGA Data. Cancers 2021, 13, 751. https://doi.org/10.3390/cancers13040751
Choe EK, Lee S, Kim SY, Shivakumar M, Park KJ, Chai YJ, Kim D. Prognostic Effect of Inflammatory Genes on Stage I–III Colorectal Cancer—Integrative Analysis of TCGA Data. Cancers. 2021; 13(4):751. https://doi.org/10.3390/cancers13040751
Chicago/Turabian StyleChoe, Eun Kyung, Sangwoo Lee, So Yeon Kim, Manu Shivakumar, Kyu Joo Park, Young Jun Chai, and Dokyoon Kim. 2021. "Prognostic Effect of Inflammatory Genes on Stage I–III Colorectal Cancer—Integrative Analysis of TCGA Data" Cancers 13, no. 4: 751. https://doi.org/10.3390/cancers13040751
APA StyleChoe, E. K., Lee, S., Kim, S. Y., Shivakumar, M., Park, K. J., Chai, Y. J., & Kim, D. (2021). Prognostic Effect of Inflammatory Genes on Stage I–III Colorectal Cancer—Integrative Analysis of TCGA Data. Cancers, 13(4), 751. https://doi.org/10.3390/cancers13040751