
Lights and Shadows in Immuno-Oncology Drug Development


{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Which Are the Current Challenges with IO?
1.2. How Can We Do Better?
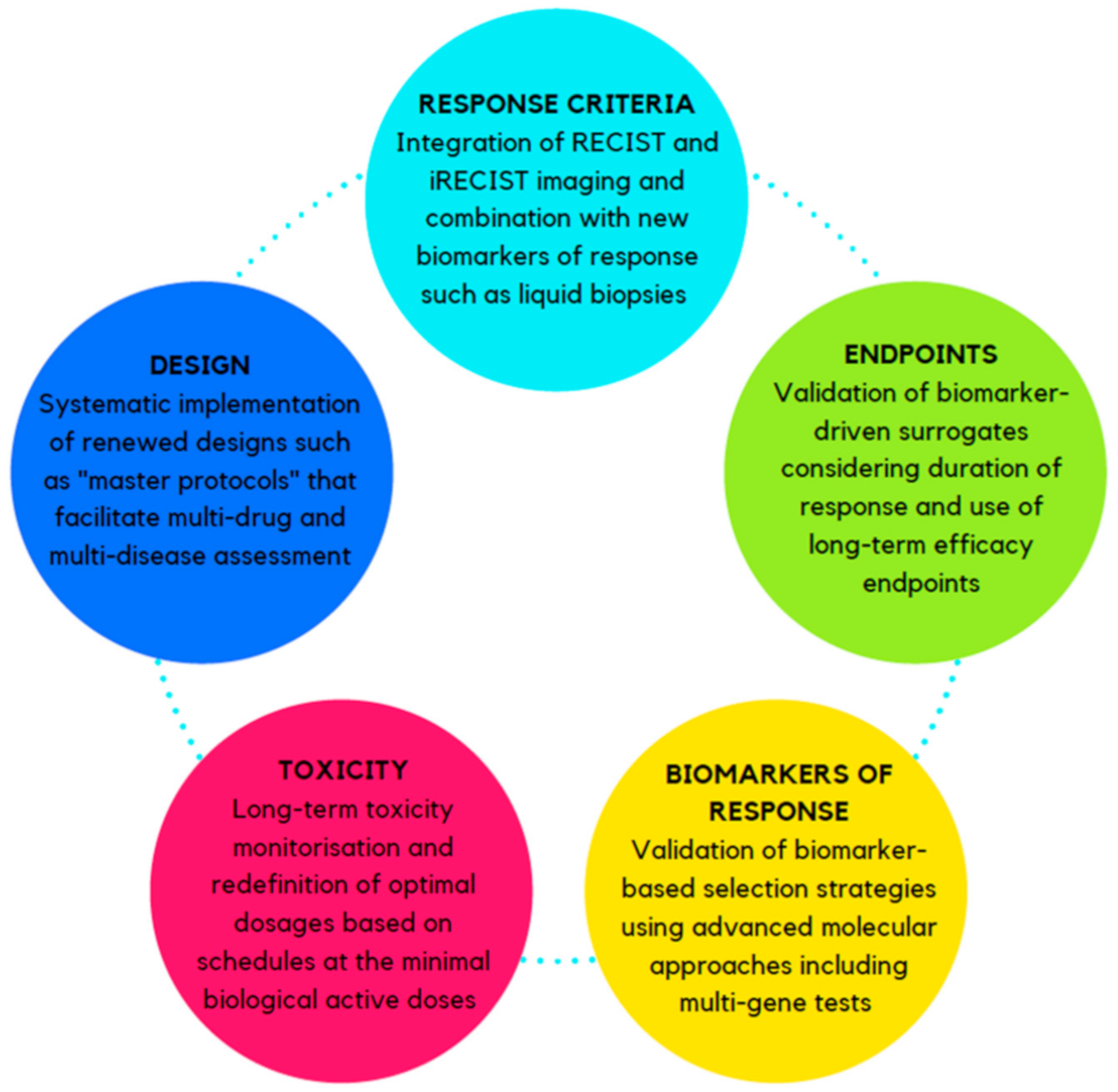
2. Challenges and Opportunities in IO Drugs Development
2.1. Response Criteria
2.2. Long-Term Efficacy Endpoints and Surrogates
2.3. Biomarkers of Response
2.4. Definition of Toxicity and Treatment Dosage
2.5. The Trial Design Itself
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Upadhaya, S.; Hubbard-Lucey, V.M.; Yu, J.X. Immuno-oncology drug development forges on despite COVID-19. Nat. Rev. Drug Discov. 2020, 19, 751–752. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Fyfe, G.; Fisher, R.I.; Rosenberg, S.A.; Sznol, M.; Parkinson, D.R.; Louie, A.C. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 1995, 13, 688–696. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.; Haanen, J.; Chen, T.-T.; Lorigan, P.; O’day, S. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20). Ann. Oncol. 2013, 24, 2694–2698. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. IMpassion130 Trial Investigators. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Van den Eynde, B.J.; Van Baren, N.; Baurain, J.F. There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256. [Google Scholar] [CrossRef]
- Anagnostou, V.; Yarchoan, M.; Hansen, A.R.; Wang, H.; Verde, F.; Sharon, E.; Collyar, D.; Chow, L.Q.M.; Forde, P.M. Immuno-oncology Trial Endpoints: Capturing Clinically Meaningful Activity. Clin. Cancer Res. 2017, 23, 4959–4969. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Atkins, M.B.; Subedi, P.; Wu, J.; Chambers, J.; Mattingly, T.J., II; Campbell, D.; Allen, J.; Ferris, A.E.; Schilsky, R.L.; et al. The promise of Immuno-oncology: Implications for defining the value of cancer treatment. J. Immunother. Cancer 2019, 7, 129. [Google Scholar] [CrossRef]
- Weber, J.S.; Hodi, F.S.; Wolchok, J.D.; Topalian, S.L.; Schadendorf, D.; Larkin, J.; Sznol, M.; Long, G.V.; Li, H.; Waxman, I.M.; et al. Safety profile of nivolumab monotherapy: A pooled analysis of patients with advanced melanoma. J. Clin. Oncol. 2017, 35, 785–792. [Google Scholar] [CrossRef]
- Johnson, D.B.; Manouchehri, A.; Haugh, A.M.; Quach, H.T.; Balko, J.M.; Lebrun-Vignes, B.; Mammen, A.; Moslehi, J.J.; Salem, J.E. Neurologic toxicity associated with immune checkpoint inhibitors: A pharmacovigilancestudy. J. Immunother. Cancer 2019, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Salem, J.E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: An observational, retrospective, pharmacovigilance study. Lancet. Oncol. 2018, 19, 1579–1589. [Google Scholar] [CrossRef]
- García-Aranda, M.; Redondo, M. Immunotherapy: A Challenge of Breast Cancer Treatment. Cancers 2019, 11, 1822. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, A.; Nanda, R. Immune Checkpoint Blockade for Breast Cancer. Cancer Treat. Res. 2018, 173, 155–165. [Google Scholar] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolcok, J.D. Immune Checkpoint Blockade in Cancer Therapy. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 34. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Barroso-Sousa, R.; Keenan, T.; Li, T.; Trippa, L.; Vaz-Luis, I.; Wulf, G.; Spring, L.; Sinclair, N.F.; Andrews, C.; et al. Effect of Eribulin With or Without Pembrolizumab on Progression-Free Survival for Patients With Hormone Receptor-Positive, ERBB2-Negative Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1598–1605. [Google Scholar] [CrossRef]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.B.; Im, S.A.; Wang, Y.; Salgado, R.; Mani, A.; et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): A phase 2, multicentre, randomised, double-blind trial. Lancet Oncol. 2020, 21, 1283–1295. [Google Scholar] [CrossRef]
- Mostafa, A.A.; Codner, D.; Hirasawa, K.; Komatsu, Y.; Young, M.N.; Steimle, V.; Drover, S. Activation of ERα signaling differentially modulates IFN-γ induced HLA-class II expression in breast cancer cells. PLoS ONE 2014, 9, e87377. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Domchek, S.M.; Clark, A.S. Immunotherapy for Breast Cancer: What Are We Missing? Clin. Cancer Res. 2017, 23, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Vafaizadeh, V.; Barekati, Z. Immuno-Oncology Biomarkers for Personalized Immunotherapy in Breast Cancer, Personalized Immunotherapy in Breast Cancer. Front. Cell. Dev. Biol. 2020, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Ben-Aharon, O.; Magnezi, R.; Leshno, M.; Goldstein, D. Association of Immunotherapy With Durable Survival as Defined by Value Frameworks for Cancer Care. JAMA Oncol. 2018, 4, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Hoos, A.; Parmiani, G.; Hege, K.; Sznol, M.; Loibner, H.; Eggermont, A.; Urba, W.; Blumenstein, B.; Sacks, N.; Keilholz, U.; et al. Vaccine Clinical Trial Working Group. A clinical development paradigm for cancer vaccines and related biologics. J. Immunother. 2007, 30, 1–15. [Google Scholar] [CrossRef]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. RECIST working group. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, 43–52. [Google Scholar] [CrossRef]
- Chai, L.F.; Prince, E.; Pillarisetty, V.; Katz, S. Challenges in assessing solid tumor responses to immunotherapy. Cancer Gene Ther. 2020, 27, 528–538. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Levit, L.A.; Schilsky, R.L.; Averbuch, S.D.; Chen, D.; Kirkwood, J.M.; McShane, L.M.; Sharon, E.; Mileham, K.F.; Postow, M.A. Trial Reporting in Immuno-Oncology (TRIO): An American Society of Clinical Oncology-Society for Immunotherapy of Cancer Statement. J. Clin. Oncol. 2019, 37, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.; Xu, W.; Lopez, J. Personalized Cancer Immunotherapy: Today’s Challenge and Tomorrow’s Promise. JIPO 2018, 1, 56–67. [Google Scholar]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Iijima, Y.; Hirotsu, Y.; Amemiya, K.; Ooka, Y.; Mochizuki, H.; Oyama, T.; Nakagomi, T.; Uchida, Y.; Kobayashi, Y.; Tsutsui, T.; et al. Very early response of circulating tumour–derived DNA in plasma predicts efficacy of nivolumab treatment in patients with non–small cell lung cancer. Eur. J. Cancer 2017, 86, 349–357. [Google Scholar] [CrossRef]
- Boland, G.M.; Flaherty, K.T. Tracking early response to immunotherapy. Nat. Cancer 2020, 1, 160–162. [Google Scholar] [CrossRef]
- Valpione, S.; Galvani, E.; Tweedy, J.; Mundra, P.A.; Banyard, A.; Middlehurst, P.; Barry, J.; Mills, S.; Salih, Z.; Weightman, J.; et al. Immune awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat. Cancer 2020, 1, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.; Kudchadkar, R.R. Predictive and on-treatment monitoring biomarkers in advanced melanoma: Moving toward personalized medicine. Cancer Treat. Rev. 2018, 71, 8–18. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Haddad, R.; Gupta, S.; Mahipal, A.; Mehra, R.; Tahara, M.; Berger, R.; Eder, J.P.; Burtness, B.; Lee, S.H.; et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef]
- Smith, I.; Robertson, J.; Kilburn, L.; Wilcox, M.; Evans, A.; Holcombe, C.; Horgan, K.; Kirwan, C.; Mallon, E.; Sibbering, M.; et al. Long-term outcome and prognostic value of Ki67 after perioperative endocrine therapy in postmenopausal women with hormone-sensitive early breast cancer (POETIC): An open-label, multicentre, parallel-group, randomised, phase 3 trial. Lancet Oncol. 2020, 21, 1443–1454. [Google Scholar] [CrossRef]
- Gyawali, B.; Hey, S.P.; Kesselheim, A.S. A comparison of response patterns for progression-free survival and overall survival following treatment for cancer with PD-1 Inhibitors. A meta-analysis of correlation and differences in effect sizes. JAMA Netw. Open 2018, 1, e180416. [Google Scholar] [CrossRef]
- Mushti, S.L.; Mulkey, F.; Sridhara, R. Evaluation of Overall Response Rate and Progression-Free Survival as Potential Surrogate Endpoints for Overall Survival in Immunotherapy Trials. Clin. Cancer Res. 2018, 24, 2268–2275. [Google Scholar] [CrossRef]
- Kok, P.S.; Cho, D.; Yoon, W.H.; Ritchie, G.; Marschner, I.; Lord, S.; Friedlander, M.; Simes, J.; Lee, C.K. Validation of Progression-Free Survival Rate at 6 Months and Objective Response for Estimating Overall Survival in Immune Checkpoint Inhibitor Trials: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2011809. [Google Scholar] [CrossRef]
- Smoragiewicz, M.; Bogaerts, J.; Calvo, E.; Marabelle, A.; Perrone, A.; Seymour, L.; Shalabi, A.; Siu, L.L.; Tabernero, J.; Giaccone, G.; et al. Design and conduct of early clinical studies of immunotherapy agent combinations: Recommendations from the task force on Methodology for the Development of Innovative Cancer Therapies. Ann. Oncol. 2018, 29, 2175–2182. [Google Scholar] [CrossRef]
- Chen, T.T. Milestone Survival: A Potential Intermediate Endpoint for Immune Checkpoint Inhibitors. J. Natl. Cancer Inst. 2015, 107, djv156. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Andtbacka, R.H.I.; Collichio, F.A.; Wolf, M.; Zhao, Z.; Shilkrut, M.; Puzanov, I.; Ross, M. Durable response rate as an endpoint in cancer immunotherapy: Insights from oncolytic virus clinical trials. J. Immunother. Cancer 2017, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- McKean, W.B.; Moser, J.C.; Rimm, D.; Hu-Lieskovan, S. Biomarkers in Precision Cancer Immunotherapy: Promise and Challenges. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e275–e291. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Lazar, V.; Leyland-Jones, B.; Schilsky, R.L.; Mendelsohn, J.; Kurzrock, R. Association of Biomarker-Based Treatment Strategies With Response Rates and Progression-Free Survival in Refractory Malignant Neoplasms: A Meta-analysis. JAMA Oncol. 2016, 2, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.E.; Rouleau, E.; Besse, B. Clinical utility of tumor mutational burden in patients with non-small cell lung cancer treated with immunotherapy. Transl. Lung Cancer Res. 2018, 7, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Dahlberg, S.E.; Adeni, A.; Sholl, L.M.; Nishino, M.; Awad, M.M. Immune Checkpoint Inhibitor Outcomes for Patients With Non-Small-Cell Lung Cancer Receiving Baseline Corticosteroids for Palliative Versus Nonpalliative Indications. J. Clin. Oncol. 2019, 31, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Crown, J. Biomarkers for Predicting Response to Immunotherapy with Immune Checkpoint Inhibitors in Cancer Patients. Clin. Chem. 2019, 65, 1228–1238. [Google Scholar] [CrossRef]
- Higgs, B.W.; Morehouse, C.A.; Streicher, K.; Brohawn, P.Z.; Pilatazi, F.; Gupta, A.; Ranade, K. Interferon Gamma Messenger RNA Signature in Tumor Biopsies Predicts Outcomes in Patients with Non-Small Cell Lung Carcinoma or Urothelial Cancer Treated with Durvalumab. Clin. Cancer Res. 2018, 24, 3857–3866. [Google Scholar] [CrossRef]
- Anurag, M.; Zhu, M.; Huang, C.; Vasaikar, S.; Wang, J.; Hoog, J.; Burugu, S.; Gao, D.; Suman, V.; Zhang, X.H.; et al. Immune Checkpoint Profiles in Luminal B Breast Cancer (Alliance). J. Natl. Cancer Inst. 2020, 112, 737–746. [Google Scholar] [CrossRef]
- Bergamino, M.; Morani, G.; Parker, J.; Schuster, G.; López-Knowles, E.; Bliss, J.; Dowsett, M.; Cheang, M.C.U. Deconvolution of Gene Expression Profiles Identifies Differentially Expressed Immune-Related Gene Profiles on Aromatase Inhibitor-Resistant Estrogen Receptor Positive Breast Cancer Tumour; National Cancer Research Institute Showcase: London, UK, 2020; Abstract number 3224. [Google Scholar]
- Terranova-Barberio, M.; Pawlowska, N.; Dhawan, M.; Moasser, M.; Chien, A.J.; Melisko, M.E.; Rugo, H.; Rahimi, R.; Deal, T.; Daud, A. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat. Commun. 2020, 11, 3584. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Wages, N.A.; Fadul, C.E. Adaptive dose-finding based on safety and feasibility in early-phase clinical trials of adoptive cell immunotherapy. Clin. Trials. 2020, 17, 157–165. [Google Scholar] [CrossRef]
- Baik, C.S.; Rubin, E.H.; Forde, P.M.; Mehnert, J.M.; Collyar, D.; Butler, M.O.; Dixon, E.L.; Chow, L.Q.M. Immuno-oncology Clinical Trial Design: Limitations, Challenges, and Opportunities. Clin. Cancer Res. 2017, 2, 4992–5002. [Google Scholar] [CrossRef]
- Mazzarella, L.; Duso, B.A.; Trapani, D.; Belli, C.; D’Amico, P.; Ferraro, E.; Viale, G.; Curigliano, G. The evolving landscape of ’next-generation’ immune checkpoint inhibitors: A review. Eur. J. Cancer 2019, 117, 14–31. [Google Scholar] [CrossRef]
- Mazzarella, L.; Morganti, S.; Marra, A.; Trapani, D.; Tini, G.; Pelicci, P.; Curigliano, G. Master protocols in immuno-oncology: Do novel drugs deserve novel designs? J. Immunother. Cancer 2020, 8, e000475. [Google Scholar] [CrossRef]
- Garralda, E.; Dienstmann, R.; Piris-Giménez, A.; Braña, I.; Rodon, J.; Tabernero, J. New clinical trial designs in the era of precision medicine. Mol. Oncol. 2019, 13, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Simon, R. Critical Review of Umbrella, Basket, and Platform Designs for Oncology Clinical Trials. Clin. Pharmacol. Ther. 2017, 102, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.H.; Siden, E.; Zoratti, M.J.; Dron, L.; Harari, O.M.; Singer, J.; Lester, R.T.; Thorlund, K.; Mills, E.J. Systematic review of basket trials, umbrella trials, and platform trials: A landscape analysis of master protocols. Trials 2020, 20, 572. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergamino Sirvén, M.; Pernas, S.; Cheang, M.C.U. Lights and Shadows in Immuno-Oncology Drug Development. Cancers 2021, 13, 691. https://doi.org/10.3390/cancers13040691
Bergamino Sirvén M, Pernas S, Cheang MCU. Lights and Shadows in Immuno-Oncology Drug Development. Cancers. 2021; 13(4):691. https://doi.org/10.3390/cancers13040691
Chicago/Turabian StyleBergamino Sirvén, Milana, Sonia Pernas, and Maggie C. U. Cheang. 2021. "Lights and Shadows in Immuno-Oncology Drug Development" Cancers 13, no. 4: 691. https://doi.org/10.3390/cancers13040691
APA StyleBergamino Sirvén, M., Pernas, S., & Cheang, M. C. U. (2021). Lights and Shadows in Immuno-Oncology Drug Development. Cancers, 13(4), 691. https://doi.org/10.3390/cancers13040691