
Photodynamic Therapy Using Hippo Pathway Inhibitor Verteporfin: A Potential Dual Mechanistic Approach in Treatment of Soft Tissue Sarcomas

 , ,
, , 

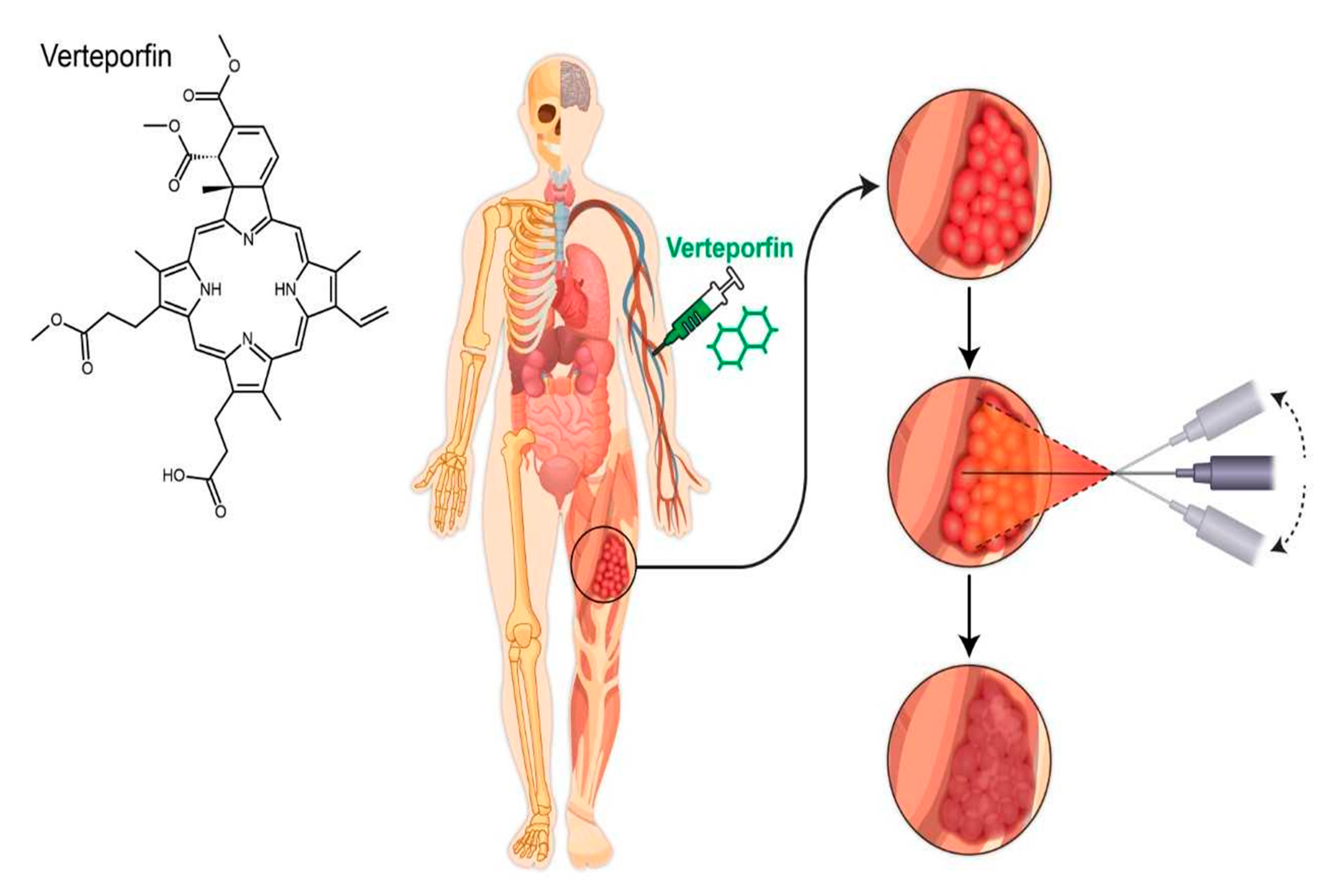{kind=link}
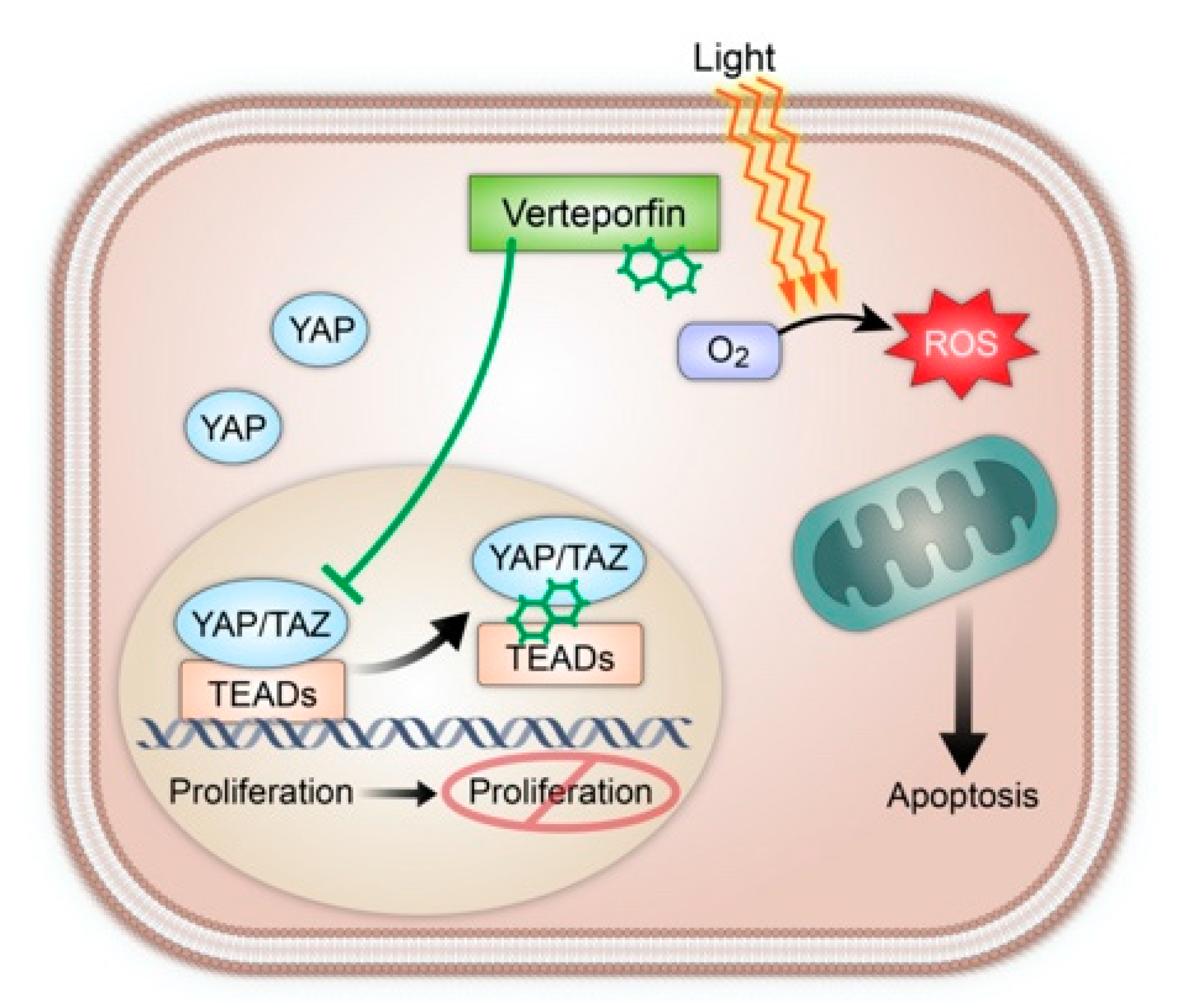{kind=link}
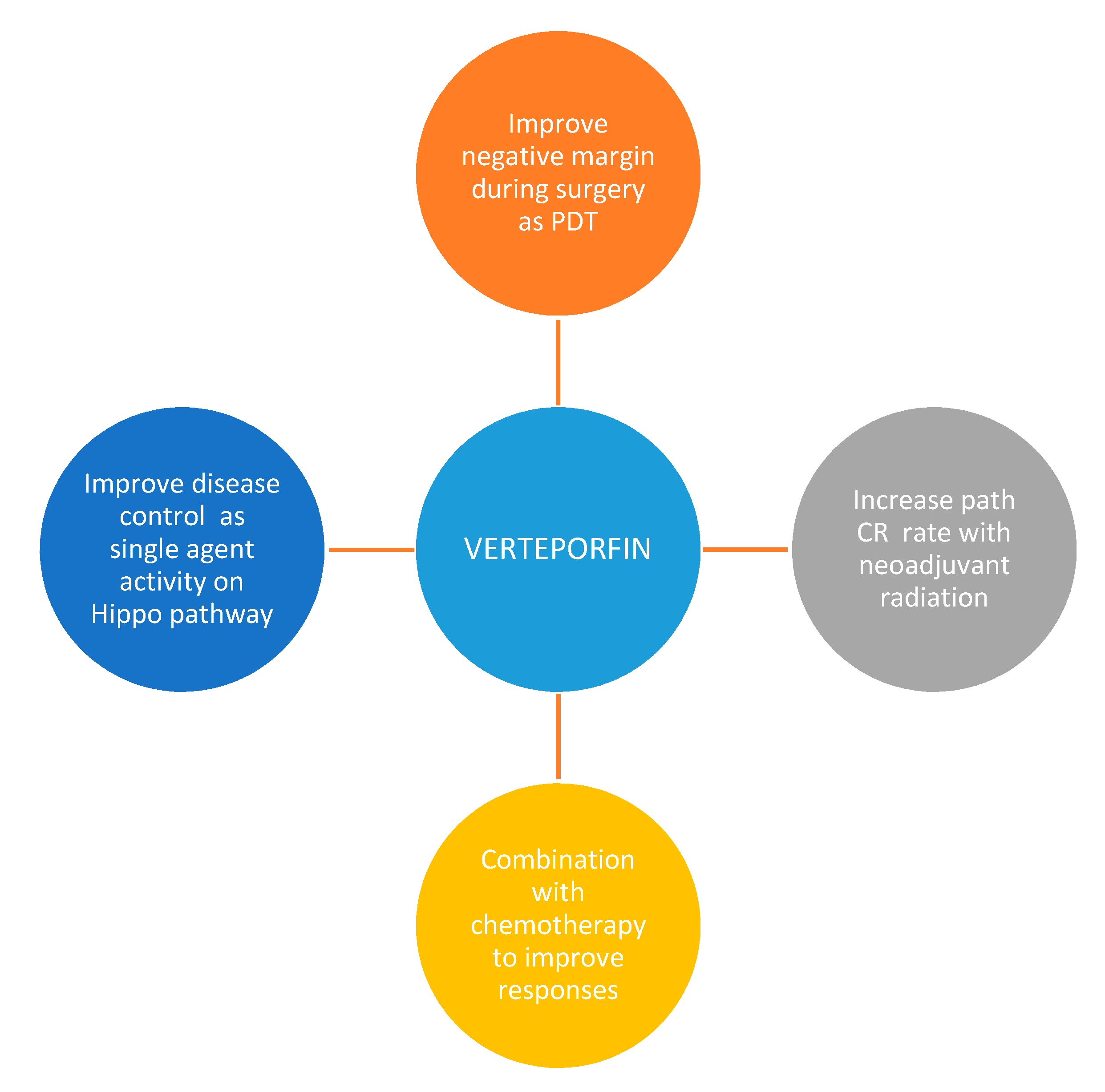{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Photodynamic Therapy in Clinical Studies
3. Considerations in Photodynamic Therapy Delivery in Soft Tissue Sarcomas
4. Why Use Photodynamic Therapy in Soft Tissue Sarcoma?
5. Hippo Pathway in Sarcoma and Role of Verteporfin
6. Proposed Delivery of Light Source in Soft Tissue Sarcoma
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daniell, M.D.; Hill, J.S. A History of Photodynamic Therapy. ANZ J. Surg. 1991, 61, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic Therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef]
- Wang, H.P.; Qian, S.Y.; Schafer, F.Q.; Domann, F.E.; Oberley, L.W.; Buettner, G.R. Phospholipid hydroperoxide glutathione peroxidase protects against singlet oxygen-induced cell damage of photodynamic therapy. Free. Radic. Biol. Med. 2001, 30, 825–835. [Google Scholar] [CrossRef]
- Gomer, C.J.; Razum, N.J. Acute Skin Response in Albino Mice Following Porphyrin Photosensitization Under Oxic and Anoxic Conditions. Photochem. Photobiol. 1984, 40, 435–439. [Google Scholar] [CrossRef]
- Kleinovink, J.W.; Van Driel, P.B.; Snoeks, T.J.; Prokopi, N.; Fransen, M.F.; Cruz, L.J.; Mezzanotte, L.; Chan, A.; Lowik, C.; Ossendorp, F. Combination of Photodynamic Therapy and Specific Immunotherapy Efficiently Eradicates Established Tumors. Clin. Cancer Res. 2016, 22, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Bown, S.G. Photodynamic therapy for photochemists. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2013, 371, 20120371. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Ortel, B.J.; Pereira, S.P.; Hasan, T.; Maytin, E.V. Biomodulatory approaches to photodynamic therapy for solid tumors. Cancer Lett. 2012, 326, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Taneja, S.S.; Bennett, J.; Coleman, J.; Grubb, R.; Andriole, G.; Reiter, R.E.; Marks, L.; Azzouzi, A.-R.; Emberton, M. Final Results of a Phase I/II Multicenter Trial of WST11 Vascular Targeted Photodynamic Therapy for Hemi-Ablation of the Prostate in Men with Unilateral Low Risk Prostate Cancer Performed in the United States. J. Urol. 2016, 196, 1096–1104. [Google Scholar] [CrossRef]
- Noweski, A.; Roosen, A.; Lebdai, S.; Barret, E.; Emberton, M.; Benzaghou, F.; Apfelbeck, M.; Gaillac, B.; Gratzke, C.; Stief, C.; et al. Medium-term Follow-up of Vascular-targeted Photodynamic Therapy of Localized Prostate Cancer Using TOOKAD Soluble WST-11 (Phase II Trials). Eur. Urol. Focus 2019, 5, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.M.; Stepp, H.; Beyer, W.; Pongratz, T.; Sroka, R.; Kriegmair, M.; Zaak, D.; Welschof, M.; Tilki, D.; Stief, C.G.; et al. Photodynamic Therapy of Bladder Cancer—A Phase I Study Using Hexaminolevulinate (HAL). Urol. Oncol. Semin. Orig. Investig. 2013, 31, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, M.A.; Kavar, B.; Hill, J.S.; Morgan, D.J.; Nation, R.L.; Stylli, S.S.; Basser, R.L.; Uren, S.; Geldard, H.; Green, M.D.; et al. Phase I and Pharmacokinetic Study of Photodynamic Therapy for High-Grade Gliomas Using a Novel Boronated Porphyrin. J. Clin. Oncol. 2001, 19, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Muto, M.; Yoshimura, K.; Niimi, M.; Ezoe, Y.; Yoda, Y.; Yamamoto, Y.; Nishisaki, H.; Higashino, K.; Iishi, H. Phase I study of photodynamic therapy using talaporfin sodium and diode laser for local failure after chemoradiotherapy for esophageal cancer. Radiat. Oncol. 2012, 7, 113. [Google Scholar] [CrossRef]
- Stoker, S.; Indrasari, S.; Herdini, C.; Hariwiyanto, B.; Karakullukcu, B.; Dhamiyati, W.; Widayati, K.; Romdhoni, A.; Fles, R.; Haryana, S.; et al. Photodynamic therapy as salvage therapy for patients with nasopharyngeal carcinoma experiencing local failures following definitive radiotherapy. Photodiagn. Photodyn. Ther. 2015, 12, 519–525. [Google Scholar] [CrossRef]
- DeWitt, J.; Sandrasegaran, K.; O’Neil, B.; House, M.G.; Zyromski, N.J.; Sehdev, A.; Perkins, S.M.; Flynn, J.; McCranor, L.; Shahda, S. Phase 1 study of EUS-guided photodynamic therapy for locally advanced pancreatic cancer. Gastrointest. Endosc. 2019, 89, 390–398. [Google Scholar] [CrossRef]
- Shafirstein, G.; Rigual, N.R.; Arshad, H.; Cooper, M.T.; Bellnier, D.A.; Wilding, G.; Tan, W.; Merzianu, M.; Henderson, B.W. Photodynamic therapy with 3-(1′-hexyloxyethyl) pyropheophorbide-a for early-stage cancer of the larynx: Phase Ib study. Head Neck 2015, 38, E377–E383. [Google Scholar] [CrossRef]
- Dhillon, S.S.; Demmy, T.L.; Yendamuri, S.; Loewen, G.M.; Nwogu, C.E.; Cooper, M.T.; Henderson, B.W. A Phase I Study of Light Dose for Photodynamic Therapy Using 2-[1-Hexyloxyethyl]-2 Devinyl Pyropheophorbide-a for the Treatment of Non–Small Cell Carcinoma In Situ or Non–Small Cell Microinvasive Bronchogenic Carcinoma: A Dose Ranging Study. J. Thorac. Oncol. 2016, 11, 234–241. [Google Scholar] [CrossRef]
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W.; et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 2014, 110, 1698–1704. [Google Scholar] [CrossRef]
- Hahn, S.M.; Fraker, D.L.; Mick, R.; Metz, J.; Busch, T.M.; Smith, D.; Zhu, T.; Rodriguez, C.; Dimofte, A.; Spitz, F.; et al. A Phase II Trial of Intraperitoneal Photodynamic Therapy for Patients with Peritoneal Carcinomatosis and Sarcomatosis. Clin. Cancer Res. 2006, 12, 2517–2525. [Google Scholar] [CrossRef]
- Park, D.H.; Lee, S.S.; Park, S.E.; Lee, J.L.; Choi, J.H.; Choi, H.J.; Jang, J.W.; Kim, H.J.; Eum, J.B.; Seo, D.-W.; et al. Randomised phase II trial of photodynamic therapy plus oral fluoropyrimidine, S-1, versus photodynamic therapy alone for unresectable hilar cholangiocarcinoma. Eur. J. Cancer 2014, 50, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Ortner, M.-A. Photodynamic therapy in cholangiocarcinomas. Best Pr. Res. Clin. Gastroenterol. 2004, 18, 147–154. [Google Scholar] [CrossRef]
- Ortner, M.E.; Liebetruth, J.; Schreiber, S.; Hanft, M.; Wruck, U.; Fusco, V.; Müller, J.M.; Hörtnagl, H.; Lochs, H. Photodynamic therapy of nonresectable cholangiocarcinoma. Gastroenterology 1998, 114, 536–542. [Google Scholar] [CrossRef]
- Berr, F.; Wiedmann, M.; Tannapfel, A.; Halm, U.; Kohlhaw, K.; Schmidt, F.; Wittekind, C.; Hauss, J.; Mössner, J. Photodynamic therapy for advanced bile duct cancer: Evidence for improved palliation and extended survival. Hepatology 2000, 31, 291–298. [Google Scholar] [CrossRef]
- Dumoulin, F.L.; Gerhardt, T.; Fuchs, S.; Scheurlen, C.; Neubrand, M.; Layer, G.; Sauerbruch, T. Phase II study of photodynamic therapy and metal stent as palliative treatment for nonresectable hilar cholangiocarcinoma. Gastrointest. Endosc. 2003, 57, 860–867. [Google Scholar] [CrossRef]
- Wagner, A.; Denzer, U.W.; Neureiter, D.; Kiesslich, T.; Puespoeck, A.; Rauws, E.A.J.; Emmanuel, K.; Degenhardt, N.; Frick, U.; Beuers, U.; et al. Temoporfin improves efficacy of photodynamic therapy in advanced biliary tract carcinoma: A multicenter prospective phase II study. Hepatology 2015, 62, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Inage, K.; Sakuma, Y.; Yamauchi, K.; Suganami, A.; Orita, S.; Kubota, G.; Oikawa, Y.; Sainoh, T.; Sato, J.; Fujimoto, K.; et al. Effect of photodynamic therapy on local muscle treatment in a rat muscle injury model: A controlled trial. J. Orthop. Surg. Res. 2015, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fekrazad, R.; Asefi, S.; Khorsandi, K.; Nejatifard, M. Photo biostimulatory effect of low dose photodynamic therapy on human mesenchymal stem cells. Photodiagn. Photodyn. Ther. 2020, 31, 101886. [Google Scholar] [CrossRef] [PubMed]
- Rigual, N.R.; Shafirstein, G.; Frustino, J.; Seshadri, M.; Cooper, M.; Wilding, G.; Sullivan, M.A.; Henderson, B. Adjuvant Intraoperative Photodynamic Therapy in Head and Neck Cancer. JAMA Otolaryngol. Neck Surg. 2013, 139, 706–711. [Google Scholar] [CrossRef]
- Borgia, F.; Giuffrida, G.; CaraDonna, E.; Vaccaro, M.; Guarneri, F.; Cannavò, S.P. Early and Late Onset Side Effects of Photodynamic Therapy. Biomedicines 2018, 6, 12. [Google Scholar] [CrossRef]
- Crompton, J.G.; Ogura, K.; Bernthal, N.M.; Kawai, A.; Eilber, F.C. Local Control of Soft Tissue and Bone Sarcomas. J. Clin. Oncol. 2018, 36, 111–117. [Google Scholar] [CrossRef]
- Survival Rates for Soft Tissue Sarcoma. American Cancer Society. 2021. Available online: https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/survival-rates.html (accessed on 2 February 2021).
- Haas, R.L.; Gronchi, A.; Van De Sande, M.A.; Baldini, E.H.; Gelderblom, H.; Messiou, C.; Wardelmann, E.; Le Cesne, A. Perioperative Management of Extremity Soft Tissue Sarcomas. J. Clin. Oncol. 2018, 36, 118–124. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, T.; Li, Z. Progress of photodynamic therapy applications in the treatment of musculoskeletal sarcoma (Review). Oncol. Lett. 2014, 8, 1403–1408. [Google Scholar] [CrossRef]
- Kusuzaki, K.; Murata, H.; Matsubara, T.; Satonaka, H.; Wakabayashi, T.; Matsumine, A.; Uchida, A. Acridine orange could be an innovative anticancer agent under photon energy. Vivo 2007, 21, 205–214. [Google Scholar]
- Schaefer, I.-M.; Hornick, J.L.; Barysauskas, C.M.; Raut, C.P.; Patel, S.A.; Royce, T.J.; Fletcher, C.D.; Baldini, E.H. Histologic Appearance After Preoperative Radiation Therapy for Soft Tissue Sarcoma: Assessment of the European Organization for Research and Treatment of Cancer–Soft Tissue and Bone Sarcoma Group Response Score. Int. J. Radiat. Oncol. 2017, 98, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Pogue, B.W.; O’Hara, J.A.; Demidenko, E.; Wilmot, C.M.; Goodwin, I.A.; Chen, B.; Swartz, H.M.; Hasan, T. Photodynamic therapy with verteporfin in the radiation-induced fibrosarcoma-1 tumor causes enhanced radiation sensitivity. Cancer Res. 2003, 63, 1025–1033. [Google Scholar] [PubMed]
- Kim, W.; Jho, E.-H. The history and regulatory mechanism of the Hippo pathway. BMB Rep. 2018, 51, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.-Y.; Yu, J.; Guan, K.-L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef]
- Pantalacci, S.; Tapon, N.; Léopold, P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 2003, 5, 921–927. [Google Scholar] [CrossRef]
- Deel, M.D.; Li, J.; Crose, L.E.S.; Linardic, C.M. A Review: Molecular Aberrations within Hippo Signaling in Bone and Soft-Tissue Sarcomas. Front. Oncol. 2015, 5, 190. [Google Scholar] [CrossRef]
- Lamar, J.M.; Xiao, Y.; Lamar, J.M. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers 2018, 10, 115. [Google Scholar] [CrossRef]
- Fullenkamp, C.A.; Hall, S.L.; Jaber, O.I.; Pakalniskis, B.L.; Savage, E.C.; Savage, J.M.; Ofori-Amanfo, G.K.; Lambertz, A.M.; Ivins, S.D.; Stipp, C.S.; et al. TAZ and YAP are frequently activated oncoproteins in sarcomas. Oncotarget 2016, 7, 30094–30108. [Google Scholar] [CrossRef]
- Isfort, I.; Elges, S.; Cyra, M.; Berthold, R.; Renner, M.; Mechtersheimer, G.; Åman, P.; Larsson, O.; Ratner, N.; Hafner, S.; et al. Prevalence of the Hippo Effectors YAP1/TAZ in Tumors of Soft Tissue and Bone. Sci. Rep. 2019, 9, 19704–19709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bai, H.; David, K.K.; Dong, J.; Zheng, Y.; Cai, J.; Giovannini, M.; Liu, P.; Anders, R.A.; Pan, D. The Merlin/NF2 Tumor Suppressor Functions through the YAP Oncoprotein to Regulate Tissue Homeostasis in Mammals. Dev. Cell 2010, 19, 27–38. [Google Scholar] [CrossRef]
- Deel, M.D.; Slemmons, K.K.; Hinson, A.R.; Genadry, K.C.; Burgess, B.A.; Crose, L.E.; Kuprasertkul, N.; Oristian, K.M.; Bentley, R.C.; Linardic, C.M. The Transcriptional Coactivator TAZ Is a Potent Mediator of Alveolar Rhabdomyosarcoma Tumorigenesis. Clin. Cancer Res. 2018, 24, 2616–2630. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The Hippo Transducer YAP1 Transforms Activated Satellite Cells and Is a Potent Effector of Embryonal Rhabdomyosarcoma Formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Eisinger-Mathason, T.S.K.; Mucaj, V.; Biju, K.M.; Nakazawa, M.S.; Gohil, M.; Cash, T.P.; Yoon, S.S.; Skuli, N.; Park, K.M.; Gerecht, S.; et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E3402–E3411. [Google Scholar] [CrossRef]
- Isfort, I.; Cyra, M.; Elges, S.; Kailayangiri, S.; Altvater, B.; Rossig, C.; Steinestel, K.; Grünewald, I.; Huss, S.; Eßeling, E.; et al. SS18-SSX–Dependent YAP/TAZ Signaling in Synovial Sarcoma. Clin. Cancer Res. 2019, 25, 3718–3731. [Google Scholar] [CrossRef]
- Trautmann, M.; Cheng, Y.; Jensen, P.; Azoitei, N.; Brunner, I.; Hüllein, J.; Slabicki, M.; Isfort, I.; Cyra, M.; Berthold, R.; et al. Requirement for YAP1 signaling in myxoid liposarcoma. EMBO Mol. Med. 2019, 11, e9889. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2017; based on November 2019 SEER data submission, posted to the SEER web site; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 2 February 2021).
- Wei, C.; Li, X. The Role of Photoactivated and Non-Photoactivated Verteporfin on Tumor. Front. Pharmacol. 2020, 11, 557429. [Google Scholar] [CrossRef]
- Morton, C.; Szeimies, R.-M.; Sidoroff, A.; Braathen, L. European guidelines for topical photodynamic therapy part 1: Treatment delivery and current indications—actinic keratoses, Bowen’s disease, basal cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Bansal, A.; Yang, F.; Xi, T.; Zhang, Y.; Ho, J.S. In vivo wireless photonic photodynamic therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 1469–1474. [Google Scholar] [CrossRef]
- Kim, A.; Zhou, J.; Samaddar, S.; Song, S.H.; Elzey, B.D.; Thompson, D.H.; Ziaie, B. An Implantable Ultrasonically-Powered Micro-Light-Source (µLight) for Photodynamic Therapy. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shafirstein, G.; Bellnier, D.A.; Oakley, E.; Hamilton, S.; Potasek, M.; Beeson, K.; Parilov, E. Interstitial Photodynamic Therapy—A Focused Review. Cancers 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rytlewski, J.D.; Scalora, N.; Garcia, K.; Tanas, M.; Toor, F.; Miller, B.; Allen, B.; Milhem, M.; Monga, V. Photodynamic Therapy Using Hippo Pathway Inhibitor Verteporfin: A Potential Dual Mechanistic Approach in Treatment of Soft Tissue Sarcomas. Cancers 2021, 13, 675. https://doi.org/10.3390/cancers13040675
Rytlewski JD, Scalora N, Garcia K, Tanas M, Toor F, Miller B, Allen B, Milhem M, Monga V. Photodynamic Therapy Using Hippo Pathway Inhibitor Verteporfin: A Potential Dual Mechanistic Approach in Treatment of Soft Tissue Sarcomas. Cancers. 2021; 13(4):675. https://doi.org/10.3390/cancers13040675
Chicago/Turabian StyleRytlewski, Jeffrey D., Nicholas Scalora, Keith Garcia, Munir Tanas, Fatima Toor, Benjamin Miller, Bryan Allen, Mohammed Milhem, and Varun Monga. 2021. "Photodynamic Therapy Using Hippo Pathway Inhibitor Verteporfin: A Potential Dual Mechanistic Approach in Treatment of Soft Tissue Sarcomas" Cancers 13, no. 4: 675. https://doi.org/10.3390/cancers13040675
APA StyleRytlewski, J. D., Scalora, N., Garcia, K., Tanas, M., Toor, F., Miller, B., Allen, B., Milhem, M., & Monga, V. (2021). Photodynamic Therapy Using Hippo Pathway Inhibitor Verteporfin: A Potential Dual Mechanistic Approach in Treatment of Soft Tissue Sarcomas. Cancers, 13(4), 675. https://doi.org/10.3390/cancers13040675