
Paediatric Gliomas: BRAF and Histone H3 as Biomarkers, Therapy and Perspective of Liquid Biopsies

 and
and
Abstract
Simple Summary
Abstract
1. Introduction
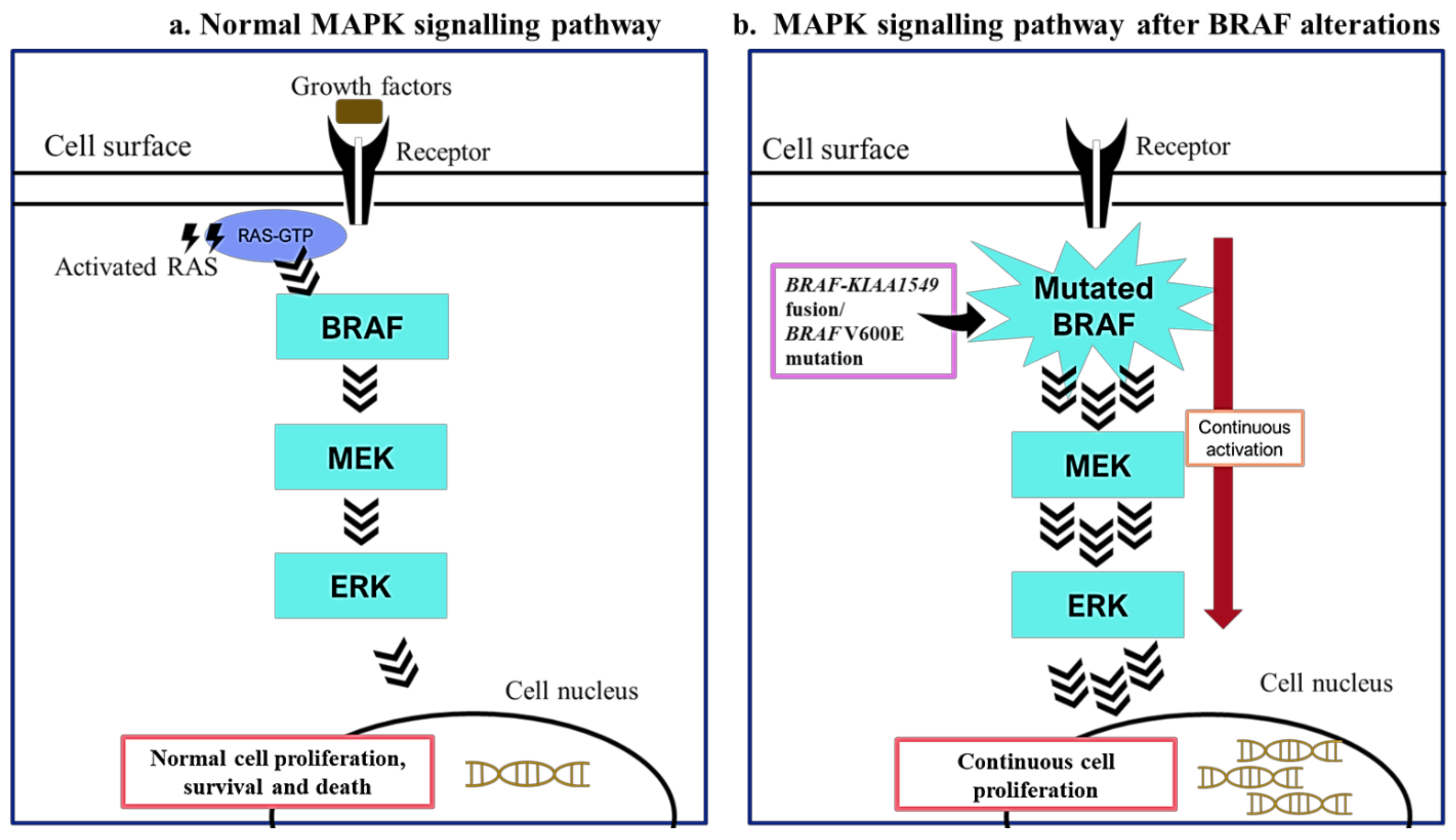
2. BRAF Mutations in Paediatric Low-Grade Gliomas
3. BRAF Mutations as Biomarkers in Paediatric Low-Grade Gliomas
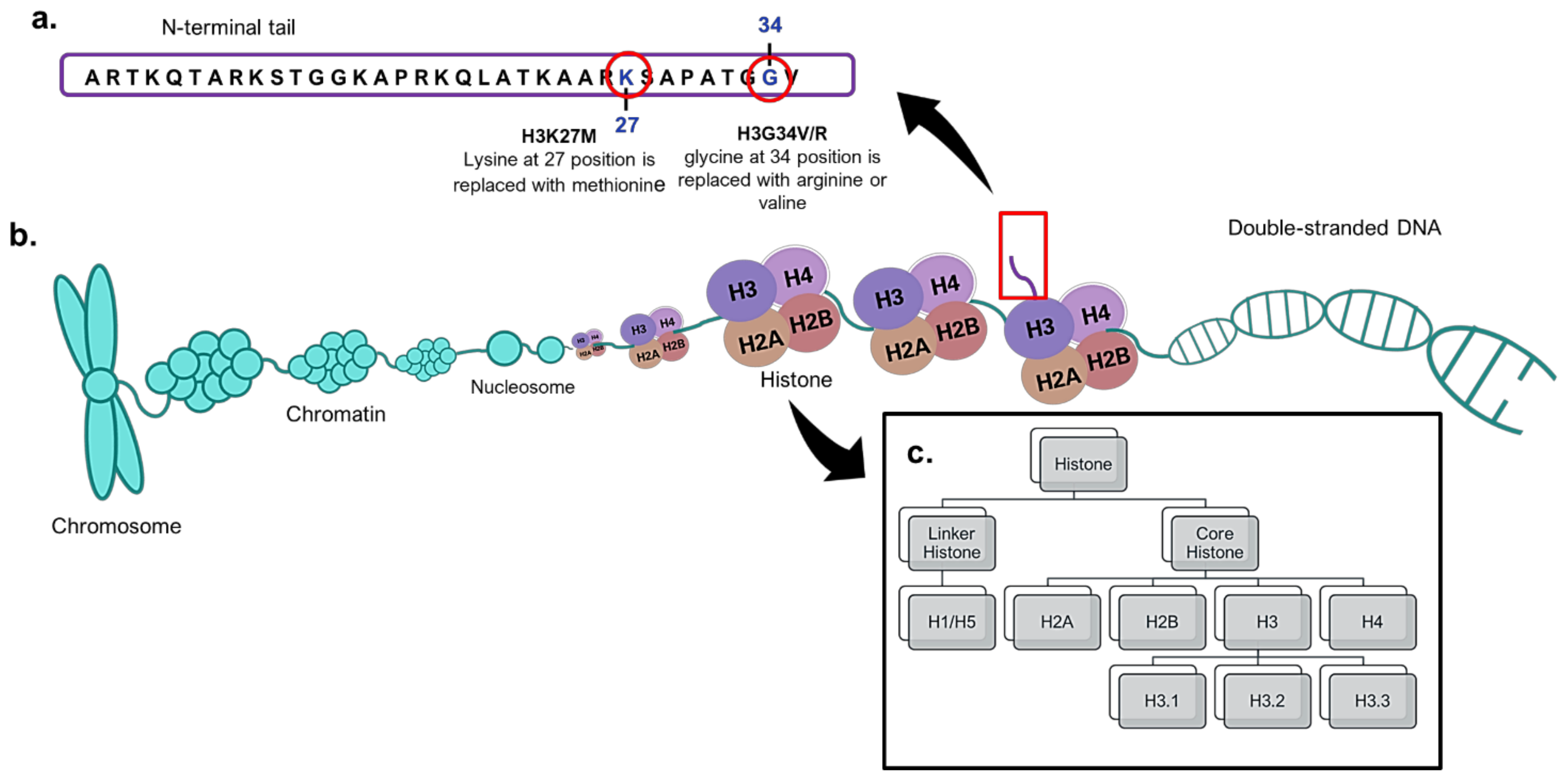
4. Histone H3 Mutations in Paediatric High-Grade Gliomas
5. H3K27M Mutations as Biomarkers in Paediatric High-Grade Gliomas
6. Perspective of Liquid Biopsy
7. Therapy in Paediatric Gliomas
BRAF Inhibitors in pLGGs with BRAF V600E
8. HDAC Therapy in pHGGs and Diffuse Intrinsic Pontine Glioma
9. Challenges in Paediatric Gliomas
10. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BBB | blood–brain barrier |
| CI | confidence interval |
| CSF | cerebrospinal fluid |
| ERK | extracellular regulated kinase |
| FISH | fluorescence in situ hybridisation |
| HDAC | histone deacetylase |
| IDH | isocitrate dehydrogenase |
| MAPK | mitogen-activated protein kinase |
| MGMT | O-6-methylguanine-DNA methyltransferase |
| pLGGs | paediatric low-grade gliomas |
| BRAF | B-Raf Proto-Oncogene or V-Raf Murine Sarcoma Viral Oncogene Homolog B |
| CNS | central nervous system |
| EGFR | epidermal growth factor receptor |
| FFPE | formalin-fixed paraffin-embedded |
| GBM | glioblastoma |
| HGGs | high-grade gliomas |
| LGGs | low-grade gliomas |
| MEK | mitogen-activated protein kinase kinase |
| pHGGs | paediatric high-grade gliomas |
| PTEN | phosphatase and tensin homologue |
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro-Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Diamandis, P.; Aldape, K. World Health Organization 2016 classification of central nervous system tumors. Neurol. Clin. 2018, 36, 439–447. [Google Scholar] [CrossRef]
- Ghizoni, E.; Naccarato, C.R.M.; Mathias, R.N. Brain Tumors in Children. In Fundamentals of Neurosurgery; Springer: Basel, Switzerland, 2019; pp. 241–261. [Google Scholar]
- Gajjar, A.; Reaman, G.H.; Racadio, J.M.; Smith, F.O. Brain Tumors in Children; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Miklja, Z.; Pasternak, A.; Stallard, S.; Nicolaides, T.; Kline-Nunnally, C.; Cole, B.; Beroukhim, R.; Bandopadhayay, P.; Chi, S.; Ramkissoon, S.H. Molecular profiling and targeted therapy in pediatric gliomas: Review and consensus recommendations. Neuro-Oncol. 2019, 21, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Collins, V.P.; Jones, D.T.; Giannini, C. Pilocytic astrocytoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 775–788. [Google Scholar] [CrossRef]
- Jones, D.T.; Gronych, J.; Lichter, P.; Witt, O.; Pfister, S.M. MAPK pathway activation in pilocytic astrocytoma. Cell. Mol. Life Sci. 2012, 69, 1799–1811. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar]
- Hawkins, C.; Walker, E.; Mohamed, N.; Zhang, C.; Jacob, K.; Shirinian, M.; Alon, N.; Kahn, D.; Fried, I.; Scheinemann, K. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin. Cancer Res. 2011, 17, 4790–4798. [Google Scholar] [CrossRef]
- Kurani, H.; Gurav, M.; Shetty, O.; Chinnaswamy, G.; Moiyadi, A.; Gupta, T.; Jalali, R.; Epari, S. Pilocytic astrocytomas: BRAFV600E and BRAF fusion expression patterns in pediatric and adult age groups. Child’s Nerv. Syst. 2019, 35, 1525–1536. [Google Scholar] [CrossRef]
- Lassaletta, A.; Zapotocky, M.; Mistry, M.; Ramaswamy, V.; Honnorat, M.; Krishnatry, R.; Stucklin, A.G.; Zhukova, N.; Arnoldo, A.; Ryall, S. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J. Clin. Oncol. 2017, 35, 2934. [Google Scholar] [CrossRef]
- Tateishi, K.; Nakamura, T.; Yamamoto, T. Molecular genetics and therapeutic targets of pediatric low-grade gliomas. Brain Tumor Pathol. 2019, 36, 74–83. [Google Scholar] [CrossRef] [PubMed]
- De Blank, P.; Bandopadhayay, P.; Haas-Kogan, D.; Fouladi, M.; Fangusaro, J. Management of pediatric low-grade glioma. Curr. Opin. Pediatr. 2019, 31, 21. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251. [Google Scholar]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3. 3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Mohammad, F.; Helin, K. Oncohistones: Drivers of pediatric cancers. Genes Dev. 2017, 31, 2313–2324. [Google Scholar] [CrossRef]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S. Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef]
- Karremann, M.; Gielen, G.H.; Hoffmann, M.; Wiese, M.; Colditz, N.; Warmuth-Metz, M.; Bison, B.; Claviez, A.; Van Vuurden, D.G.; Von Bueren, A.O. Diffuse high-grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro-Oncol. 2018, 20, 123–131. [Google Scholar] [CrossRef]
- Kasper, L.H.; Baker, S.J. Invited Review: Emerging functions of histone H3 mutations in paediatric diffuse high-grade gliomas. Neuropathol. Appl. Neurobiol. 2020, 46, 73–85. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Liu, X.Y.; Sturm, D.; Jabado, N. Chromatin Remodeling Defects in Pediatric and Young Adult Glioblastoma: A Tale of a Variant H istone 3 Tail. Brain Pathol. 2013, 23, 210–216. [Google Scholar] [CrossRef]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef]
- Braunstein, S.; Raleigh, D.; Bindra, R.; Mueller, S.; Haas-Kogan, D. Pediatric high-grade glioma: Current molecular landscape and therapeutic approaches. J. Neuro-Oncol. 2017, 134, 541–549. [Google Scholar] [CrossRef]
- Robison, N.J.; Kieran, M.W. Diffuse intrinsic pontine glioma: A reassessment. J. Neuro-Oncol. 2014, 119, 7–15. [Google Scholar] [CrossRef]
- Gupta, N.; Goumnerova, L.C.; Manley, P.; Chi, S.N.; Neuberg, D.; Puligandla, M.; Fangusaro, J.; Goldman, S.; Tomita, T.; Alden, T. Prospective feasibility and safety assessment of surgical biopsy for patients with newly diagnosed diffuse intrinsic pontine glioma. Neuro-Oncol. 2018, 20, 1547–1555. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Monje, M. Diffuse intrinsic pontine glioma: From diagnosis to next-generation clinical trials. Curr. Treat. Options Neurol. 2019, 21, 37. [Google Scholar] [CrossRef]
- Cin, H.; Meyer, C.; Herr, R.; Janzarik, W.G.; Lambert, S.; Jones, D.T.; Jacob, K.; Benner, A.; Witt, H.; Remke, M. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 763–774. [Google Scholar] [CrossRef]
- Myall, N.J.; Padda, S.K. BRAF: Novel Therapies for an Emerging Target. In Targeted Therapies for Lung Cancer; Springer: Basel, Switzerland, 2019; pp. 79–100. [Google Scholar]
- Penman, C.L.; Faulkner, C.; Lowis, S.P.; Kurian, K.M. Current understanding of BRAF alterations in diagnosis, prognosis, and therapeutic targeting in pediatric low-grade gliomas. Front. Oncol. 2015, 5, 54. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, T.-W.; Hashizume, R.; Hariono, S.; Barkovich, K.J.; Fan, Q.-W.; Prados, M.; James, C.D.; Weiss, W.A.; Nicolaides, T. Combined BRAF V600E and MEK blockade for BRAF V600E-mutant gliomas. J. Neuro-Oncol. 2017, 131, 495–505. [Google Scholar] [CrossRef]
- Hsiao, S.J.; Karajannis, M.A.; Diolaiti, D.; Mansukhani, M.M.; Bender, J.G.; Kung, A.L.; Garvin, J.H. A novel, potentially targetable TMEM106B-BRAF fusion in pleomorphic xanthoastrocytoma. Mol. Case Stud. 2017, 3, a001396. [Google Scholar] [CrossRef]
- Tutuka, C.S.; Andrews, M.C.; Mariadason, J.M.; Ioannidis, P.; Hudson, C.; Cebon, J.; Behren, A. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol. Cancer 2017, 16, 112. [Google Scholar] [CrossRef]
- See, W.L.; Mukherjee, J. Targeting the RAS-RAF-MEK-ERK signaling pathway in gliomas. In Handbook of Brain Tumor Chemotherapy, Molecular Therapeutics, and Immunotherapy; Elsevier: Amsterdam, The Netherlands, 2018; pp. 323–332. [Google Scholar]
- Maraka, S.; Janku, F. BRAF alterations in primary brain tumors. Discov. Med. 2018, 26, 51–60. [Google Scholar] [PubMed]
- Myung, J.K.; Cho, H.; Park, C.-K.; Kim, S.-K.; Lee, S.-H.; Park, S.-H. Analysis of the BRAFV600E mutation in central nervous system tumors. Transl. Oncol. 2012, 5, 430. [Google Scholar] [CrossRef]
- Gierke, M.; Sperveslage, J.; Schwab, D.; Beschorner, R.; Ebinger, M.; Schuhmann, M.U.; Schittenhelm, J. Analysis of IDH1-R132 mutation, BRAF V600 mutation and KIAA1549–BRAF fusion transcript status in central nervous system tumors supports pediatric tumor classification. J. Cancer Res. Clin. Oncol. 2016, 142, 89–100. [Google Scholar] [CrossRef]
- Behling, F.; Schittenhelm, J. Oncogenic BRAF alterations and their role in brain tumors. Cancers 2019, 11, 794. [Google Scholar] [CrossRef]
- Tian, Y.; Rich, B.E.; Vena, N.; Craig, J.M.; MacConaill, L.E.; Rajaram, V.; Goldman, S.; Taha, H.; Mahmoud, M.; Ozek, M. Detection of KIAA1549-BRAF fusion transcripts in formalin-fixed paraffin-embedded pediatric low-grade gliomas. J. Mol. Diagn. 2011, 13, 669–677. [Google Scholar] [CrossRef]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for carcinogenesis and molecular therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 2020, 8, 30. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Xue, Y.; Martelotto, L.; Baslan, T.; Vides, A.; Solomon, M.; Mai, T.T.; Chaudhary, N.; Riely, G.J.; Li, B.T.; Scott, K. An approach to suppress the evolution of resistance in BRAF V600E-mutant cancer. Nat. Med. 2017, 23, 929. [Google Scholar] [CrossRef]
- Ryall, S.; Arnoldo, A.; Krishnatry, R.; Mistry, M.; Khor, K.; Sheth, J.; Ling, C.; Leung, S.; Zapotocky, M.; Guerreiro Stucklin, A. Multiplex detection of pediatric low-grade glioma signature fusion transcripts and duplications using the NanoString nCounter system. J. Neuropathol. Exp. Neurol. 2017, 76, 562–570. [Google Scholar] [CrossRef]
- Appay, R.; Fina, F.; Macagno, N.; Padovani, L.; Colin, C.; Barets, D.; Ordioni, J.; Scavarda, D.; Giangaspero, F.; Badiali, M. Duplications of KIAA1549 and BRAF screening by Droplet Digital PCR from formalin-fixed paraffin-embedded DNA is an accurate alternative for KIAA1549-BRAF fusion detection in pilocytic astrocytomas. Mod. Pathol. 2018, 31, 1490–1501. [Google Scholar] [CrossRef]
- Merlin, M.-S.; Gilson, P.; Rouyer, M.; Chastagner, P.; Doz, F.; Varlet, P.; Leroux, A.; Gauchotte, G.; Merlin, J.-L. Rapid fully-automated assay for routine molecular diagnosis of BRAF mutations for personalized therapy of low grade gliomas. Pediatr. Hematol. Oncol. 2020, 37, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, C.; Soria, E.; Ramírez, N. The identification and isolation of CTCs: A biological Rubik’s cube. Crit. Rev. Oncol. Hematol. 2018, 126, 129–134. [Google Scholar] [CrossRef]
- Bonner, E.R.; Bornhorst, M.; Packer, R.J.; Nazarian, J. Liquid biopsy for pediatric central nervous system tumors. NPJ Precis. Oncol. 2018, 2, 29. [Google Scholar] [CrossRef]
- García-Romero, N.; Carrión-Navarro, J.; Areal-Hidalgo, P.; Ortiz de Mendivil, A.; Asensi-Puig, A.; Madurga, R.; Núñez-Torres, R.; González-Neira, A.; Belda-Iniesta, C.; González-Rumayor, V. BRAF V600E Detection in Liquid Biopsies from Pediatric Central Nervous System Tumors. Cancers 2020, 12, 66. [Google Scholar] [CrossRef]
- Horbinski, C.; Nikiforova, M.N.; Hagenkord, J.M.; Hamilton, R.L.; Pollack, I.F. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro-Oncol. 2012, 14, 777–789. [Google Scholar] [CrossRef]
- Colin, C.; Padovani, L.; Chappe, C.; Mercurio, S.; Scavarda, D.; Loundou, A.; Frassineti, F.; Andre, N.; Bouvier, C.; Korshunov, A. Outcome analysis of childhood pilocytic astrocytomas: A retrospective study of 148 cases at a single institution. Neuropathol. Appl. Neurobiol. 2013, 39, 693–705. [Google Scholar] [CrossRef]
- Vuong, H.G.; Altibi, A.M.; Duong, U.N.; Ngo, H.T.; Pham, T.Q.; Fung, K.-M.; Hassell, L. BRAF mutation is associated with an improved survival in glioma—a systematic review and meta-analysis. Mol. Neurobiol. 2018, 55, 3718–3724. [Google Scholar] [CrossRef]
- Mistry, M.; Zhukova, N.; Merico, D.; Rakopoulos, P.; Krishnatry, R.; Shago, M.; Stavropoulos, J.; Alon, N.; Pole, J.D.; Ray, P.N. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J. Clin. Oncol. 2015, 33, 1015. [Google Scholar] [CrossRef]
- Workman, J.L.; Abmayr, S.M. Fundamentals of Chromatin; Springer: New York, NY, USA, 2014. [Google Scholar]
- Crabtree, J.S. Fundamentals of epigenetics. In Clinical Precision Medicine; Elsevier: Amsterdam, The Netherlands, 2020; pp. 27–37. [Google Scholar]
- Huang, T.; Garcia, R.; Qi, J.; Lulla, R.; Horbinski, C.; Behdad, A.; Wadhwani, N.; Shilatifard, A.; James, C.; Saratsis, A. Detection of histone H3 K27M mutation and post-translational modifications in pediatric diffuse midline glioma via tissue immunohistochemistry informs diagnosis and clinical outcomes. Oncotarget 2018, 9, 37112. [Google Scholar] [CrossRef]
- Wan, Y.C.E.; Liu, J.; Chan, K.M. Histone H3 mutations in cancer. Curr. Pharmacol. Rep. 2018, 4, 292–300. [Google Scholar] [CrossRef]
- Bechet, D.; Gielen, G.G.; Korshunov, A.; Pfister, S.M.; Rousso, C.; Faury, D.; Fiset, P.-O.; Benlimane, N.; Lewis, P.W.; Lu, C. Specific detection of methionine 27 mutation in histone 3 variants (H3K27M) in fixed tissue from high-grade astrocytomas. Acta Neuropathol. 2014, 128, 733–741. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L. K27M mutation in histone H3. 3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.-Y.; Kambhampati, M.; Hwang, E.I.; Faury, D. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016, 7, 11185. [Google Scholar] [CrossRef]
- Dufour, C.; Perbet, R.; Leblond, P.; Vasseur, R.; Stechly, L.; Pierache, A.; Reyns, N.; Touzet, G.; Le Rhun, E.; Vinchon, M. Identification of prognostic markers in diffuse midline gliomas H3K27M-mutant. Brain Pathol. 2020, 30, 179–190. [Google Scholar] [CrossRef]
- Stallard, S.; Savelieff, M.G.; Wierzbicki, K.; Mullan, B.; Miklja, Z.; Bruzek, A.; Garcia, T.; Siada, R.; Anderson, B.; Singer, B.H.; et al. CSF H3F3A K27M circulating tumor DNA copy number quantifies tumor growth and in vitro treatment response. Acta Neuropathol. Commun. 2018, 6, 80. [Google Scholar] [CrossRef]
- Huang, T.Y.; Piunti, A.; Lulla, R.R.; Qi, J.; Horbinski, C.M.; Tomita, T.; James, C.D.; Shilatifard, A.; Saratsis, A.M. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol. Commun. 2017, 5, 28. [Google Scholar] [CrossRef]
- Pan, C.; Diplas, B.H.; Chen, X.; Wu, Y.; Xiao, X.; Jiang, L.; Geng, Y.; Xu, C.; Sun, Y.; Zhang, P. Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta Neuropathol. 2019, 137, 297–306. [Google Scholar] [CrossRef]
- Panditharatna, E.; Kilburn, L.B.; Aboian, M.S.; Kambhampati, M.; Gordish-Dressman, H.; Magge, S.N.; Gupta, N.; Myseros, J.S.; Hwang, E.I.; Kline, C. Clinically relevant and minimally invasive tumor surveillance of pediatric diffuse midline gliomas using patient-derived liquid biopsy. Clin. Cancer Res. 2018, 24, 5850–5859. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef]
- Sareen, H.; Garrett, C.; Lynch, D.; Powter, B.; Brungs, D.; Cooper, A.; Po, J.; Koh, E.-S.; Vessey, J.Y.; McKechnie, S. The role of liquid biopsies in detecting molecular tumor biomarkers in brain cancer patients. Cancers 2020, 12, 1831. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Wisoff, J.H.; Sanford, R.A.; Heier, L.A.; Sposto, R.; Burger, P.C.; Yates, A.J.; Holmes, E.J.; Kun, L.E. Primary neurosurgery for pediatric low-grade gliomas: A prospective multi-institutional study from the Children’s Oncology Group. Neurosurgery 2011, 68, 1548–1555. [Google Scholar] [CrossRef]
- Hargrave, D.R.; Bouffet, E.; Tabori, U.; Broniscer, A.; Cohen, K.J.; Hansford, J.R.; Geoerger, B.; Hingorani, P.; Dunkel, I.J.; Russo, M.W. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation–positive relapsed or refractory low-grade glioma: Results from a phase I/IIa study. Clin. Cancer Res. 2019, 25, 7303–7311. [Google Scholar] [CrossRef]
- Bandopadhayay, P.; Bergthold, G.; London, W.B.; Goumnerova, L.C.; Morales La Madrid, A.; Marcus, K.J.; Guo, D.; Ullrich, N.J.; Robison, N.J.; Chi, S.N. Long-term outcome of 4040 children diagnosed with pediatric low-grade gliomas: An analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr. Blood Cancer 2014, 61, 1173–1179. [Google Scholar] [CrossRef]
- Rao, A.A.N.; Packer, R.J. Advances in the management of low-grade gliomas. Curr. Oncol. Rep. 2014, 16, 398. [Google Scholar]
- Packer, R.J.; Ater, J.; Allen, J.; Phillips, P.; Geyer, R.; Nicholson, H.S.; Jakacki, R.; Kurczynski, E.; Needle, M.; Finlay, J. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J. Neurosurg. 1997, 86, 747–754. [Google Scholar] [CrossRef]
- Raikar, S.S.; Halloran, D.R.; Elliot, M.; McHugh, M.; Patel, S.; Gauvain, K.M. Outcomes of pediatric low-grade gliomas treated with radiation therapy: A single-institution study. J. Pediatr. Hematol. Oncol. 2014, 36, e366. [Google Scholar] [CrossRef]
- Merchant, T.E.; Conklin, H.M.; Wu, S.; Lustig, R.H.; Xiong, X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: Prospective evaluation of cognitive, endocrine, and hearing deficits. J. Clin. Oncol. 2009, 27, 3691. [Google Scholar] [CrossRef]
- Merchant, T.E.; Pollack, I.F.; Loeffler, J.S. Brain tumors across the age spectrum: Biology, therapy, and late effects. Semin. Radiat. Oncol. 2010, 20, 58–66. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600–mutant anaplastic thyroid cancer. J. Clin. Oncol. 2018, 36, 7. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Hargrave, D. The first study of dabrafenib in pediatric patients with BRAF V600–mutant relapsed or refractory low-grade gliomas. Ann. Oncol. 2016, 27, LBA19_PR. [Google Scholar]
- Del Bufalo, F.; Ceglie, G.; Cacchione, A.; Alessi, I.; Colafati, G.S.; Carai, A.; Diomedi-Camassei, F.; De Billy, E.; Agolini, E.; Mastronuzzi, A.; et al. BRAF V600E inhibitor (Vemurafenib) for BRAF V600E mutated low grade gliomas. Front. Oncol. 2018, 8, 526. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Gajjar, A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer 2014, 14, 258. [Google Scholar] [CrossRef]
- Chi, A.S.; Tarapore, R.S.; Hall, M.D.; Shonka, N.; Gardner, S.; Umemura, Y.; Sumrall, A.; Khatib, Z.; Mueller, S.; Kline, C. Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J. Neuro-Oncol. 2019, 145, 97–105. [Google Scholar] [CrossRef]
- Cohen, K.J.; Pollack, I.F.; Zhou, T.; Buxton, A.; Holmes, E.J.; Burger, P.C.; Brat, D.J.; Rosenblum, M.K.; Hamilton, R.L.; Lavey, R.S. Temozolomide in the treatment of high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro-Oncol. 2011, 13, 317–323. [Google Scholar] [CrossRef]
- Jones, C.; Karajannis, M.A.; Jones, D.T.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.-J.; Gupta, N.; Hawkins, C. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro-Oncol. 2017, 19, 153–161. [Google Scholar] [CrossRef]
- New, M.; Olzscha, H.; La Thangue, N.B. HDAC inhibitor-based therapies: Can we interpret the code? Mol. Oncol. 2012, 6, 637–656. [Google Scholar] [CrossRef]
- Anne, M.; Sammartino, D.; Barginear, M.F.; Budman, D. Profile of panobinostat and its potential for treatment in solid tumors: An update. Oncotargets Ther. 2013, 6, 1613. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.-A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Allen, J.E.; Prabhu, V.V.; Talekar, M.; Van Den Heuvel, A.P.J.; Lim, B.; Dicker, D.T.; Fritz, J.L.; Beck, A.; El-Deiry, W.S. Genetic and pharmacological screens converge in identifying FLIP, BCL2, and IAP proteins as key regulators of sensitivity to the TRAIL-inducing anticancer agent ONC201/TIC10. Cancer Res. 2015, 75, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolloff, N.G.; Messaris, E.; Scata, K.A.; Wang, W. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci. Transl. Med. 2013, 5, 117ra17. [Google Scholar] [CrossRef]
- Li, J.; Zhu, S.; Kozono, D.; Ng, K.; Futalan, D.; Shen, Y.; Akers, J.C.; Steed, T.; Kushwaha, D.; Schlabach, M. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget 2014, 5, 882. [Google Scholar] [CrossRef]
- Caragher, S.P.; Hall, R.R.; Ahsan, R.; Ahmed, A.U. Monoamines in glioblastoma: Complex biology with therapeutic potential. Neuro-Oncol. 2018, 20, 1014–1025. [Google Scholar] [CrossRef]
- Ishida, C.T.; Zhang, Y.; Bianchetti, E.; Shu, C.; Nguyen, T.T.; Kleiner, G.; Sanchez-Quintero, M.J.; Quinzii, C.M.; Westhoff, M.-A.; Karpel-Massler, G. Metabolic reprogramming by dual AKT/ERK inhibition through imipridones elicits unique vulnerabilities in glioblastoma. Clin. Cancer Res. 2018, 24, 5392–5406. [Google Scholar] [CrossRef]
- Kline, C.L.B.; Van den Heuvel, A.P.J.; Allen, J.E.; Prabhu, V.V.; Dicker, D.T.; El-Deiry, W.S. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2α kinases. Sci. Signal. 2016, 9, ra18. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef]
- Rajesh, Y.; Pal, I.; Banik, P.; Chakraborty, S.; Borkar, S.A.; Dey, G.; Mukherjee, A.; Mandal, M. Insights into molecular therapy of glioma: Current challenges and next generation blueprint. Acta Pharmacol. Sin. 2017, 38, 591–613. [Google Scholar] [CrossRef]
- Toll, S.A.; Tran, H.N.; Cotter, J.; Judkins, A.R.; Tamrazi, B.; Biegel, J.A.; Dhall, G.; Robison, N.J.; Waters, K.; Patel, P. Sustained response of three pediatric BRAFV600E mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget 2019, 10, 551. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef]
- Kieran, M.W.; Geoerger, B.; Dunkel, I.J.; Broniscer, A.; Hargrave, D.; Hingorani, P.; Aerts, I.; Bertozzi, A.-I.; Cohen, K.J.; Hummel, T.R.; et al. A phase I and pharmacokinetic study of oral dabrafenib in children and adolescent patients with recurrent or refractory BRAF V600 mutation–positive solid tumors. Clin. Cancer Res. 2019, 25, 7294–7302. [Google Scholar] [CrossRef]
- Hargrave, D.; Witt, O.; Cohen, K.; Packer, R.; Lissat, A.; Kordes, U.; Laetsch, T.; Hoffman, L.; Lassaletta, A.; Gerber, N. 407TiP Phase II open-label, global study evaluating dabrafenib in combination with trametinib in pediatric patients with BRAF V600–mutant high-grade glioma (HGG) or low-grade glioma (LGG). Ann. Oncol. 2018, 29, 395. [Google Scholar] [CrossRef]
- Marks, A.M.; Bindra, R.S.; DiLuna, M.L.; Huttner, A.; Jairam, V.; Kahle, K.T.; Kieran, M.W. Response to the BRAF/MEK inhibitors dabrafenib/trametinib in an adolescent with a BRAF V600E mutated anaplastic ganglioglioma intolerant to vemurafenib. Pediatr. Blood Cancer 2018, 65, e26969. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Paediatric Gliomas | Biomaterial | Biosources | Diagnostic Methods | Genetic Mutations and Findings | References |
|---|---|---|---|---|---|
| Paediatric brain tumour (n = 11), including diffuse midline gliomas (n = 6) | Circulating tumour DNA and genomic tumour DNA | CSF | Sanger sequencing and nested PCR | H3F3AK27M mutations were detected in four out of six samples using the newly established methods. | [63] |
| Diffuse intrinsic pontine glioma | Circulating tumour DNA | CSF | Droplet digital PCR | H3F3AK27M mutations were detected. | [62] |
| Brainstem gliomas (n = 57) and diffuse intrinsic pontine glioma (n = 23) | Circulating tumour DNA | CSF | Deep sequencing | Multiple tumour-specific mutations such as H3F3A, TP53, ATRX, PDGFRA, HIST1H3B, FAT1, PPM1D, ACVR1, NF1, IDH1 and PIK3CA were detected. H3F3A was the highest mutated gene, with a percentage of approximately 48%. Approximately 9% of altered HIST1H3B genes were detected. | [64] |
| Diffuse midline glioma (n = 48) | Circulating tumour DNA | 79 plasma samples and 30 CSF samples | Droplet digital PCR | BRAF, PPM1D, H3F3A, HIST1H3B, ACVR1 and PIK3R1 mutations were detected. Eighty-eight percent of the CFS and plasma circulating tumour DNA(ctDNA) harboured H3K27M mutations. The amount of ctDNA in plasma was lower than that of the CSF. | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, J.Y.; Wijesinghe, I.V.S.; Alfarizal Kamarudin, M.N.; Parhar, I. Paediatric Gliomas: BRAF and Histone H3 as Biomarkers, Therapy and Perspective of Liquid Biopsies. Cancers 2021, 13, 607. https://doi.org/10.3390/cancers13040607
Tan JY, Wijesinghe IVS, Alfarizal Kamarudin MN, Parhar I. Paediatric Gliomas: BRAF and Histone H3 as Biomarkers, Therapy and Perspective of Liquid Biopsies. Cancers. 2021; 13(4):607. https://doi.org/10.3390/cancers13040607
Chicago/Turabian StyleTan, Jean Yin, Ipalawattage Vindya Stephnie Wijesinghe, Muhamad Noor Alfarizal Kamarudin, and Ishwar Parhar. 2021. "Paediatric Gliomas: BRAF and Histone H3 as Biomarkers, Therapy and Perspective of Liquid Biopsies" Cancers 13, no. 4: 607. https://doi.org/10.3390/cancers13040607
APA StyleTan, J. Y., Wijesinghe, I. V. S., Alfarizal Kamarudin, M. N., & Parhar, I. (2021). Paediatric Gliomas: BRAF and Histone H3 as Biomarkers, Therapy and Perspective of Liquid Biopsies. Cancers, 13(4), 607. https://doi.org/10.3390/cancers13040607