
Bispecific Antibodies in Prostate Cancer Therapy: Current Status and Perspectives



{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Checkpoint Inhibition in Prostate Cancer
3. CAR-T Cells in Prostate Cancer
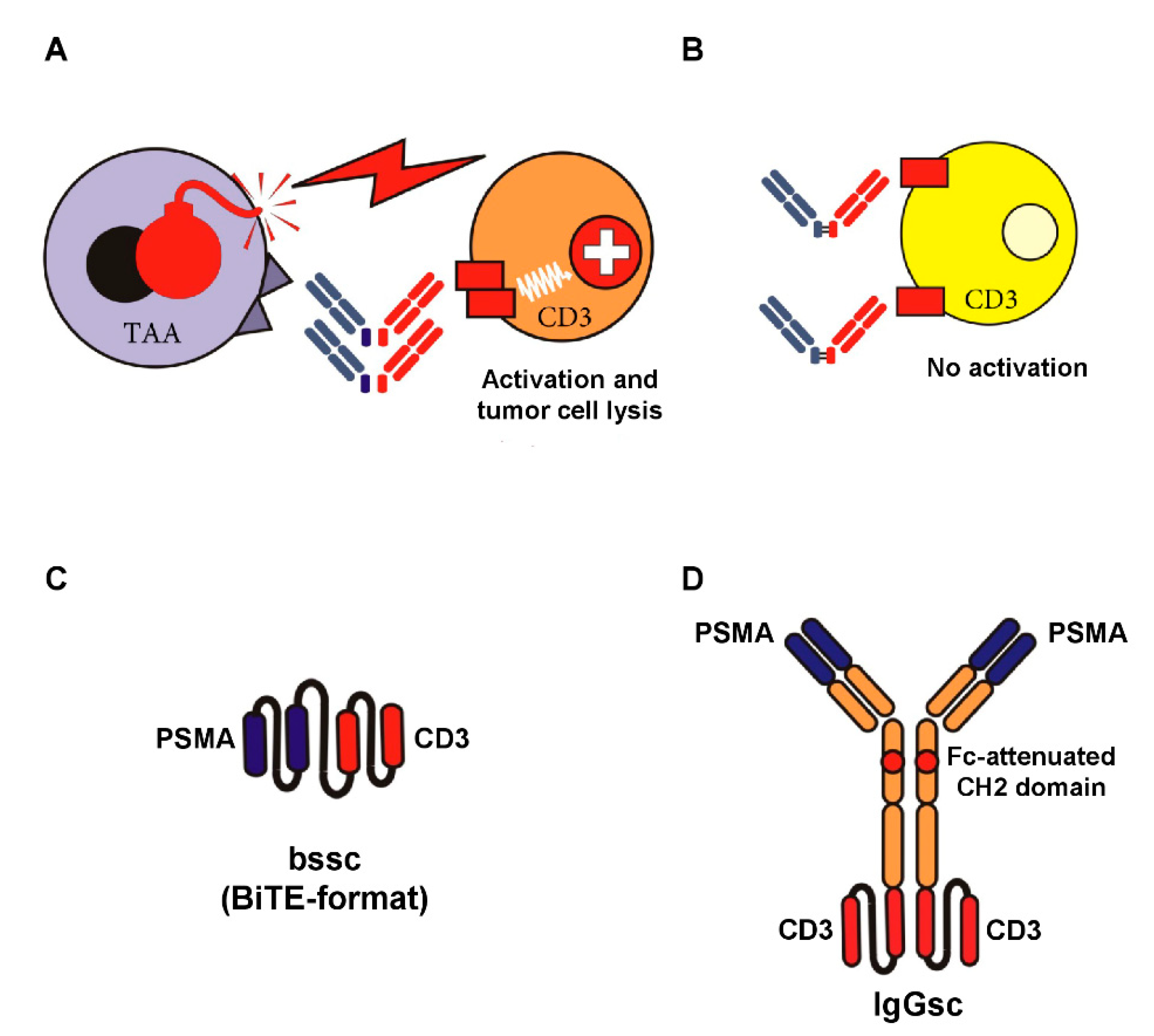
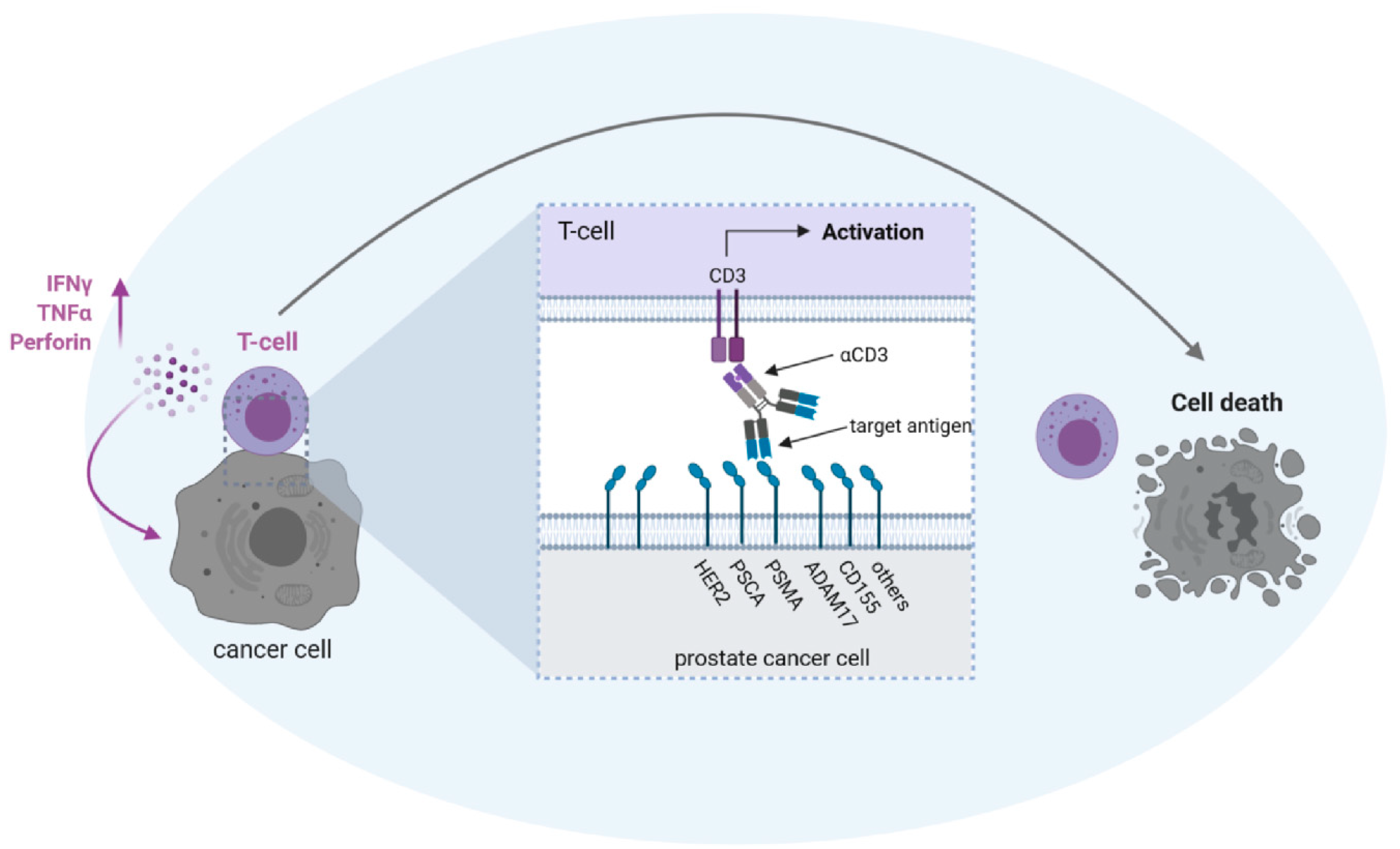
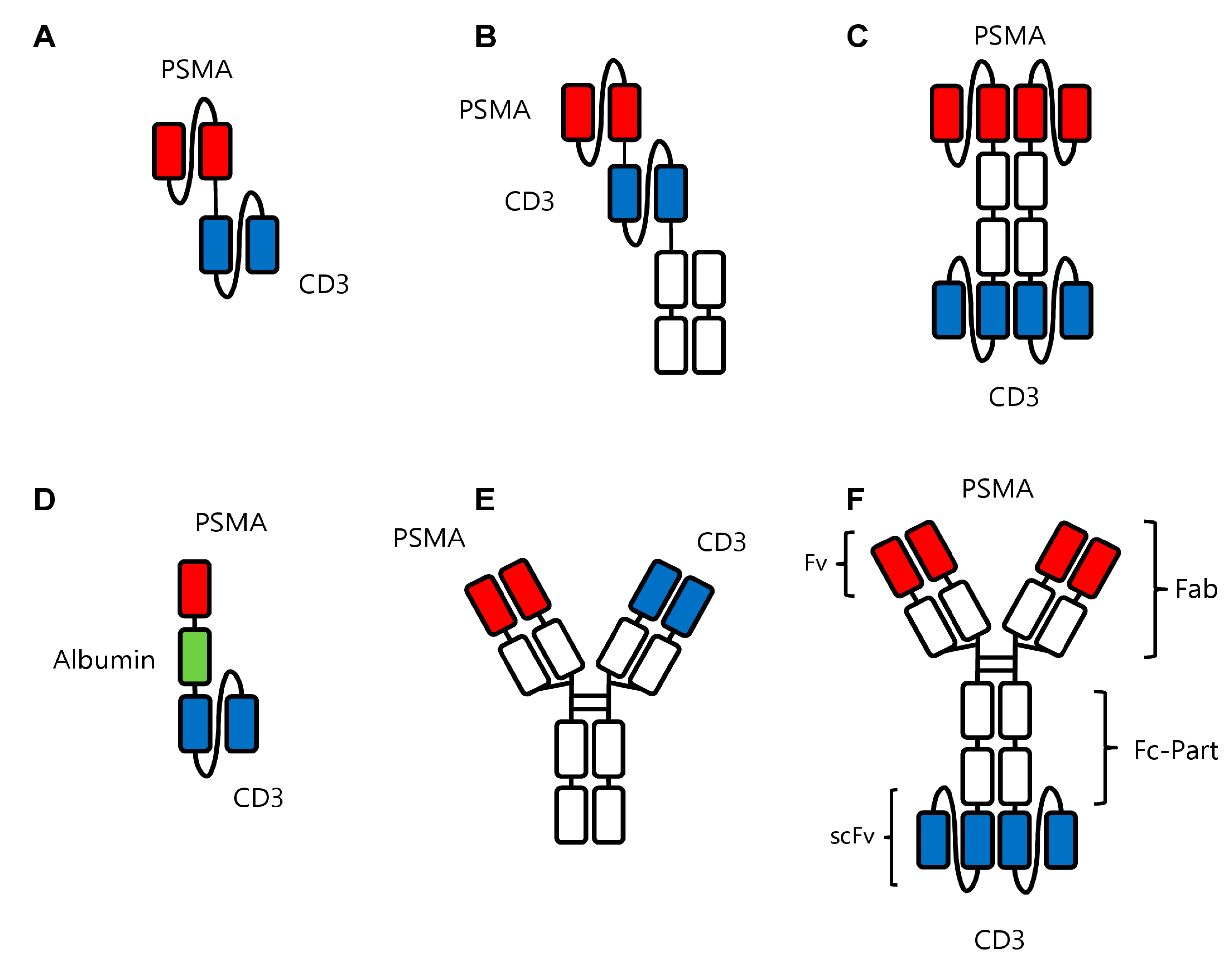
4. BsAbs in Prostate Cancer
4.1. Background
4.2. Target Antigens for bsAbs in PC
4.2.1. PSMA
4.2.2. Other Targets
5. Toxicity Considerations
- (i)
- desired “on-target on-tumor” T cell activation;
- (ii)
- (iii)
- undesired “on-target off-tumor” effects occurring upon targeting of target antigens that are not expressed in a highly tumor-restricted manner, as exemplified with Blinatumomab, where the target CD19 is expressed on healthy B cells.
6. Current bsAbs under Clinical Evaluation
6.1. Pasotuxizumab/BAY 2010112/AMG 212
6.2. AMG-160
6.3. APVO414/MOR209/ES414
6.4. HPN424
6.5. JNJ-63898081
6.6. CC-1
7. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Methods
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Berthold, D.R.; Pond, G.R.; Soban, F.; de Wit, R.; Eisenberger, M.; Tannock, I.F. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: Updated survival in the TAX 327 study. J. Clin. Oncol. 2008, 26, 242–245. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Kelly, P.N. The Cancer Immunotherapy Revolution. Science 2018, 359, 1344–1345. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.B.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.P.; Aggarwal, R.; et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Koo, K.C. Current Status and Future Perspectives of Checkpoint Inhibitor Immunotherapy for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 5484. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Shaukat, F.; Velho, P.I.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Scholz, M.; Yep, S.; Chancey, M.; Kelly, C.; Chau, K.; Turner, J.; Lam, R.; Drake, C.G. Phase I clinical trial of sipuleucel-T combined with escalating doses of ipilimumab in progressive metastatic castrate-resistant prostate cancer. Immunotargets Ther. 2017, 6, 11–16. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 17 December 2020).
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef]
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6 (Suppl. 10), S13–S18. [Google Scholar]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Slovin, S.F.; Wang, X.Y.; Hullings, M.; Arauz, G.; Bartido, S.; Lewis, J.S.; Schoder, H.; Zanzonico, P.; Scher, H.I.; Sadelain, M.; et al. Chimeric antigen receptor (CAR(+)) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). J. Clin. Oncol. 2013, 31. [Google Scholar] [CrossRef]
- Narayan, V.; Gladney, W.; Plesa, G.; Vapiwala, N.; Carpenter, E.; Maude, S.L.; Lal, P.; Lacey, S.F.; Melenhorst, J.J.; Sebro, R.; et al. A phase I clinical trial of PSMA-directed/TGF13-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; Weiner, L.M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- EMA. Blinatumomab. Available online: www.ema.europa.eu/documents/overview/blincyto-epar-medicine-overview_de.pdf (accessed on 22 December 2020).
- Portell, C.A.; Wenzell, C.M.; Advani, A.S. Clinical and pharmacologic aspects of blinatumomab in the treatment of B-cell acute lymphoblastic leukemia. Clin. Pharmacol. 2013, 5, 5–11. [Google Scholar] [CrossRef]
- Demarest, S.J.; Glaser, S.M. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr. Opin. Drug Discov. Dev. 2008, 11, 675–687. [Google Scholar]
- Perchiacca, J.M.; Tessier, P.M. Engineering aggregation-resistant antibodies. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 263–286. [Google Scholar] [CrossRef]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef]
- Jung, G.; Ledbetter, J.A.; Muller-Eberhard, H.J. Induction of cytotoxicity in resting human T lymphocytes bound to tumor cells by antibody heteroconjugates. Proc. Natl. Acad. Sci. USA 1987, 84, 4611–4615. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Brandl, M.; Eisner, W.; Fraunberger, P.; Reifenberger, G.; Schlegel, U.; Wiestler, O.D.; Reulen, H.J.; Wilmanns, W. Local immunotherapy of glioma patients with a combination of 2 bispecific antibody fragments and resting autologous lymphocytes: Evidence for in situ t-cell activation and therapeutic efficacy. Int. J. Cancer 2001, 91, 225–230. [Google Scholar] [CrossRef]
- Hunig, T. The storm has cleared: Lessons from the CD28 superagonist TGN1412 trial. Nat. Rev. Immunol. 2012, 12, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634. [Google Scholar]
- Chang, S.S.; Reuter, V.E.; Heston, W.D.; Bander, N.H.; Grauer, L.S.; Gaudin, P.B. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999, 59, 3192–3198. [Google Scholar]
- Ross, J.S.; Gray, K.E.; Webb, I.J.; Gray, G.S.; Rolfe, M.; Schenkein, D.P.; Nanus, D.M.; Millowsky, M.I.; Bander, N.H. Antibody-based therapeutics: Focus on prostate cancer. Cancer Metastasis Rev. 2005, 24, 521–537. [Google Scholar] [CrossRef]
- Israeli, R.S.; Powell, C.T.; Corr, J.G.; Fair, W.R.; Heston, W.D. Expression of the prostate-specific membrane antigen. Cancer Res. 1994, 54, 1807–1811. [Google Scholar]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J., Jr.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Seaman, S.; Zhu, Z.; Saha, S.; Zhang, X.M.; Yang, M.Y.; Hilton, M.B.; Morris, K.; Szot, C.; Morris, H.; Swing, D.A.; et al. Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer Cell 2017, 31, 501–515.e8. [Google Scholar] [CrossRef]
- Ceci, F.; Castellucci, P.; Graziani, T.; Farolfi, A.; Fonti, C.; Lodi, F.; Fanti, S. (68)Ga-PSMA-11 PET/CT in recurrent prostate cancer: Efficacy in different clinical stages of PSA failure after radical therapy. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.P.; Macura, K.J.; Mena, E.; Blackford, A.L.; Nadal, R.; Antonarakis, E.S.; Eisenberger, M.; Carducci, M.; Fan, H.; Dannals, R.F.; et al. PSMA-Based [(18)F]DCFPyL PET/CT Is Superior to Conventional Imaging for Lesion Detection in Patients with Metastatic Prostate Cancer. Mol. Imaging Biol. 2016, 18, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.; Papa, N.; Christidis, D.; Wetherell, D.; Hofman, M.S.; Murphy, D.G.; Bolton, D.; Lawrentschuk, N. Sensitivity, Specificity, and Predictors of Positive (68)Ga-Prostate-specific Membrane Antigen Positron Emission Tomography in Advanced Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. 2016, 70, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Bander, N.H.; Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J. Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J. Clin. Oncol. 2005, 23, 4591–4601. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.K.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Ng, S.; et al. TheraP: A randomised phase II trial of Lu-177-PSMA-617 (LuPSMA) theranostic versus cabazitaxel in metastatic castration resistant prostate cancer (mCRPC) progressing after docetaxel: Initial results (ANZUP protocol 1603). J. Clin. Oncol. 2020, 38, 5500. [Google Scholar] [CrossRef]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [(177)Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Beltran, H.; Vallabhajosula, S.; Goldsmith, S.J.; Osborne, J.; Matulich, D.; Petrillo, K.; Parmar, S.; Nanus, D.M.; Bander, N.H. Anti-prostate-specific membrane antigen-based radioimmunotherapy for prostate cancer. Cancer 2010, 116, 1075–1083. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Vallabhajosula, S.; Christos, P.J.; Jhanwar, Y.S.; Batra, J.S.; Lam, L.; Osborne, J.; Beltran, H.; Molina, A.M.; Goldsmith, S.J.; et al. Phase 1/2 study of fractionated dose lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 ((177) Lu-J591) for metastatic castration-resistant prostate cancer. Cancer 2019, 125, 2561–2569. [Google Scholar] [CrossRef]
- Hummel, H.D.; Kufer, P.; Grullich, C.; Seggewiss-Bernhardt, R.; Deschler-Baier, B.; Chatterjee, M.; Goebeler, M.E.; Miller, K.; de Santis, M.; Loidl, W.; et al. Pasotuxizumab, a BiTE((R)) immune therapy for castration-resistant prostate cancer: Phase I, dose-escalation study findings. Immunotherapy 2020. [Google Scholar] [CrossRef]
- Heitmann, J.S.; Walz, J.S.; Pflugler, M.; Kauer, J.; Schlenk, R.F.; Jung, G.; Salih, H.R. Protocol of a prospective, multicentre phase I study to evaluate the safety, tolerability and preliminary efficacy of the bispecific PSMAxCD3 antibody CC-1 in patients with castration-resistant prostate carcinoma. BMJ Open 2020, 10, e039639. [Google Scholar] [CrossRef]
- Bendell, J.C.; Fong, L.; Stein, M.N.; Beer, T.M.; Ross, A.; Gao, X.; Weitzman, A.; Austin, R.; Ganti, V.; Law, C.L.; et al. First-in-human phase I study of HPN424, a tri-specific half-life extended PSMA-targeting T-cell engager in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 5552. [Google Scholar] [CrossRef]
- Leconet, W.; Liu, H.; Guo, M.; Le Lamer-Dechamps, S.; Molinier, C.; Kim, S.; Vrlinic, T.; Oster, M.; Liu, F.; Navarro, V.; et al. Anti-PSMA/CD3 Bispecific Antibody Delivery and Antitumor Activity Using a Polymeric Depot Formulation. Mol. Cancer Ther. 2018, 17, 1927–1940. [Google Scholar] [CrossRef] [PubMed]
- Raff, A.B.; Gray, A.; Kast, W.M. Prostate stem cell antigen: A prospective therapeutic and diagnostic target. Cancer Lett. 2009, 277, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.E.; Gu, Z.; Watabe, T.; Thomas, G.; Szigeti, K.; Davis, E.; Wahl, M.; Nisitani, S.; Yamashiro, J.; Le Beau, M.M.; et al. Prostate stem cell antigen: A cell surface marker overexpressed in prostate cancer. Proc. Natl. Acad. Sci. USA 1998, 95, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Thomas, G.; Yamashiro, J.; Shintaku, I.P.; Dorey, F.; Raitano, A.; Witte, O.N.; Said, J.W.; Loda, M.; Reiter, R.E. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000, 19, 1288–1296. [Google Scholar] [CrossRef]
- Lam, J.S.; Yamashiro, J.; Shintaku, I.P.; Vessella, R.L.; Jenkins, R.B.; Horvath, S.; Said, J.W.; Reiter, R.E. Prostate stem cell antigen is overexpressed in prostate cancer metastases. Clin. Cancer Res. 2005, 11, 2591–2596. [Google Scholar] [CrossRef]
- Argani, P.; Rosty, C.; Reiter, R.E.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Ryu, B.; Skinner, H.G.; Goggins, M.; Jaffee, E.M.; et al. Discovery of new markers of cancer through serial analysis of gene expression: Prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001, 61, 4320–4324. [Google Scholar]
- Elsamman, E.; Fukumori, T.; Kasai, T.; Nakatsuji, H.; Nishitani, M.A.; Toida, K.; Ali, N.; Kanayama, H.O. Prostate stem cell antigen predicts tumour recurrence in superficial transitional cell carcinoma of the urinary bladder. BJU Int. 2006, 97, 1202–1207. [Google Scholar] [CrossRef]
- Waeckerle-Men, Y.; Uetz-von Allmen, E.; Fopp, M.; von Moos, R.; Bohme, C.; Schmid, H.P.; Ackermann, D.; Cerny, T.; Ludewig, B.; Groettrup, M.; et al. Dendritic cell-based multi-epitope immunotherapy of hormone-refractory prostate carcinoma. Cancer Immunol. Immunother. 2006, 55, 1524–1533. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Topfer, K.; Stamova, S.; Krone, F.; Cartellieri, M.; Koristka, S.; Michalk, I.; Lindemann, D.; Schmitz, M.; et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T cells. J. Immunol. 2012, 189, 3249–3259. [Google Scholar] [CrossRef]
- Masson, D.; Jarry, A.; Baury, B.; Blanchardie, P.; Laboisse, C.; Lustenberger, P.; Denis, M.G. Overexpression of the CD155 gene in human colorectal carcinoma. Gut 2001, 49, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Lachmann, S.; Rosenfeld, M.R.; Gutin, P.H.; Wimmer, E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc. Natl. Acad. Sci. USA 2000, 97, 6803–6808. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, C.L.; Wimmer, E.; Racaniello, V.R. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 1989, 56, 855–865. [Google Scholar] [CrossRef]
- Zhao, H.; Ma, J.; Lei, T.; Ma, W.; Zhang, M. The bispecific anti-CD3 x anti-CD155 antibody mediates T cell immunotherapy for human prostate cancer. Investig. New Drugs 2019, 37, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, C.; Jackson, M.W.; Choong, C.S.; Stahl, J.; Marshall, V.R.; Horsfall, D.J.; Tilley, W.D. Elevated levels of HER-2/neu and androgen receptor in clinically localized prostate cancer identifies metastatic potential. Prostate 2008, 68, 830–838. [Google Scholar] [CrossRef]
- Nishio, Y.; Yamada, Y.; Kokubo, H.; Nakamura, K.; Aoki, S.; Taki, T.; Honda, N.; Nakagawa, A.; Saga, S.; Hara, K. Prognostic significance of immunohistochemical expression of the HER-2/neu oncoprotein in bone metastatic prostate cancer. Urology 2006, 68, 110–115. [Google Scholar] [CrossRef]
- Vaishampayan, U.; Thakur, A.; Rathore, R.; Kouttab, N.; Lum, L.G. Phase I Study of Anti-CD3 x Anti-Her2 Bispecific Antibody in Metastatic Castrate Resistant Prostate Cancer Patients. Prostate Cancer 2015, 2015, 285193. [Google Scholar] [CrossRef]
- Moss, M.L.; Jin, S.L.; Milla, M.E.; Bickett, D.M.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Arribas, J.; Esselens, C. ADAM17 as a therapeutic target in multiple diseases. Curr. Pharm. Des. 2009, 15, 2319–2335. [Google Scholar] [CrossRef]
- Sinnathamby, G.; Zerfass, J.; Hafner, J.; Block, P.; Nickens, Z.; Hobeika, A.; Secord, A.A.; Lyerly, H.K.; Morse, M.A.; Philip, R. ADAM metallopeptidase domain 17 (ADAM17) is naturally processed through major histocompatibility complex (MHC) class I molecules and is a potential immunotherapeutic target in breast, ovarian and prostate cancers. Clin. Exp. Immunol. 2011, 163, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Trad, A.; Baumgart, A.; Huske, L.; Lorenzen, I.; Chalaris, A.; Grotzinger, J.; Dechow, T.; Scheller, J.; Rose-John, S. A novel bispecific single-chain antibody for ADAM17 and CD3 induces T-cell-mediated lysis of prostate cancer cells. Biochem. J. 2012, 445, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Worn, A.; Pluckthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef]
- Durben, M.; Schmiedel, D.; Hofmann, M.; Vogt, F.; Nubling, T.; Pyz, E.; Buhring, H.J.; Rammensee, H.G.; Salih, H.R.; Grosse-Hovest, L.; et al. Characterization of a bispecific FLT3 X CD3 antibody in an improved, recombinant format for the treatment of leukemia. Mol. Ther. 2015, 23, 648–655. [Google Scholar] [CrossRef]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Mhawech-Fauceglia, P.; Zhang, S.; Terracciano, L.; Sauter, G.; Chadhuri, A.; Herrmann, F.R.; Penetrante, R. Prostate-specific membrane antigen (PSMA) protein expression in normal and neoplastic tissues and its sensitivity and specificity in prostate adenocarcinoma: An immunohistochemical study using mutiple tumour tissue microarray technique. Histopathology 2007, 50, 472–483. [Google Scholar] [CrossRef]
- Emmett, L.; Willowson, K.; Violet, J.; Shin, J.; Blanksby, A.; Lee, J. Lutetium (177) PSMA radionuclide therapy for men with prostate cancer: A review of the current literature and discussion of practical aspects of therapy. J. Med. Radiat. Sci. 2017, 64, 52–60. [Google Scholar] [CrossRef]
- Yu, S.; Li, A.; Liu, Q.; Yuan, X.; Xu, H.; Jiao, D.; Pestell, R.G.; Han, X.; Wu, K. Recent advances of bispecific antibodies in solid tumors. J. Hematol. Oncol. 2017, 10, 155. [Google Scholar] [CrossRef]
- Jurichson, J. Aptevo Therapeutics and MorphoSys End Joint Development and Commercialization Agreement for MOR209/ES414. Available online: https://www.globenewswire.com/news-release/2017/08/31/1106420/0/en/Aptevo-Therapeutics-and-MorphoSys-End-Joint-Development-and-Commercialization58Agreement-for-MOR209-ES414.html (accessed on 1 December 2020).
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Lorenczewski, G.; Friedrich, M.; Kischel, R.; Dahlhoff, C.; Anlahr, J.; Balazs, M.; Rock, D.; Boyle, M.C.; Goldstein, R.; Coxon, A.; et al. Generation of a Half-Life Extended Anti-CD19 BiTE (R) Antibody Construct Compatible with Once-Weekly Dosing for Treatment of CD19-Positive Malignancies. Blood 2017, 130. [Google Scholar] [CrossRef]
- Tran, B.; Horvath, L.; Dorff, T.B.; Greil, R.; Machiels, J.P.H.; Roncolato, F.; Autio, K.A.; Rettig, M.; Fizazi, K.; Lolkema, M.P.; et al. Phase I study of AMG 160, a half-life extended bispecific T-cell engager (HLE BiTE) immune therapy targeting prostate-specific membrane antigen (PSMA), in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Hernandez-Hoyos, G.; Sewell, T.; Bader, R.; Bannink, J.; Chenault, R.A.; Daugherty, M.; Dasovich, M.; Fang, H.; Gottschalk, R.; Kumer, J.; et al. MOR209/ES414, a Novel Bispecific Antibody Targeting PSMA for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Jurchison, S. Aptevo Therapeutics Reports Third Quarter 2018 Financial Results. Available online: https://aptevotherapeutics.gcs-web.com/news-releases/news-release-details/aptevo-therapeutics-reports-third-quarter-2018-financial-results (accessed on 17 December 2020).
- Lemon, B.; Aaron, W.; Austin, R.; Baeuerle, P.; Barath, M.; Jones, A.; Jones, S.D.; Kwant, K.; Law, C.L.; Muchnik, A.; et al. Abstract 1773: HPN424, a half-life extended, PSMA/CD3-specific TriTAC for the treatment of metastatic prostate cancer. Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Lemon, B.; Aaron, W.; Austin, R.; Baeuerle, P.; Barath, M.; Jones, A.; Jones, S.D.; Kwant, K.; Law, C.L.; Muchnik, A.; et al. HPN424, a half-life extended, PSMA/CD3-specific TriTAC for the treatment of metastatic prostate cancer. Available online: https://www.harpoontx.com/file.cfm/43/docs/AACR_2018_Poster_HPN424.pdf (accessed on 17 December 2020).
- Wesche, H.; Aaron, W.; Austin, R.J.; Baeuerle, P.A.; Jones, A.; Lemon, B.; Sexton, K. TriTACs are novel T cell-engaging therapeutic proteins optimized for the treatment of solid tumors and for long serum half-life. Cancer Res. 2018, 78, 3814. [Google Scholar] [CrossRef]
- Klaassen, Z. ASCO 2020: First-in-Human Phase I Study of HPN424, a Tri-Specific Half-Life Extended PSMA-Targeting T-Cell Engager in Patients with mCRPC. Available online: https://www.urotoday.com/conference-highlights/asco-2020/asco-2020-prostate-cancer/121856-asco-2020-first-in-human-phase-i-study-of-hpn424-a-tri-specific-half-life-extended-psma-targeting-t-cell-engager-in-patients-with-mcrpc.html (accessed on 17 December 2020).
- Annual Report 2019. Innovating Antibodies, Improving Lives. Available online: https://2019overview.genmab.com/Genmab_AR19_Web.pdf (accessed on 1 December 2020).
- Coloma, M.J.; Morrison, S.L. Design and production of novel tetravalent bispecific antibodies. Nat. Biotechnol 1997, 15, 159–163. [Google Scholar] [CrossRef]
- Higano, C.S.; Armstrong, A.J.; Sartor, A.O.; Vogelzang, N.J.; Kantoff, P.W.; McLeod, D.G.; Pieczonka, C.M.; Penson, D.F.; Shore, N.D.; Vacirca, J.; et al. Real-world outcomes of sipuleucel-T treatment in PROCEED, a prospective registry of men with metastatic castration-resistant prostate cancer. Cancer 2019, 125, 4172–4180. [Google Scholar] [CrossRef]
- Pishvaian, M.; Morse, M.A.; McDevitt, J.; Norton, J.D.; Ren, S.; Robbie, G.J.; Ryan, P.C.; Soukharev, S.; Bao, H.; Denlinger, C.S. Phase 1 Dose Escalation Study of MEDI-565, a Bispecific T-Cell Engager that Targets Human Carcinoembryonic Antigen, in Patients With Advanced Gastrointestinal Adenocarcinomas. Clin. Colorectal. Cancer 2016, 15, 345–351. [Google Scholar] [CrossRef]
- Kebenko, M.; Goebeler, M.E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology 2018, 7, e1450710. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Babich, J.W.; Kratochwil, C.; Giesel, F.L.; Eisenhut, M.; Kopka, K.; Haberkorn, U. The Rise of PSMA Ligands for Diagnosis and Therapy of Prostate Cancer. J. Nucl. Med. 2016, 57, 79s–89s. [Google Scholar] [CrossRef] [PubMed]
- Zekri, L.; Vogt, F. An IgG-based bispecific PSMAxCD3 antibody for improved dual targeting of tumor- and vascular cells in PSMA positive cancer. EMBO 2020, in press. [Google Scholar]
- Ahmed, N.; Caimi, P.; Reese, J.S.; Otegbeye, F.; Patel, S.; Schneider, D.; Boughan, K.M.; Cooper, B.; Gallogly, M.; Kruger, W.; et al. Prophylactic Tocilizumab in Patients with Relapsed Refractory Lymphoma Treated with Anti CD19 Chimeric Antigen Receptor T-Cell Therapy. Biol. Blood Marrow Transplant. 2020, 26, S275–S276. [Google Scholar] [CrossRef]
- Kauer, J.; Horner, S.; Osburg, L.; Muller, S.; Marklin, M.; Heitmann, J.S.; Zekri, L.; Rammensee, H.G.; Salih, H.R.; Jung, G. Tocilizumab, but not dexamethasone, prevents CRS without affecting antitumor activity of bispecific antibodies. J. Immunother. Cancer 2020, 8, e000621. [Google Scholar] [CrossRef]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef]
- Rathi, C.; Meibohm, B. Clinical pharmacology of bispecific antibody constructs. J. Clin. Pharmacol. 2015, 55 (Suppl. 3), S21–S28. [Google Scholar] [CrossRef]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R.; et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017, 129, 609–618. [Google Scholar] [CrossRef]
- Liu, L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell 2018, 9, 15–32. [Google Scholar] [CrossRef]
- Kohnke, T.; Krupka, C.; Tischer, J.; Knosel, T.; Subklewe, M. Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J. Hematol. Oncol. 2015, 8, 111. [Google Scholar] [CrossRef]
- Feucht, J.; Kayser, S.; Gorodezki, D.; Hamieh, M.; Doring, M.; Blaeschke, F.; Schlegel, P.; Bosmuller, H.; Quintanilla-Fend, L.; Ebinger, M.; et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget 2016, 7, 76902–76919. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Kohnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Wang, Y.; Li, R.; Rossi, D.L.; Liu, D.; Rossi, E.A.; Cardillo, T.M.; Goldenberg, D.M. Combination Therapy with Bispecific Antibodies and PD-1 Blockade Enhances the Antitumor Potency of T Cells. Cancer Res. 2017, 77, 5384–5394. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Borchmann, P.; Wagner-Johnston, N.D.; Provencio, M.; Cordoba, R.; Papadopoulos, K.; Martin, A.; Grande, C.; Jagadeesh, D.; Lakhani, N.; et al. Safety and Preliminary Antitumor Activity of the Anti-PD-1 Monoclonal Antibody REGN2810 Alone or in Combination with REGN1979, an Anti-CD20 x Anti-CD3 Bispecific Antibody, in Patients with B-Lymphoid Malignancies. Blood 2017, 130. [Google Scholar] [CrossRef]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and lb studies of the novel carcinoembryonic antigen (CEA) 1-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Dao, T.; Pankov, D.; Scott, A.; Korontsvit, T.; Zakhaleva, V.; Xu, Y.; Xiang, J.; Yan, S.; de Morais Guerreiro, M.D.; Veomett, N.; et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 2015, 33, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Adami, A.A. Antitumor immunity: Easy as 1, 2, 3 with monoclonal bispecific trifunctional antibodies? Cancer Res. 2013, 73, 5613–5617. [Google Scholar] [CrossRef] [PubMed]
- Memarnejadian, A.; Meilleur, C.E.; Shaler, C.R.; Khazaie, K.; Bennink, J.R.; Schell, T.D.; Haeryfar, S.M.M. PD-1 Blockade Promotes Epitope Spreading in Anticancer CD8(+) T Cell Responses by Preventing Fratricidal Death of Subdominant Clones To Relieve Immunodomination. J. Immunol. 2017, 199, 3348–3359. [Google Scholar] [CrossRef]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; Vom Berg, J. Rationale for Combining Bispecific T Cell Activating Antibodies with Checkpoint Blockade for Cancer Therapy. Front Oncol. 2018, 8, 285. [Google Scholar] [CrossRef]
- Manger, B.; Weiss, A.; Weyand, C.; Goronzy, J.; Stobo, J.D. T cell activation: Differences in the signals required for IL 2 production by nonactivated and activated T cells. J. Immunol. 1985, 135, 3669–3673. [Google Scholar]
- Weiss, A.; Manger, B.; Imboden, J. Synergy between the T3/antigen receptor complex and Tp44 in the activation of human T cells. J. Immunol. 1986, 137, 819–825. [Google Scholar] [PubMed]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Correnti, C.E.; Laszlo, G.S.; de van der Schueren, W.J.; Godwin, C.D.; Bandaranayake, A.; Busch, M.A.; Gudgeon, C.J.; Bates, O.M.; Olson, J.M.; Mehlin, C.; et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018, 32, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nature Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heitmann, J.S.; Pfluegler, M.; Jung, G.; Salih, H.R. Bispecific Antibodies in Prostate Cancer Therapy: Current Status and Perspectives. Cancers 2021, 13, 549. https://doi.org/10.3390/cancers13030549
Heitmann JS, Pfluegler M, Jung G, Salih HR. Bispecific Antibodies in Prostate Cancer Therapy: Current Status and Perspectives. Cancers. 2021; 13(3):549. https://doi.org/10.3390/cancers13030549
Chicago/Turabian StyleHeitmann, Jonas S., Martin Pfluegler, Gundram Jung, and Helmut R. Salih. 2021. "Bispecific Antibodies in Prostate Cancer Therapy: Current Status and Perspectives" Cancers 13, no. 3: 549. https://doi.org/10.3390/cancers13030549
APA StyleHeitmann, J. S., Pfluegler, M., Jung, G., & Salih, H. R. (2021). Bispecific Antibodies in Prostate Cancer Therapy: Current Status and Perspectives. Cancers, 13(3), 549. https://doi.org/10.3390/cancers13030549