Role of Hypoxia-Mediated Autophagy in Tumor Cell Death and Survival

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
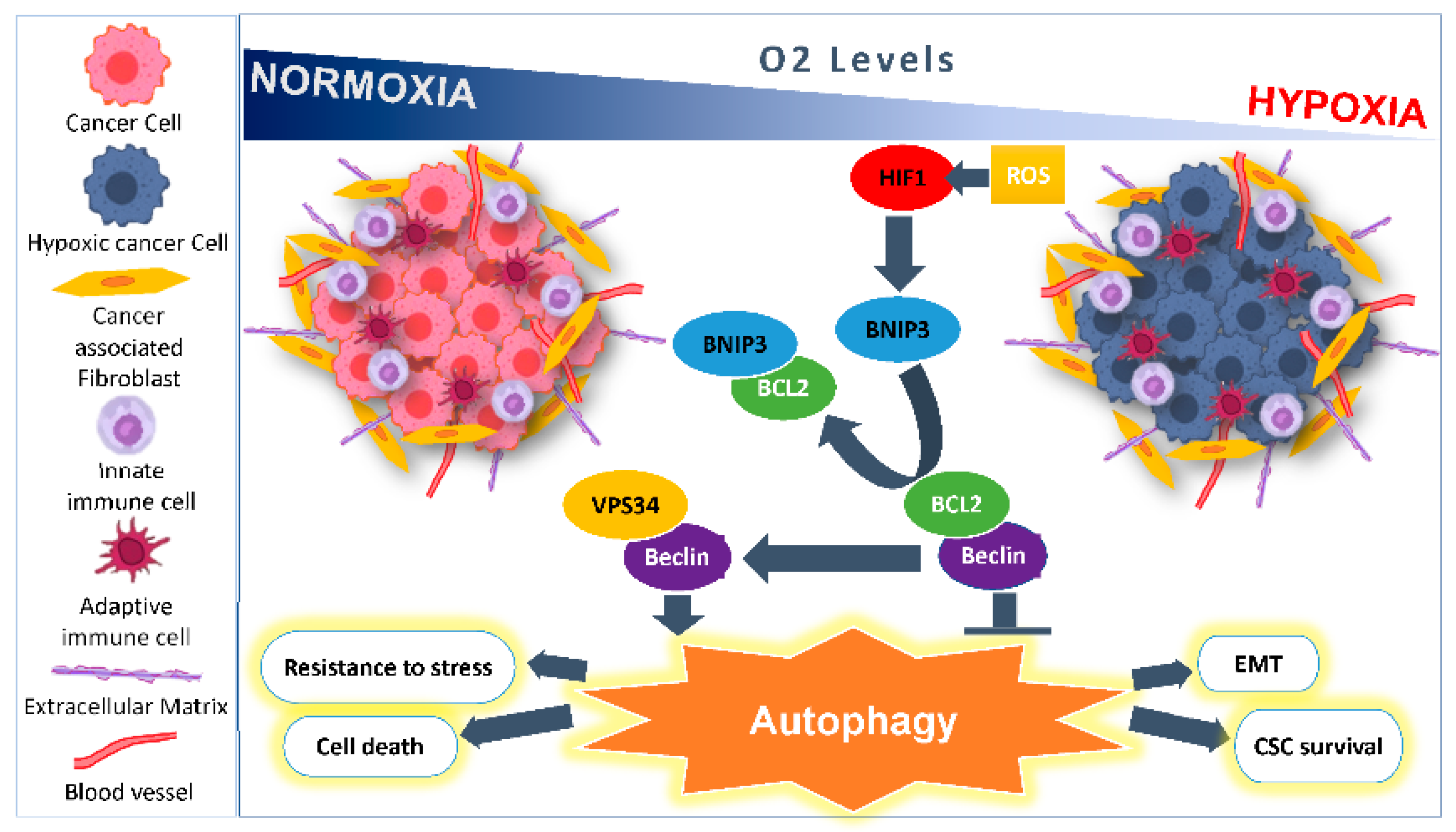
2. Microenvironmental Hypoxic Stress Induced Autophagy
2.1. As a Survival Mechanism for Hypoxic Cells
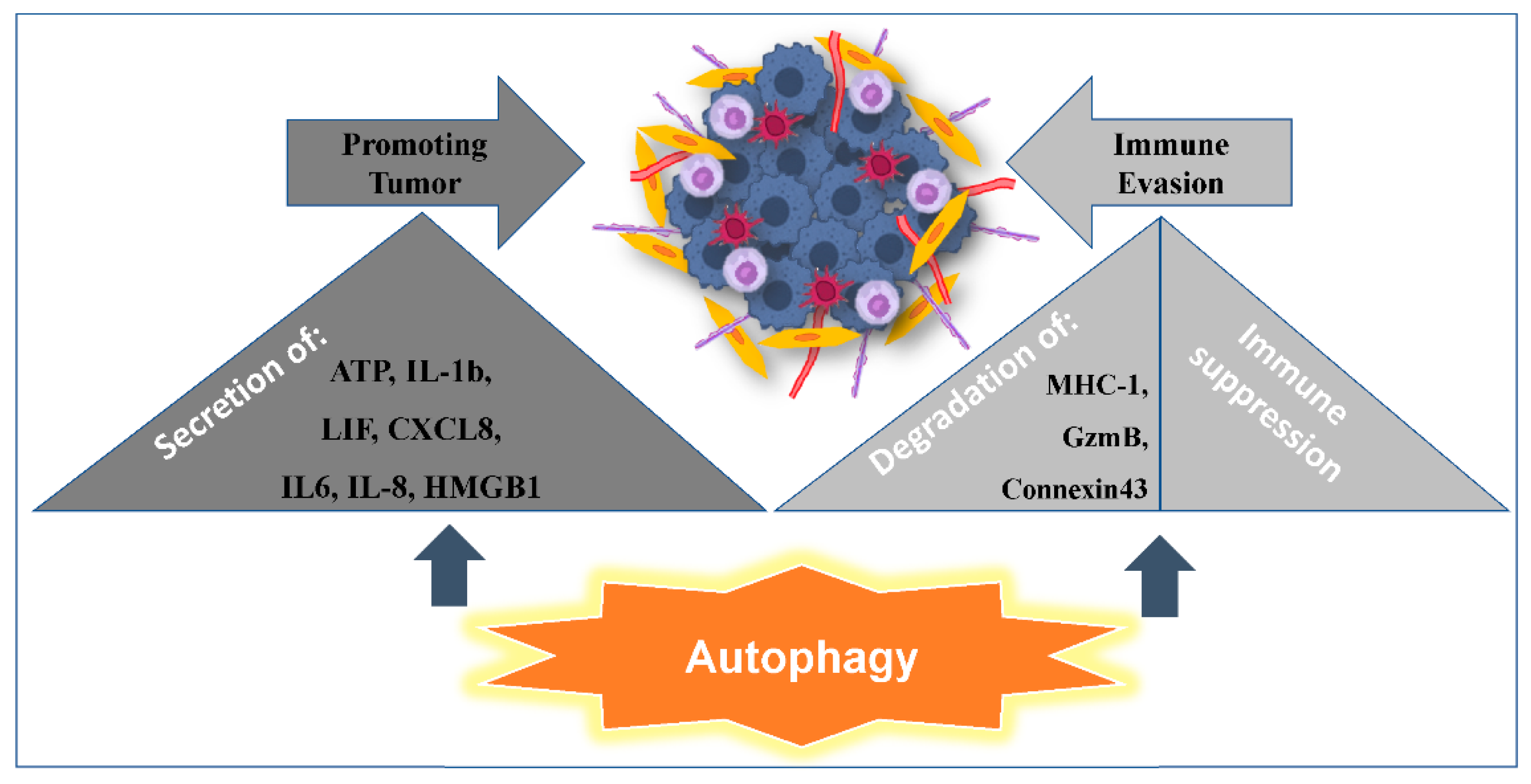
2.2. Autophagy Promoting Immunosuppression
2.3. Autophagy Promoting Tumor Resistance to Immune Cell-Mediated Cytotoxicity
2.4. Microenvironmental Hypoxic Stress Induced Autophagy as a Modulator of Tumor Immunogenicity and Adjuvanticity
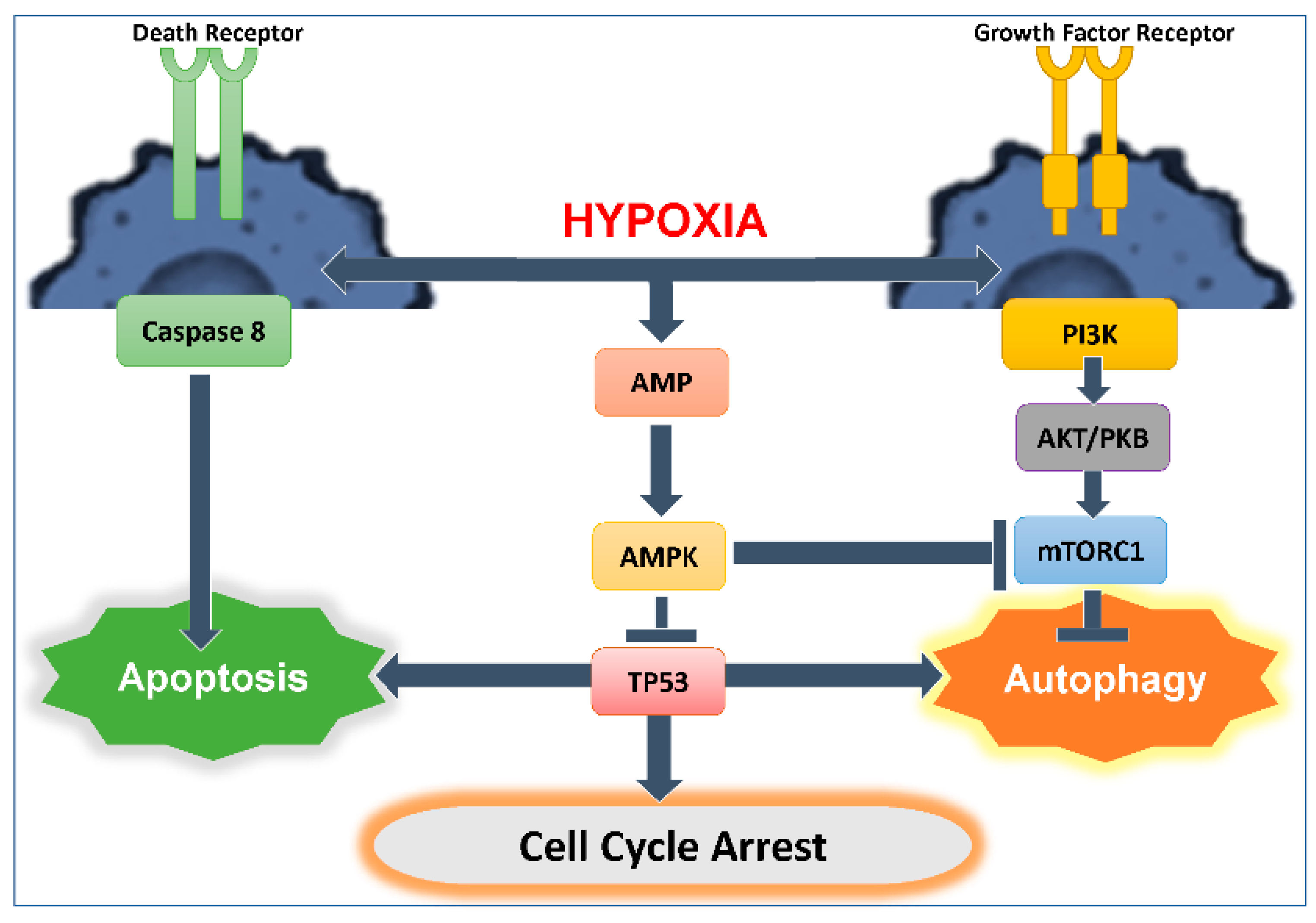
3. Autophagy and Apoptosis Relationship
3.1. Interplay between Autophagy and Apoptosis
3.2. Role of TP53 in Autophagy Induction and Regulation: Influence on Anti-Tumor Cytotoxic Immune Response
4. Hypoxia Role in Linking Autophagy and Cancer Stem Cells
5. Key Signaling Pathways Impacted by Autophagy in CSCs
6. Autophagy and Promotion of Inflammation
7. Targeting Autophagy to Improve Current Cutting-Edge Therapy Approaches
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vomero, M.; Manganelli, V.; Barbati, C.; Colasanti, T.; Capozzi, A.; Finucci, A.; Spinelli, F.R.; Ceccarelli, F.; Perricone, C.; Truglia, S.; et al. Reduction of autophagy and increase in apoptosis correlates with a favorable clinical outcome in patients with rheumatoid arthritis treated with anti-TNF drugs. Arthritis Res. Ther. 2019, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The lysosome. Sci. Am. 1963, 208, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Cojocari, D.; Keulers, T.G.; Jutten, B.; Starmans, M.H.; de Jong, M.C.; Begg, A.C.; Savelkouls, K.G.; Bussink, J.; Vooijs, M.; et al. The autophagy associated gene, ULK1, promotes tolerance to chronic and acute hypoxia. Radiother. Oncol. 2013, 108, 529–534. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Q.; Tan, J.; Fang, Y.; An, X.; Chen, B. Autophagy and hippocampal neuronal injury. Sleep Breath 2014, 18, 243–249. [Google Scholar] [CrossRef]
- Liu, H.; He, Z.; Simon, H.U. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin. Cancer Biol. 2013, 23, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, H.L. Death and survival of neuronal and astrocytic cells in ischemic brain injury: A role of autophagy. Acta Pharmacol. Sin. 2011, 32, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Azad, M.B.; Gibson, S.B. Role of BNIP3 in proliferation and hypoxia-induced autophagy: Implications for personalized cancer therapies. Ann. N. Y. Acad. Sci. 2010, 1210, 8–16. [Google Scholar] [CrossRef]
- Cerrada, I.; Ruiz-Sauri, A.; Carrero, R.; Trigueros, C.; Dorronsoro, A.; Sanchez-Puelles, J.M.; Diez-Juan, A.; Montero, J.A.; Sepulveda, P. Hypoxia-inducible factor 1 alpha contributes to cardiac healing in mesenchymal stem cells-mediated cardiac repair. Stem Cells Dev. 2013, 22, 501–511. [Google Scholar] [CrossRef]
- Gui, L.; Liu, B.; Lv, G. Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp Ther Med 2016, 11, 2233–2239. [Google Scholar] [CrossRef]
- Abdul Rahim, S.A.; Dirkse, A.; Oudin, A.; Schuster, A.; Bohler, J.; Barthelemy, V.; Muller, A.; Vallar, L.; Janji, B.; Golebiewska, A.; et al. Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br. J. Cancer 2017, 117, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.; Yao, W.; Alsiddig, M.C.; Huo, L.; Liu, H.; Miao, Y.L. Hypoxia-inducible factor-1alpha-dependent autophagy plays a role in glycolysis switch in mouse granulosa cells. Biol. Reprod. 2018, 99, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Moschetta, M.; Reale, A.; Marasco, C.; Vacca, A.; Carratu, M.R. Therapeutic targeting of the mTOR-signalling pathway in cancer: Benefits and limitations. Br. J. Pharmacol. 2014, 171, 3801–3813. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- DePavia, A.; Jonasch, E.; Liu, X.D. Autophagy degrades hypoxia inducible factors. Mol. Cell Oncol. 2016, 3, e1104428. [Google Scholar] [CrossRef]
- Wu, H.M.; Jiang, Z.F.; Ding, P.S.; Shao, L.J.; Liu, R.Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef]
- An, H.J.; Maeng, O.; Kang, K.H.; Lee, J.O.; Kim, Y.S.; Paik, S.G.; Lee, H. Activation of Ras up-regulates pro-apoptotic BNIP3 in nitric oxide-induced cell death. J. Biol. Chem. 2006, 281, 33939–33948. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Yurkova, N.; Zhang, T.; Gang, H.; Aguilar, F.; Weidman, D.; Scramstad, C.; Weisman, H.; Kirshenbaum, L.A. Antagonism of E2F-1 regulated Bnip3 transcription by NF-kappaB is essential for basal cell survival. Proc. Natl. Acad. Sci. USA 2008, 105, 20734–20739. [Google Scholar] [CrossRef] [PubMed]
- Yurkova, N.; Shaw, J.; Blackie, K.; Weidman, D.; Jayas, R.; Flynn, B.; Kirshenbaum, L.A. The cell cycle factor E2F-1 activates Bnip3 and the intrinsic death pathway in ventricular myocytes. Circ. Res. 2008, 102, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Burton, T.R.; Henson, E.S.; Baijal, P.; Eisenstat, D.D.; Gibson, S.B. The pro-cell death Bcl-2 family member, BNIP3, is localized to the nucleus of human glial cells: Implications for glioblastoma multiforme tumor cell survival under hypoxia. Int. J. Cancer 2006, 118, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Rouleau, M.; Gounon, P.; Brahimi-Horn, M.C.; Pouyssegur, J.; Mazure, N.M. Hypoxic enlarged mitochondria protect cancer cells from apoptotic stimuli. J. Cell Physiol. 2010, 222, 648–657. [Google Scholar] [CrossRef]
- Ma, Y.; Zhou, Y.; Zhu, Y.C.; Wang, S.Q.; Ping, P.; Chen, X.F. Lipophagy Contributes to Testosterone Biosynthesis in Male Rat Leydig Cells. Endocrinology 2018, 159, 1119–1129. [Google Scholar] [CrossRef]
- Buart, S.; Terry, S.; Noman, M.Z.; Lanoy, E.; Boutros, C.; Fogel, P.; Dessen, P.; Meurice, G.; Gaston-Mathe, Y.; Vielh, P.; et al. Transcriptional response to hypoxic stress in melanoma and prognostic potential of GBE1 and BNIP3. Oncotarget 2017, 8, 108786–108801. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Macleod, K.F. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 2015, 11, 1937–1938. [Google Scholar] [CrossRef]
- Okami, J.; Simeone, D.M.; Logsdon, C.D. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 2004, 64, 5338–5346. [Google Scholar] [CrossRef]
- He, J.; Pei, L.; Jiang, H.; Yang, W.; Chen, J.; Liang, H. Chemoresistance of colorectal cancer to 5-fluorouracil is associated with silencing of the BNIP3 gene through aberrant methylation. J. Cancer 2017, 8, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Liu, Z.; Liu, J.; Wang, H.; Huang, L.; Lin, T.; Liu, J.; Wei, Q.; Zeng, H.; He, G.; et al. Expression and epigenetic regulatory mechanism of BNIP3 in clear cell renal cell carcinoma. Int. J. Oncol. 2019, 54, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Toyota, M.; Suzuki, H.; Murai, M.; Akino, K.; Ueno, M.; Nojima, M.; Yawata, A.; Miyakawa, H.; Suga, T.; et al. Upregulation of BNIP3 by 5-aza-2’-deoxycytidine sensitizes pancreatic cancer cells to hypoxia-mediated cell death. J. Gastroenterol. 2005, 40, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Koop, E.A.; van Laar, T.; van Wichen, D.F.; de Weger, R.A.; Wall, E.; van Diest, P.J. Expression of BNIP3 in invasive breast cancer: Correlations with the hypoxic response and clinicopathological features. BMC Cancer 2009, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Blanco, C.; Fondevila, F.; Fernández-Palanca, P.; García-Palomo, A.; Pelt, J.V.; Verslype, C.; González-Gallego, J.; Mauriz, J.L. Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells. Cancers 2019, 11, 1984. [Google Scholar] [CrossRef]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/eIF2alpha arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15, R2. [Google Scholar] [CrossRef]
- Meijer, T.W.; Kaanders, J.H.; Span, P.N.; Bussink, J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin. Cancer Res. 2012, 18, 5585–5594. [Google Scholar] [CrossRef]
- Roy, S.; Leidal, A.M.; Ye, J.; Ronen, S.M.; Debnath, J. Autophagy-Dependent Shuttling of TBC1D5 Controls Plasma Membrane Translocation of GLUT1 and Glucose Uptake. Mol. Cell 2017, 67, 84–95. [Google Scholar] [CrossRef]
- Grohm, J.; Plesnila, N.; Culmsee, C. Bid mediates fission, membrane permeabilization and peri-nuclear accumulation of mitochondria as a prerequisite for oxidative neuronal cell death. Brain Behav. Immun. 2010, 24, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.T.; Shyue, S.K.; Lai, M.K. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation 2007, 84, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besancon, F.; Bauvy, C.; Souquere, S.; Pierron, G.; Codogno, P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef]
- Jeong, J.K.; Gurunathan, S.; Kang, M.H.; Han, J.W.; Das, J.; Choi, Y.J.; Kwon, D.N.; Cho, S.G.; Park, C.; Seo, H.G.; et al. Hypoxia-mediated autophagic flux inhibits silver nanoparticle-triggered apoptosis in human lung cancer cells. Sci. Rep. 2016, 6, 21688. [Google Scholar] [CrossRef] [PubMed]
- Rikka, S.; Quinsay, M.N.; Thomas, R.L.; Kubli, D.A.; Zhang, X.; Murphy, A.N.; Gustafsson, A.B. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011, 18, 721–731. [Google Scholar] [CrossRef]
- Janji, B.; Viry, E.; Moussay, E.; Paggetti, J.; Arakelian, T.; Mgrditchian, T.; Messai, Y.; Noman, M.Z.; Van Moer, K.; Hasmim, M.; et al. The multifaceted role of autophagy in tumor evasion from immune surveillance. Oncotarget 2016, 7, 17591–17607. [Google Scholar] [CrossRef]
- Wen, Z.F.; Liu, H.; Gao, R.; Zhou, M.; Ma, J.; Zhang, Y.; Zhao, J.; Chen, Y.; Zhang, T.; Huang, F.; et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J. Immun. Cancer 2018, 6, 151. [Google Scholar] [CrossRef]
- Connell, P.P.; Weichselbaum, R.R. A downside to apoptosis in cancer therapy? Med. 2011, 17, 780–782. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef]
- Garg, A.D.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.V.; Mathieu, C.; Agostinis, P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S.; et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, E9271–E9279. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Alissafi, T.; Hatzioannou, A.; Mintzas, K.; Barouni, R.M.; Banos, A.; Sormendi, S.; Polyzos, A.; Xilouri, M.; Wielockx, B.; Gogas, H.; et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Invest. 2018, 128, 3840–3852. [Google Scholar] [CrossRef] [PubMed]
- Lawson, K.A.; Sousa, C.M.; Zhang, X.; Kim, E.; Akthar, R.; Caumanns, J.J.; Yao, Y.; Mikolajewicz, N.; Ross, C.; Brown, K.R.; et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 2020, 586, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Demachi-Okamura, A.; Torikai, H.; Akatsuka, Y.; Miyoshi, H.; Yoshimori, T.; Kuzushima, K. Autophagy creates a CTL epitope that mimics tumor-associated antigens. PLoS ONE 2012, 7, e47126. [Google Scholar] [CrossRef]
- Akalay, I.; Tan, T.Z.; Kumar, P.; Janji, B.; Mami-Chouaib, F.; Charpy, C.; Vielh, P.; Larsen, A.K.; Thiery, J.P.; Sabbah, M.; et al. Targeting WNT1-inducible signaling pathway protein 2 alters human breast cancer cell susceptibility to specific lysis through regulation of KLF-4 and miR-7 expression. Oncogene 2015, 34, 2261–2271. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Tittarelli, A.; Janji, B.; Van Moer, K.; Noman, M.Z.; Chouaib, S. The Selective Degradation of Synaptic Connexin 43 Protein by Hypoxia-induced Autophagy Impairs Natural Killer Cell-mediated Tumor Cell Killing. J. Biol. Chem. 2015, 290, 23670–23679. [Google Scholar] [CrossRef]
- Messai, Y.; Noman, M.Z.; Hasmim, M.; Janji, B.; Tittarelli, A.; Boutet, M.; Baud, V.; Viry, E.; Billot, K.; Nanbakhsh, A.; et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. 2014, 74, 6820–6832. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Engelsen, A.S.T.; Buart, S.; Elsayed, W.S.; Venkatesh, G.H.; Chouaib, S. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett. 2020, 492, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Autophagy in Immune Response: Impact on Cancer Immunotherapy; Chouaib, S., Ed.; Academic Press: London, UK, 2020; pp. i–iii. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Bloy, N.; Garcia, P.; Laumont, C.M.; Pitt, J.M.; Sistigu, A.; Stoll, G.; Yamazaki, T.; Bonneil, E.; Buque, A.; Humeau, J.; et al. Immunogenic stress and death of cancer cells: Contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol. Rev. 2017, 280, 165–174. [Google Scholar] [CrossRef]
- Gao, D. Compound-therapy based on cancer-immunity cycle: Promising prospects for antitumor regimens. Am. J. Cancer Res. 2019, 9, 212–218. [Google Scholar]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0 s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Hou, W.; Zhang, Q.; Yan, Z.; Chen, R.; Zeh Iii, H.J.; Kang, R.; Lotze, M.T.; Tang, D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013, 4, e966. [Google Scholar] [CrossRef]
- Zhang, Q.; Kang, R.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. DAMPs and autophagy: Cellular adaptation to injury and unscheduled cell death. Autophagy 2013, 9, 451–458. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct Target Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H. [Immunohistochemical study of schwannoma--with respect to the histogenesis of Antoni B area]. Nihon Seikeigeka Gakkai Zasshi 1989, 63, 800–809. [Google Scholar]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Kim, I.; Lemasters, J.J. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox Signal 2011, 14, 1919–1928. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, T.; Wang, J.; Zhang, Z.; Zhai, Y.; Yang, G.Y.; Sun, X. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem. Biophys. Res. Commun. 2014, 444, 182–188. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.R.; Wang, X.; Hu, W.W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef]
- Zhang, R.; Zhu, F.; Ren, J.; Huang, L.; Liu, P.; Wu, G. Beclin1/PI3K-mediated autophagy prevents hypoxia-induced apoptosis in EAhy926 cell line. Cancer Biother. Radiopharm. 2011, 26, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010, 6, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Shravage, B.V.; Sagona, A.P.; Lamark, T.; Bjorkoy, G.; Johansen, T.; Rusten, T.E.; Brech, A.; Baehrecke, E.H.; Stenmark, H. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J. Cell Biol. 2010, 190, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009, 16, 1093–1107. [Google Scholar] [CrossRef]
- Betin, V.M.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef]
- Oral, O.; Oz-Arslan, D.; Itah, Z.; Naghavi, A.; Deveci, R.; Karacali, S.; Gozuacik, D. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 2012, 17, 810–820. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Puyal, J.; Vaslin, A.; Mottier, V.; Clarke, P.G. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann. Neurol. 2009, 66, 378–389. [Google Scholar] [CrossRef]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef]
- Shen, H.M.; Codogno, P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011, 7, 457–465. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kepp, O.; Kroemer, G. The end of autophagic cell death? Autophagy 2012, 8, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Abrams, J. p53: The Janus of autophagy? Nat. Cell Biol. 2008, 10, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Di, J.; Cao, H.; Bai, J.; Zheng, J. p53-mediated autophagic regulation: A prospective strategy for cancer therapy. Cancer Lett. 2015, 363, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Malik, S.A.; Morselli, E.; Kepp, O.; Criollo, A.; Mouchel, P.L.; Carnuccio, R.; Kroemer, G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 2009, 8, 1571–1576. [Google Scholar] [CrossRef]
- Gao, W.; Shen, Z.; Shang, L.; Wang, X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011, 18, 1598–1607. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Frohlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef]
- Chollat-Namy, M.; Ben Safta-Saadoun, T.; Haferssas, D.; Meurice, G.; Chouaib, S.; Thiery, J. The pharmalogical reactivation of p53 function improves breast tumor cell lysis by granzyme B and NK cells through induction of autophagy. Cell Death Dis. 2019, 10, 695. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Buart, S.; Tan, T.Z.; Gros, G.; Noman, M.Z.; Lorens, J.B.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: Consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology 2017, 6, e1271858. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1alpha Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef]
- Nakuluri, K.; Mukhi, D.; Nishad, R.; Saleem, M.A.; Mungamuri, S.K.; Menon, R.K.; Pasupulati, A.K. Hypoxia induces ZEB2 in podocytes: Implications in the pathogenesis of proteinuria. J. Cell Physiol. 2019, 234, 6503–6518. [Google Scholar] [CrossRef]
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. Snail Is a Direct Target of Hypoxia-inducible Factor 1alpha (HIF1alpha) in Hypoxia-induced Endothelial to Mesenchymal Transition of Human Coronary Endothelial Cells. J. Biol. Chem. 2015, 290, 16653–16664. [Google Scholar] [CrossRef]
- Terry, S.; Faouzi Zaarour, R.; Hassan Venkatesh, G.; Francis, A.; El-Sayed, W.; Buart, S.; Bravo, P.; Thiery, J.; Chouaib, S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. Int. J. Mol. Sci. 2018, 19, 44. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Tanaka, F.; Morita, H.; Hiraki, A.; Hashimoto, S. Hypoxia-induced HIF-1alpha and ZEB1 are critical for the malignant transformation of ameloblastoma via TGF-beta-dependent EMT. Cancer Med. 2019, 8, 7822–7832. [Google Scholar] [CrossRef]
- Tong, W.W.; Tong, G.H.; Liu, Y. Cancer stem cells and hypoxia-inducible factors (Review). Int. J. Oncol. 2018, 53, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Dynamic regulation of stem cell specification and maintenance by hypoxia-inducible factors. Mol. Aspects Med. 2016, 47–48, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.; Lotze, M.; Tang, D. The Multifaceted Effects of Autophagy on the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1225, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Li, X.; Tan, R.; An, J.; Cai, Z.; Hu, X.; Wang, F.; Wang, H.; Lu, C.; Lu, H. HIF-1alpha/Beclin1-Mediated Autophagy Is Involved in Neuroprotection Induced by Hypoxic Preconditioning. J. Mol. Neurosci. 2018, 66, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Hasmim, M.; Janji, B.; Khaled, M.; Noman, M.Z.; Louache, F.; Bordereaux, D.; Abderamane, A.; Baud, V.; Mami-Chouaib, F.; Chouaib, S. Cutting Edge: NANOG Activates Autophagy under Hypoxic Stress by Binding to BNIP3L Promoter. J. Immunol. 2017, 198, 1423–1428. [Google Scholar] [CrossRef]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef]
- Yan, W.; Chen, Z.; Chen, J.; Chen, H. Isoflurane preconditioning protects rat brain from ischemia reperfusion injury via up-regulating the HIF-1alpha expression through Akt/mTOR/s6K activation. Cell Mol. Biol. (Noisy-le-grand) 2016, 62, 38–44. [Google Scholar]
- Agani, F.; Jiang, B.H. Oxygen-independent regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr. Cancer Drug Targets 2013, 13, 245–251. [Google Scholar] [CrossRef]
- Matsumoto, K.; Arao, T.; Tanaka, K.; Kaneda, H.; Kudo, K.; Fujita, Y.; Tamura, D.; Aomatsu, K.; Tamura, T.; Yamada, Y.; et al. mTOR signal and hypoxia-inducible factor-1 alpha regulate CD133 expression in cancer cells. Cancer Res. 2009, 69, 7160–7164. [Google Scholar] [CrossRef]
- Matsubara, S.; Ding, Q.; Miyazaki, Y.; Kuwahata, T.; Tsukasa, K.; Takao, S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci. Rep. 2013, 3, 3230. [Google Scholar] [CrossRef] [PubMed]
- Losuwannarak, N.; Maiuthed, A.; Kitkumthorn, N.; Leelahavanichkul, A.; Roytrakul, S.; Chanvorachote, P. Gigantol Targets Cancer Stem Cells and Destabilizes Tumors via the Suppression of the PI3K/AKT and JAK/STAT Pathways in Ectopic Lung Cancer Xenografts. Cancers 2019, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ding, X.; Ye, H.; Wang, J.; Shao, J.; Huang, T. Hypoxia enhances the migration and invasion of human glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1alpha pathway. Neuroreport 2018, 29, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Shi, B.; Ma, M.; Zheng, Y.; Pan, Y.; Lin, X. mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J. Cell Physiol. 2019, 234, 12562–12568. [Google Scholar] [CrossRef]
- Seong, M.; Lee, J.; Kang, H. Hypoxia-induced regulation of mTOR signaling by miR-7 targeting REDD1. J. Cell Biochem. 2019, 120, 4523–4532. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.; Xu, Y.; Liu, J.; Xie, M.; Ni, W.; Chen, S. KLF5 promotes hypoxia-induced survival and inhibits apoptosis in non-small cell lung cancer cells via HIF-1alpha. Int. J. Oncol. 2014, 45, 1507–1514. [Google Scholar] [CrossRef]
- Gong, T.; Cui, L.; Wang, H.; Wang, H.; Han, N. Knockdown of KLF5 suppresses hypoxia-induced resistance to cisplatin in NSCLC cells by regulating HIF-1alpha-dependent glycolysis through inactivation of the PI3K/Akt/mTOR pathway. J. Transl. Med. 2018, 16, 164. [Google Scholar] [CrossRef]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011, 10, 3871–3885. [Google Scholar] [CrossRef]
- Yan, C.; Zhao, J.; Qin, Y.; Zhao, F.; Ji, L.; Zhang, J. Overexpression of ATG4a promotes autophagy and proliferation, and inhibits apoptosis in lens epithelial cells via the AMPK and Akt pathways. Mol. Med. Rep. 2020, 22, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Chen, B.; Mok, H.; Zhang, G.; Ren, C.; Liao, N. Comprehensive analysis of autophagy-related prognostic genes in breast cancer. J. Cell Mol. Med. 2020, 24, 9145–9153. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Zhu, G.; Rong, X.; Zhou, Y.; Jiang, P.; Chen, P. Upregulation of ATG4A promotes osteosarcoma cell epithelial-to-mesenchymal transition through the Notch signaling pathway. Int. J. Clin. Exp. Pathol. 2017, 10, 7975–7982. [Google Scholar] [PubMed]
- Yang, S.W.; Ping, Y.F.; Jiang, Y.X.; Luo, X.; Zhang, X.; Bian, X.W.; Yu, P.W. ATG4A promotes tumor metastasis by inducing the epithelial-mesenchymal transition and stem-like properties in gastric cells. Oncotarget 2016, 7, 39279–39292. [Google Scholar] [CrossRef] [PubMed]
- Song, B.Q.; Chi, Y.; Li, X.; Du, W.J.; Han, Z.B.; Tian, J.J.; Li, J.J.; Chen, F.; Wu, H.H.; Han, L.X.; et al. Inhibition of Notch Signaling Promotes the Adipogenic Differentiation of Mesenchymal Stem Cells Through Autophagy Activation and PTEN-PI3K/AKT/mTOR Pathway. Cell Physiol. Biochem. 2015, 36, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Muller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15, R109. [Google Scholar] [CrossRef]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Shen, H.; Yin, L.; Deng, G.; Guo, C.; Han, Y.; Li, Y.; Cai, C.; Fu, Y.; Liu, S.; Zeng, S. Knockdown of Beclin-1 impairs epithelial-mesenchymal transition of colon cancer cells. J. Cell Biochem. 2018, 119, 7022–7031. [Google Scholar] [CrossRef]
- Hu, F.; Li, G.; Huang, C.; Hou, Z.; Yang, X.; Luo, X.; Feng, Y.; Wang, G.; Hu, J.; Cao, Z. The autophagy-independent role of BECN1 in colorectal cancer metastasis through regulating STAT3 signaling pathway activation. Cell Death Dis. 2020, 11, 304. [Google Scholar] [CrossRef]
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell Mol. Biol. Lett. 2018, 23, 12. [Google Scholar] [CrossRef]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Larrue, C.; Heydt, Q.; Saland, E.; Boutzen, H.; Kaoma, T.; Sarry, J.E.; Joffre, C.; Recher, C. Oncogenic KIT mutations induce STAT3-dependent autophagy to support cell proliferation in acute myeloid leukemia. Oncogenesis 2019, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Min, Y.; Im, J.S.; Son, J.; Lee, J.S.; Lee, K.Y. p62 is Negatively Implicated in the TRAF6-BECN1 Signaling Axis for Autophagy Activation and Cancer Progression by Toll-Like Receptor 4 (TLR4). Cells 2020, 9, 1142. [Google Scholar] [CrossRef]
- Xu, L.Z.; Li, S.S.; Zhou, W.; Kang, Z.J.; Zhang, Q.X.; Kamran, M.; Xu, J.; Liang, D.P.; Wang, C.L.; Hou, Z.J.; et al. p62/SQSTM1 enhances breast cancer stem-like properties by stabilizing MYC mRNA. Oncogene 2017, 36, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.G.; Choi, B.H.; Ku, S.K.; Kwak, M.K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef]
- Tamura, K.; Watanabe, K.; Matsushita, Y.; Watanabe, H.; Motoyama, D.; Ito, T.; Sugiyama, T.; Otsuka, A.; Miyake, H. Enhanced Sensitivity to NVP-BEZ235 by Inhibition of p62/SQSTM1 in Human Bladder Cancer KoTCC-1 Cells Both In Vitro and In Vivo. In Vivo 2020, 34, 1001–1008. [Google Scholar] [CrossRef]
- Kraya, A.A.; Piao, S.; Xu, X.; Zhang, G.; Herlyn, M.; Gimotty, P.; Levine, B.; Amaravadi, R.K.; Speicher, D.W. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy 2015, 11, 60–74. [Google Scholar] [CrossRef]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Lin, Z.; Flomenberg, N.; Howell, A.; Pestell, R.G.; Lisanti, M.P.; Sotgia, F. Cytokine production and inflammation drive autophagy in the tumor microenvironment: Role of stromal caveolin-1 as a key regulator. Cell Cycle 2011, 10, 1784–1793. [Google Scholar] [CrossRef]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, J.; Horita, H.; Redzic, J.; Hansen, K.; Frankel, A.E.; Thorburn, A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009, 16, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.X.; Wong, S.H.; Chan, M.T.; Yu, L.; Yu, S.S.; Wu, F.; Xiao, Z.; Wang, X.; Zhang, L.; Cheng, A.S.; et al. Autophagy Mediates HBx-Induced Nuclear Factor-kappaB Activation and Release of IL-6, IL-8, and CXCL2 in Hepatocytes. J. Cell Physiol. 2015, 230, 2382–2389. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; Ottewell, P. The role of IL-1B in breast cancer bone metastasis. Endocr. Relat. Cancer 2018, 25, R421–R434. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.C.; Bellamkonda, K.; Alex Molina, L.; Xiang, S.; Liska, D.; Sarvestani, S.K.; Chakrabarti, S.; Berg, A.; Jorgensen, M.L.; Hatala, D.; et al. Disrupting Inflammation-Associated CXCL8-CXCR1 Signaling Inhibits Tumorigenicity Initiated by Sporadic- and Colitis-Colon Cancer Stem Cells. Neoplasia 2019, 21, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.C.; Tsang, N.M.; Chiang, W.C.; Chang, K.P.; Hsueh, C.; Liang, Y.; Juang, J.L.; Chow, K.P.; Chang, Y.S. Leukemia inhibitory factor promotes nasopharyngeal carcinoma progression and radioresistance. J. Clin. Investig. 2013, 123, 5269–5283. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Alfaro, C.; Sanmamed, M.F.; Rodriguez-Ruiz, M.E.; Teijeira, A.; Onate, C.; Gonzalez, A.; Ponz, M.; Schalper, K.A.; Perez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Song, M.; Ping, Y.; Zhang, K.; Yang, L.; Li, F.; Zhang, C.; Cheng, S.; Yue, D.; Maimela, N.R.; Qu, J.; et al. Low-Dose IFNgamma Induces Tumor Cell Stemness in Tumor Microenvironment of Non-Small Cell Lung Cancer. Cancer Res. 2019, 79, 3737–3748. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.S. Stimulatory versus suppressive effects of GM-CSF on tumor progression in multiple cancer types. Exp. Mol. Med. 2016, 48, e242. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Cheng, J.; Sun, L.; Wang, Y.; Wang, C.; Liu, X.; Zhang, Z.; Zhao, M.; Luo, Y.; Tian, L.; et al. HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via paracrine effect. Cell Death Dis. 2018, 9, 648. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Wang, X.; Li, Y.; Cao, Y.; Chen, X. Extracellular ATP a New Player in Cancer Metabolism: NSCLC Cells Internalize ATP In Vitro and In Vivo Using Multiple Endocytic Mechanisms. Mol. Cancer Res. 2016, 14, 1087–1096. [Google Scholar] [CrossRef]
- Townsend, K.N.; Hughson, L.R.; Schlie, K.; Poon, V.I.; Westerback, A.; Lum, J.J. Autophagy inhibition in cancer therapy: Metabolic considerations for antitumor immunity. Immunol. Rev. 2012, 249, 176–194. [Google Scholar] [CrossRef]
- Rao, S.; Yang, H.; Penninger, J.M.; Kroemer, G. Autophagy in non-small cell lung carcinogenesis: A positive regulator of antitumor immunosurveillance. Autophagy 2014, 10, 529–531. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Zizkovsky, V.; Koubova, J.; Havranova, M.; Chudy, D.; Fuchs, V. [Alpha-1-fetoprotein in the amniotic fluid and blood of pregnant women]. Cesk. Gynekol. 1988, 53, 330–339. [Google Scholar]
- Rebecca, V.W.; Nicastri, M.C.; McLaughlin, N.; Fennelly, C.; McAfee, Q.; Ronghe, A.; Nofal, M.; Lim, C.Y.; Witze, E.; Chude, C.I.; et al. A Unified Approach to Targeting the Lysosome’s Degradative and Growth Signaling Roles. Cancer Discov. 2017, 7, 1266–1283. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol. 2020, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Dong, R. Transcriptional regulation of autophagy-lysosomal pathway in cancer. Thorac. Cancer 2020, 11, 216–223. [Google Scholar] [CrossRef]
- Qureshi-Baig, K.; Kuhn, D.; Viry, E.; Pozdeev, V.I.; Schmitz, M.; Rodriguez, F.; Ullmann, P.; Koncina, E.; Nurmik, M.; Frasquilho, S.; et al. Hypoxia-induced autophagy drives colorectal cancer initiation and progression by activating the PRKC/PKC-EZR (ezrin) pathway. Autophagy 2020, 16, 1436–1452. [Google Scholar] [CrossRef] [PubMed]
- Koom, W.S.; Sai, S.; Suzuki, M.; Fujimori, A.; Yamada, S.; Tsujii, H. Superior Effect of the Combination of Carbon-Ion Beam Irradiation and 5-Fluorouracil on Colorectal Cancer Stem Cells in vitro and in vivo. Onco. Targets Ther. 2020, 13, 12625–12635. [Google Scholar] [CrossRef]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaarour, R.F.; Azakir, B.; Hajam, E.Y.; Nawafleh, H.; Zeinelabdin, N.A.; Engelsen, A.S.T.; Thiery, J.; Jamora, C.; Chouaib, S. Role of Hypoxia-Mediated Autophagy in Tumor Cell Death and Survival. Cancers 2021, 13, 533. https://doi.org/10.3390/cancers13030533
Zaarour RF, Azakir B, Hajam EY, Nawafleh H, Zeinelabdin NA, Engelsen AST, Thiery J, Jamora C, Chouaib S. Role of Hypoxia-Mediated Autophagy in Tumor Cell Death and Survival. Cancers. 2021; 13(3):533. https://doi.org/10.3390/cancers13030533
Chicago/Turabian StyleZaarour, Rania F., Bilal Azakir, Edries Y. Hajam, Husam Nawafleh, Nagwa A. Zeinelabdin, Agnete S.T. Engelsen, Jérome Thiery, Colin Jamora, and Salem Chouaib. 2021. "Role of Hypoxia-Mediated Autophagy in Tumor Cell Death and Survival" Cancers 13, no. 3: 533. https://doi.org/10.3390/cancers13030533
APA StyleZaarour, R. F., Azakir, B., Hajam, E. Y., Nawafleh, H., Zeinelabdin, N. A., Engelsen, A. S. T., Thiery, J., Jamora, C., & Chouaib, S. (2021). Role of Hypoxia-Mediated Autophagy in Tumor Cell Death and Survival. Cancers, 13(3), 533. https://doi.org/10.3390/cancers13030533