
Diagnosis of Lynch Syndrome and Strategies to Distinguish Lynch-Related Tumors from Sporadic MSI/dMMR Tumors


Simple Summary
Abstract
1. Introduction
2. Tumor Characteristics in Lynch Syndrome
2.1. Histopathology
2.1.1. Colorectal Cancers
2.1.2. Endometrial Cancers
2.1.3. Ovarian Cancers
2.1.4. Urothelial Carcinomas
2.1.5. Non-Colorectal Digestive Cancers
2.1.6. Sebaceous Tumors
2.1.7. Brain Tumors
2.1.8. Others Cancers
2.2. MSI and Immunohistochemistry in LS
2.2.1. MSI
- Gene. The sensitivity of MSI has been shown to be lower in MSH6 mutation carriers (77% with a pentaplex panel) compared to MLH1 and MSH2 mutation carriers (~90%). This is due to fewer unstable markers and a shorter instability length (in bp) observed for MSH6 deficiency [21,114,117,118,119]. In contrast, no particular difficulties have been described for the three other genes, including PMS2 [120].
- Tumor type. The sensitivity of MSI has been shown to be lower in non-CRC tumors, especially in endometrium, brain, and urothelial tumors, as a consequence of fewer unstable markers and a shorter instability length [25,69,117,118,121,122,123,124,125]. For example, MSI is particularly difficult to detect in brain tumors, with less than 20% of brain tumors from patients with LS (or CMMRD) exhibiting MSI [69,105,126,127,128]. These observations have led to the recommendation to analyze normal DNA in parallel for non-CRC tumors [114,122,124,125].
- MSI-High vs. MSI-Low. Two categories of MSI were initially defined, based on the number of positive markers, i.e., MSI-high (MSI-H), corresponding to an instability at two or more out of the five markers tested, and MSI-low (MSI-L), corresponding to an instability at only one out of the five markers tested) [110,114]. MSI-L colorectal tumors were not shown to differ in their clinicopathologic features or in most molecular features from MSS tumors, leading to consider MSI-L and MSS tumors together and to regard as MSI only MSI-H tumors [129]. However, the sensitivity for LS detection generally increases when MSI-L is considered to be a positive test result (>90% vs. 67–100%) but with lower specificity (45–85% vs. 61–93%) [12,123,130,131,132,133]. As MSI testing is a screening test and not a final diagnostic test, some authors suggest to take into account the MSI-L results to perform constitutional genetic testing [131].
2.2.2. Immunohistochemistry
2.2.3. Comparison of MSI and IHC for Identification of LS
2.3. Focus on Colorectal Carcinogenesis and Adenomas in LS
3. Diagnosis of Lynch Syndrome
3.1. Clinical Criteria
3.2. Molecular Genetic Diagnosis
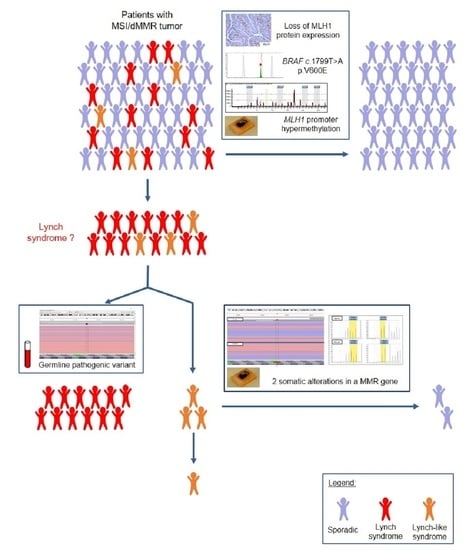
4. Differential Diagnosis of LS-Related vs. Sporadic MSI/dMMR Tumors
4.1. Clinical Presentation
4.2. Molecular Presentation
4.2.1. MLH1 Promoter Methylation
4.2.2. BRAF Mutations
4.2.3. BRAF Mutation Analysis vs. MLH1 Methylation Analysis
4.2.4. Other Potential Markers
4.2.5. MMR Gene Analysis in Tumors
4.3. Optimal Strategy for Discrimination of LS-Related and Sporadic MSI/dMMR Tumors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Moller, P.; Seppala, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppala, T.T.; Ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Bucksch, K.; Zachariae, S.; Aretz, S.; Buttner, R.; Holinski-Feder, E.; Holzapfel, S.; Huneburg, R.; Kloor, M.; von Knebel Doeberitz, M.; Morak, M.; et al. Cancer risks in Lynch syndrome, Lynch-like syndrome, and familial colorectal cancer type X: A prospective cohort study. BMC Cancer 2020, 20, 460. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; Mangu, P.B.; Gruber, S.B.; Hamilton, S.R.; Kalady, M.F.; Lau, M.W.; Lu, K.H.; Roach, N.; Limburg, P.J. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J. Clin. Oncol. 2015, 33, 209–217. [Google Scholar] [CrossRef]
- Vangala, D.B.; Cauchin, E.; Balmana, J.; Wyrwicz, L.; van Cutsem, E.; Guller, U.; Castells, A.; Carneiro, F.; Hammel, P.; Ducreux, M.; et al. Screening and surveillance in hereditary gastrointestinal cancers: Recommendations from the European Society of Digestive Oncology (ESDO) expert discussion at the 20th European Society for Medical Oncology (ESMO)/World Congress on Gastrointestinal Cancer, Barcelona, June 2018. Eur. J. Cancer 2018, 104, 91–103. [Google Scholar]
- Seppala, T.T.; Latchford, A.; Negoi, I.; Sampaio Soares, A.; Jimenez-Rodriguez, R.; Sanchez-Guillen, L.; Evans, D.G.; Ryan, N.; Crosbie, E.J.; Dominguez-Valentin, M.; et al. European guidelines from the EHTG and ESCP for Lynch syndrome: An updated third edition of the Mallorca guidelines based on gene and gender. Br. J. Surg. 2020. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Zaanan, A.; Shi, Q.; Taieb, J.; Alberts, S.R.; Meyers, J.P.; Smyrk, T.C.; Julie, C.; Zawadi, A.; Tabernero, J.; Mini, E.; et al. Clinical Outcomes in Patients with Colon Cancer with Microsatellite Instability of Sporadic or Familial Origin Treated with Adjuvant FOLFOX With or without Cetuximab: A Pooled Analysis of the PETACC8 and N0147 Trials. JCO Precis. Oncol. 2020, 4, 116–127. [Google Scholar] [CrossRef]
- Ten Broeke, S.W.; van der Klift, H.M.; Tops, C.M.J.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; de la Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2-Associated Lynch Syndrome. J. Clin. Oncol. 2018, 36, 2961–2968. [Google Scholar] [CrossRef]
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Barnetson, R.A.; Tenesa, A.; Farrington, S.M.; Nicholl, I.D.; Cetnarskyj, R.; Porteous, M.E.; Campbell, H.; Dunlop, M.G. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N. Engl. J. Med. 2006, 354, 2751–2763. [Google Scholar] [CrossRef]
- Moreira, L.; Balaguer, F.; Lindor, N.; de la Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Ruschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Clendenning, M.; Sotamaa, K.; Prior, T.; Westman, J.A.; et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J. Clin. Oncol. 2008, 26, 5783–5788. [Google Scholar] [CrossRef]
- Kahn, R.M.; Gordhandas, S.; Maddy, B.P.; Baltich Nelson, B.; Askin, G.; Christos, P.J.; Caputo, T.A.; Chapman-Davis, E.; Holcomb, K.; Frey, M.K. Universal endometrial cancer tumor typing: How much has immunohistochemistry, microsatellite instability, and MLH1 methylation improved the diagnosis of Lynch syndrome across the population? Cancer 2019, 125, 3172–3183. [Google Scholar] [CrossRef]
- Mills, A.M.; Liou, S.; Ford, J.M.; Berek, J.S.; Pai, R.K.; Longacre, T.A. Lynch syndrome screening should be considered for all patients with newly diagnosed endometrial cancer. Am. J. Surg. Pathol. 2014, 38, 1501–1509. [Google Scholar] [CrossRef]
- Goodfellow, P.J.; Billingsley, C.C.; Lankes, H.A.; Ali, S.; Cohn, D.E.; Broaddus, R.J.; Ramirez, N.; Pritchard, C.C.; Hampel, H.; Chassen, A.S.; et al. Combined Microsatellite Instability, MLH1 Methylation Analysis, and Immunohistochemistry for Lynch Syndrome Screening in Endometrial Cancers From GOG210: An NRG Oncology and Gynecologic Oncology Group Study. J. Clin. Oncol. 2015, 33, 4301–4308. [Google Scholar] [CrossRef]
- Palomaki, G.E.; McClain, M.R.; Melillo, S.; Hampel, H.L.; Thibodeau, S.N. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet. Med. 2009, 11, 42–65. [Google Scholar] [CrossRef]
- Provenzale, D.; Gupta, S.; Ahnen, D.J.; Bray, T.; Cannon, J.A.; Cooper, G.; David, D.S.; Early, D.S.; Erwin, D.; Ford, J.M.; et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 1010–1030. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef]
- Vasen, H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef]
- Ju, J.Y.; Mills, A.M.; Mahadevan, M.S.; Fan, J.; Culp, S.H.; Thomas, M.H.; Cathro, H.P. Universal Lynch Syndrome Screening Should be Performed in All Upper Tract Urothelial Carcinomas. Am. J. Surg. Pathol. 2018, 42, 1549–1555. [Google Scholar] [CrossRef]
- Orta, L.; Klimstra, D.S.; Qin, J.; Mecca, P.; Tang, L.H.; Busam, K.J.; Shia, J. Towards identification of hereditary DNA mismatch repair deficiency: Sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am. J. Surg. Pathol. 2009, 33, 934–944. [Google Scholar] [CrossRef]
- Young, J.; Simms, L.A.; Biden, K.G.; Wynter, C.; Whitehall, V.; Karamatic, R.; George, J.; Goldblatt, J.; Walpole, I.; Robin, S.A.; et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: Parallel pathways of tumorigenesis. Am. J. Pathol. 2001, 159, 2107–2116. [Google Scholar] [CrossRef]
- Halvarsson, B.; Anderson, H.; Domanska, K.; Lindmark, G.; Nilbert, M. Clinicopathologic factors identify sporadic mismatch repair-defective colon cancers. Am. J. Clin. Pathol. 2008, 129, 238–244. [Google Scholar] [CrossRef]
- Hartman, D.J.; Brand, R.E.; Hu, H.; Bahary, N.; Dudley, B.; Chiosea, S.I.; Nikiforova, M.N.; Pai, R.K. Lynch syndrome-associated colorectal carcinoma: Frequent involvement of the left colon and rectum and late-onset presentation supports a universal screening approach. Hum. Pathol. 2013, 44, 2518–2528. [Google Scholar] [CrossRef]
- Yamada, R.; Yamaguchi, T.; Iijima, T.; Wakaume, R.; Takao, M.; Koizumi, K.; Hishima, T.; Horiguchi, S.I. Differences in histological features and PD-L1 expression between sporadic microsatellite instability and Lynch-syndrome-associated disease in Japanese patients with colorectal cancer. Int. J. Clin. Oncol. 2018, 23, 504–513. [Google Scholar] [CrossRef]
- Moller, P.; Seppala, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef]
- Mas-Moya, J.; Dudley, B.; Brand, R.E.; Thull, D.; Bahary, N.; Nikiforova, M.N.; Pai, R.K. Clinicopathological comparison of colorectal and endometrial carcinomas in patients with Lynch-like syndrome versus patients with Lynch syndrome. Hum. Pathol. 2015, 46, 1616–1625. [Google Scholar] [CrossRef]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef]
- Shia, J.; Ellis, N.A.; Paty, P.B.; Nash, G.M.; Qin, J.; Offit, K.; Zhang, X.M.; Markowitz, A.J.; Nafa, K.; Guillem, J.G.; et al. Value of histopathology in predicting microsatellite instability in hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer. Am. J. Surg. Pathol. 2003, 27, 1407–1417. [Google Scholar] [CrossRef]
- Gologan, A.; Sepulveda, A.R. Microsatellite instability and DNA mismatch repair deficiency testing in hereditary and sporadic gastrointestinal cancers. Clin. Lab. Med. 2005, 25, 179–196. [Google Scholar] [CrossRef]
- Jenkins, M.A.; Hayashi, S.; O’Shea, A.M.; Burgart, L.J.; Smyrk, T.C.; Shimizu, D.; Waring, P.M.; Ruszkiewicz, A.R.; Pollett, A.F.; Redston, M.; et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: A population-based study. Gastroenterology 2007, 133, 48–56. [Google Scholar] [CrossRef]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef]
- Yearsley, M.; Hampel, H.; Lehman, A.; Nakagawa, H.; de la Chapelle, A.; Frankel, W.L. Histologic features distinguish microsatellite-high from microsatellite-low and microsatellite-stable colorectal carcinomas, but do not differentiate germline mutations from methylation of the MLH1 promoter. Hum. Pathol. 2006, 37, 831–838. [Google Scholar] [CrossRef]
- Hemminger, J.A.; Pearlman, R.; Haraldsdottir, S.; Knight, D.; Jonasson, J.G.; Pritchard, C.C.; Hampel, H.; Frankel, W.L. Histology of colorectal adenocarcinoma with double somatic mismatch-repair mutations is indistinguishable from those caused by Lynch syndrome. Hum. Pathol. 2018, 78, 125–130. [Google Scholar] [CrossRef]
- Cosgrove, C.M.; Cohn, D.E.; Hampel, H.; Frankel, W.L.; Jones, D.; McElroy, J.P.; Suarez, A.A.; Zhao, W.; Chen, W.; Salani, R.; et al. Epigenetic silencing of MLH1 in endometrial cancers is associated with larger tumor volume, increased rate of lymph node positivity and reduced recurrence-free survival. Gynecol. Oncol. 2017, 146, 588–595. [Google Scholar] [CrossRef]
- Rossi, L.; Le Frere-Belda, M.A.; Laurent-Puig, P.; Buecher, B.; De Pauw, A.; Stoppa-Lyonnet, D.; Canlorbe, G.; Caron, O.; Borghese, B.; Colas, C.; et al. Clinicopathologic Characteristics of Endometrial Cancer in Lynch Syndrome: A French Multicenter Study. Int. J. Gynecol. Cancer 2017, 27, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Broaddus, R.R.; Lynch, H.T.; Chen, L.M.; Daniels, M.S.; Conrad, P.; Munsell, M.F.; White, K.G.; Luthra, R.; Lu, K.H. Pathologic features of endometrial carcinoma associated with HNPCC: A comparison with sporadic endometrial carcinoma. Cancer 2006, 106, 87–94. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.; Panescu, J.; Lockman, J.; Sotamaa, K.; Fix, D.; Comeras, I.; La Jeunesse, J.; Nakagawa, H.; Westman, J.A.; et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006, 66, 7810–7817. [Google Scholar] [CrossRef] [PubMed]
- Leenen, C.H.; van Lier, M.G.; van Doorn, H.C.; van Leerdam, M.E.; Kooi, S.G.; de Waard, J.; Hoedemaeker, R.F.; van den Ouweland, A.M.; Hulspas, S.M.; Dubbink, H.J.; et al. Prospective evaluation of molecular screening for Lynch syndrome in patients with endometrial cancer </= 70 years. Gynecol. Oncol. 2012, 125, 414–420. [Google Scholar] [PubMed]
- Buchanan, D.D.; Tan, Y.Y.; Walsh, M.D.; Clendenning, M.; Metcalf, A.M.; Ferguson, K.; Arnold, S.T.; Thompson, B.A.; Lose, F.A.; Parsons, M.T.; et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J. Clin. Oncol. 2014, 32, 90–100. [Google Scholar] [CrossRef]
- Bruegl, A.S.; Djordjevic, B.; Urbauer, D.L.; Westin, S.N.; Soliman, P.T.; Lu, K.H.; Luthra, R.; Broaddus, R.R. Utility of MLH1 methylation analysis in the clinical evaluation of Lynch Syndrome in women with endometrial cancer. Curr. Pharm. Des. 2014, 20, 1655–1663. [Google Scholar] [CrossRef]
- Shia, J.; Black, D.; Hummer, A.J.; Boyd, J.; Soslow, R.A. Routinely assessed morphological features correlate with microsatellite instability status in endometrial cancer. Hum. Pathol. 2008, 39, 116–125. [Google Scholar] [CrossRef]
- Garg, K.; Soslow, R.A. Lynch syndrome (hereditary non-polyposis colorectal cancer) and endometrial carcinoma. J. Clin. Pathol. 2009, 62, 679–684. [Google Scholar] [CrossRef]
- Honore, L.H.; Hanson, J.; Andrew, S.E. Microsatellite instability in endometrioid endometrial carcinoma: Correlation with clinically relevant pathologic variables. Int. J. Gynecol. Cancer 2006, 16, 1386–1392. [Google Scholar] [CrossRef]
- Walsh, M.D.; Cummings, M.C.; Buchanan, D.D.; Dambacher, W.M.; Arnold, S.; McKeone, D.; Byrnes, R.; Barker, M.A.; Leggett, B.A.; Gattas, M.; et al. Molecular, pathologic, and clinical features of early-onset endometrial cancer: Identifying presumptive Lynch syndrome patients. Clin. Cancer Res. 2008, 14, 1692–1700. [Google Scholar] [CrossRef]
- Ramchander, N.C.; Ryan, N.A.J.; Walker, T.D.J.; Harries, L.; Bolton, J.; Bosse, T.; Evans, D.G.; Crosbie, E.J. Distinct Immunological Landscapes Characterize Inherited and Sporadic Mismatch Repair Deficient Endometrial Cancer. Front. Immunol. 2019, 10, 3023. [Google Scholar] [CrossRef]
- Djordjevic, B.; Barkoh, B.A.; Luthra, R.; Broaddus, R.R. Relationship between PTEN, DNA mismatch repair, and tumor histotype in endometrial carcinoma: Retained positive expression of PTEN preferentially identifies sporadic non-endometrioid carcinomas. Mod. Pathol. 2013, 26, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.A.; Moskaluk, C.A.; Mills, A.M. Mucinous Differentiation with Tumor Infiltrating Lymphocytes Is a Feature of Sporadically Methylated Endometrial Carcinomas. Int. J. Gynecol. Pathol. 2017, 36, 205–216. [Google Scholar] [CrossRef] [PubMed]
- ACOG Practice Bulletin No. 147: Lynch syndrome. Obs. Gynecol. 2014, 124, 1042–1054. [CrossRef]
- Lancaster, J.M.; Powell, C.B.; Chen, L.M.; Richardson, D.L. Society of Gynecologic Oncology statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol. Oncol. 2015, 136, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Leskela, S.; Romero, I.; Cristobal, E.; Perez-Mies, B.; Rosa-Rosa, J.M.; Gutierrez-Pecharroman, A.; Caniego-Casas, T.; Santon, A.; Ojeda, B.; Lopez-Reig, R.; et al. Mismatch Repair Deficiency in Ovarian Carcinoma: Frequency, Causes, and Consequences. Am. J. Surg. Pathol. 2020, 44, 649–656. [Google Scholar] [CrossRef]
- Aysal, A.; Karnezis, A.; Medhi, I.; Grenert, J.P.; Zaloudek, C.J.; Rabban, J.T. Ovarian endometrioid adenocarcinoma: Incidence and clinical significance of the morphologic and immunohistochemical markers of mismatch repair protein defects and tumor microsatellite instability. Am. J. Surg. Pathol. 2012, 36, 163–172. [Google Scholar] [CrossRef]
- Xiao, X.; Dong, D.; He, W.; Song, L.; Wang, Q.; Yue, J.; Xie, L. Mismatch repair deficiency is associated with MSI phenotype, increased tumor-infiltrating lymphocytes and PD-L1 expression in immune cells in ovarian cancer. Gynecol. Oncol. 2018, 149, 146–154. [Google Scholar] [CrossRef]
- Watson, P.; Butzow, R.; Lynch, H.T.; Mecklin, J.P.; Jarvinen, H.J.; Vasen, H.F.; Madlensky, L.; Fidalgo, P.; Bernstein, I. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol. Oncol. 2001, 82, 223–228. [Google Scholar] [CrossRef]
- Ketabi, Z.; Bartuma, K.; Bernstein, I.; Malander, S.; Gronberg, H.; Bjorck, E.; Holck, S.; Nilbert, M. Ovarian cancer linked to Lynch syndrome typically presents as early-onset, non-serous epithelial tumors. Gynecol. Oncol. 2011, 121, 462–465. [Google Scholar] [CrossRef]
- Grindedal, E.M.; Renkonen-Sinisalo, L.; Vasen, H.; Evans, G.; Sala, P.; Blanco, I.; Gronwald, J.; Apold, J.; Eccles, D.M.; Sanchez, A.A.; et al. Survival in women with MMR mutations and ovarian cancer: A multicentre study in Lynch syndrome kindreds. J. Med. Genet. 2010, 47, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Niskakoski, A.; Kaur, S.; Renkonen-Sinisalo, L.; Lassus, H.; Jarvinen, H.J.; Mecklin, J.P.; Butzow, R.; Peltomaki, P. Distinct molecular profiles in Lynch syndrome-associated and sporadic ovarian carcinomas. Int. J. Cancer 2013, 133, 2596–2608. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Helder-Woolderink, J.M.; Blok, E.A.; Vasen, H.F.; Hollema, H.; Mourits, M.J.; De Bock, G.H. Ovarian cancer in Lynch syndrome; a systematic review. Eur. J. Cancer 2016, 55, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Woolderink, J.M.; De Bock, G.H.; de Hullu, J.A.; Hollema, H.; Zweemer, R.P.; Slangen, B.F.M.; Gaarenstroom, K.N.; van Beurden, M.; van Doorn, H.C.; Sijmons, R.H.; et al. Characteristics of Lynch syndrome associated ovarian cancer. Gynecol. Oncol. 2018, 150, 324–330. [Google Scholar] [CrossRef]
- Urakami, S.; Inoshita, N.; Oka, S.; Miyama, Y.; Nomura, S.; Arai, M.; Sakaguchi, K.; Kurosawa, K.; Okaneya, T. Clinicopathological characteristics of patients with upper urinary tract urothelial cancer with loss of immunohistochemical expression of the DNA mismatch repair proteins in universal screening. Int. J. Urol. 2018, 25, 151–156. [Google Scholar] [CrossRef]
- Harper, H.L.; McKenney, J.K.; Heald, B.; Stephenson, A.; Campbell, S.C.; Plesec, T.; Magi-Galluzzi, C. Upper tract urothelial carcinomas: Frequency of association with mismatch repair protein loss and lynch syndrome. Mod. Pathol. 2017, 30, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Gayhart, M.G.; Johnson, N.; Paul, A.; Quillin, J.M.; Hampton, L.J.; Idowu, M.O.; Smith, S.C. Universal Mismatch Repair Protein Screening in Upper Tract Urothelial Carcinoma. Am. J. Clin. Pathol. 2020, 154, 792–801. [Google Scholar] [CrossRef]
- Metcalfe, M.J.; Petros, F.G.; Rao, P.; Mork, M.E.; Xiao, L.; Broaddus, R.R.; Matin, S.F. Universal Point of Care Testing for Lynch Syndrome in Patients with Upper Tract Urothelial Carcinoma. J. Urol. 2018, 199, 60–65. [Google Scholar] [CrossRef]
- Gylling, A.H.; Nieminen, T.T.; Abdel-Rahman, W.M.; Nuorva, K.; Juhola, M.; Joensuu, E.I.; Jarvinen, H.J.; Mecklin, J.P.; Aarnio, M.; Peltomaki, P.T. Differential cancer predisposition in Lynch syndrome: Insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis 2008, 29, 1351–1359. [Google Scholar] [CrossRef]
- Crockett, D.G.; Wagner, D.G.; Holmang, S.; Johansson, S.L.; Lynch, H.T. Upper urinary tract carcinoma in Lynch syndrome cases. J. Urol. 2011, 185, 1627–1630. [Google Scholar] [CrossRef]
- Roupret, M.; Yates, D.R.; Comperat, E.; Cussenot, O. Upper urinary tract urothelial cell carcinomas and other urological malignancies involved in the hereditary nonpolyposis colorectal cancer (lynch syndrome) tumor spectrum. Eur. Urol. 2008, 54, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Dietmaier, W.; Hofstadter, F.; Burgart, L.J.; Cheville, J.C.; Blaszyk, H. Urothelial carcinoma of the upper urinary tract: Inverted growth pattern is predictive of microsatellite instability. Hum. Pathol. 2003, 34, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Joost, P.; Therkildsen, C.; Dominguez-Valentin, M.; Jonsson, M.; Nilbert, M. Urinary Tract Cancer in Lynch Syndrome; Increased Risk in Carriers of MSH2 Mutations. Urology 2015, 86, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; An, J.Y.; Noh, S.H.; Shin, S.K.; Lee, Y.C. High microsatellite instability predicts good prognosis in intestinal-type gastric cancers. J. Gastroenterol. Hepatol. 2011, 26, 585–592. [Google Scholar] [CrossRef]
- Polom, K.; Marano, L.; Marrelli, D.; De Luca, R.; Roviello, G.; Savelli, V.; Tan, P.; Roviello, F. Meta-analysis of microsatellite instability in relation to clinicopathological characteristics and overall survival in gastric cancer. Br. J. Surg. 2018, 105, 159–167. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, T.H.; Cho, N.Y.; Yang, H.K.; Kim, W.H.; Kang, G.H. Differential clinicopathologic features in microsatellite-unstable gastric cancers with and without MLH1 methylation. Hum. Pathol. 2013, 44, 1055–1064. [Google Scholar] [CrossRef]
- Fornasarig, M.; Magris, R.; De Re, V.; Bidoli, E.; Canzonieri, V.; Maiero, S.; Viel, A.; Cannizzaro, R. Molecular and Pathological Features of Gastric Cancer in Lynch Syndrome and Familial Adenomatous Polyposis. Int. J. Mol. Sci. 2018, 19, 1682. [Google Scholar] [CrossRef]
- Kim, J.; Braun, D.; Ukaegbu, C.; Dhingra, T.G.; Kastrinos, F.; Parmigiani, G.; Syngal, S.; Yurgelun, M.B. Clinical Factors Associated With Gastric Cancer in Individuals With Lynch Syndrome. Clin. Gastroenterol. Hepatol. 2020, 18, 830–837.e831. [Google Scholar] [CrossRef]
- Ju, J.Y.; Dibbern, M.E.; Mahadevan, M.S.; Fan, J.; Kunk, P.R.; Stelow, E.B. Mismatch Repair Protein Deficiency/Microsatellite Instability Is Rare in Cholangiocarcinomas and Associated With Distinctive Morphologies. Am. J. Clin. Pathol. 2020, 153, 598–604. [Google Scholar] [CrossRef]
- Silva, V.W.; Askan, G.; Daniel, T.D.; Lowery, M.; Klimstra, D.S.; Abou-Alfa, G.K.; Shia, J. Biliary carcinomas: Pathology and the role of DNA mismatch repair deficiency. Chin. Clin. Oncol. 2016, 5, 62. [Google Scholar] [CrossRef]
- Aparicio, T.; Svrcek, M.; Zaanan, A.; Beohou, E.; Laforest, A.; Afchain, P.; Mitry, E.; Taieb, J.; Di Fiore, F.; Gornet, J.M.; et al. Small bowel adenocarcinoma phenotyping, a clinicobiological prognostic study. Br. J. Cancer 2013, 109, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Ten Kate, G.L.; Kleibeuker, J.H.; Nagengast, F.M.; Craanen, M.; Cats, A.; Menko, F.H.; Vasen, H.F. Is surveillance of the small bowel indicated for Lynch syndrome families? Gut 2007, 56, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Lupinacci, R.M.; Goloudina, A.; Buhard, O.; Bachet, J.B.; Marechal, R.; Demetter, P.; Cros, J.; Bardier-Dupas, A.; Collura, A.; Cervera, P.; et al. Prevalence of Microsatellite Instability in Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2018, 154, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.; Syngal, S. Hereditary pancreatic cancer. Gastroenterology 2010, 139, 1076–1080.e2. [Google Scholar] [CrossRef] [PubMed]
- Banville, N.; Geraghty, R.; Fox, E.; Leahy, D.T.; Green, A.; Keegan, D.; Geoghegan, J.; O’Donoghue, D.; Hyland, J.; Sheahan, K. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum. Pathol. 2006, 37, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Bujanda, L.; Herreros-Villanueva, M. Pancreatic Cancer in Lynch Syndrome Patients. J. Cancer 2017, 8, 3667–3674. [Google Scholar] [CrossRef]
- Flanagan, M.R.; Jayaraj, A.; Xiong, W.; Yeh, M.M.; Raskind, W.H.; Pillarisetty, V.G. Pancreatic intraductal papillary mucinous neoplasm in a patient with Lynch syndrome. World J. Gastroenterol. 2015, 21, 2820–2825. [Google Scholar] [CrossRef]
- Sparr, J.A.; Bandipalliam, P.; Redston, M.S.; Syngal, S. Intraductal papillary mucinous neoplasm of the pancreas with loss of mismatch repair in a patient with Lynch syndrome. Am. J. Surg. Pathol. 2009, 33, 309–312. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, W.Y.; Hwang, D.Y.; Han, H.S. Intraductal papillary mucinous neoplasm of the ileal heterotopic pancreas in a patient with hereditary non-polyposis colorectal cancer: A case report. World J. Gastroenterol. 2015, 21, 7916–7920. [Google Scholar] [CrossRef]
- Bhaijee, F.; Brown, A.S. Muir-Torre syndrome. Arch. Pathol. Lab. Med. 2014, 138, 1685–1689. [Google Scholar] [CrossRef]
- Ferreira, I.; Wiedemeyer, K.; Demetter, P.; Adams, D.J.; Arends, M.J.; Brenn, T. Update on the pathology, genetics and somatic landscape of sebaceous tumours. Histopathology 2020, 76, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.N.; Raymond, V.M.; Dandapani, M.; Marvin, M.; Kohlmann, W.; Chittenden, A.; Koeppe, E.; Gustafson, S.L.; Else, T.; Fullen, D.R.; et al. Screening for germline mismatch repair mutations following diagnosis of sebaceous neoplasm. JAMA Derm. 2014, 150, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Plocharczyk, E.F.; Frankel, W.L.; Hampel, H.; Peters, S.B. Mismatch repair protein deficiency is common in sebaceous neoplasms and suggests the importance of screening for Lynch syndrome. Am. J. Derm. 2013, 35, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Riegert-Johnson, D.L.; Thomas, B.C.; Thomas, C.S.; Heckman, M.G.; Krishna, M.; DiCaudo, D.J.; Bridges, A.G.; Hunt, K.S.; Rumilla, K.M.; et al. Screening for Muir-Torre syndrome using mismatch repair protein immunohistochemistry of sebaceous neoplasms. J. Genet. Couns 2013, 22, 393–405. [Google Scholar] [CrossRef]
- Kruse, R.; Rutten, A.; Schweiger, N.; Jakob, E.; Mathiak, M.; Propping, P.; Mangold, E.; Bisceglia, M.; Ruzicka, T. Frequency of microsatellite instability in unselected sebaceous gland neoplasias and hyperplasias. J. Investig. Derm. 2003, 120, 858–864. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Navi, D.; Wadhera, A.; Fung, M.A.; Fazel, N. Muir-Torre syndrome. Derm. Online J. 2006, 12, 4. [Google Scholar]
- South, C.D.; Hampel, H.; Comeras, I.; Westman, J.A.; Frankel, W.L.; de la Chapelle, A. The frequency of Muir-Torre syndrome among Lynch syndrome families. J. Natl. Cancer Inst. 2008, 100, 277–281. [Google Scholar] [CrossRef]
- Ponti, G.; Losi, L.; Di Gregorio, C.; Roncucci, L.; Pedroni, M.; Scarselli, A.; Benatti, P.; Seidenari, S.; Pellacani, G.; Lembo, L.; et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: Role of clinical features, microsatellite instability, and immunohistochemistry. Cancer 2005, 103, 1018–1025. [Google Scholar] [CrossRef]
- Akhtar, S.; Oza, K.K.; Khan, S.A.; Wright, J. Muir-Torre syndrome: Case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J. Am. Acad. Derm. 1999, 41, 681–686. [Google Scholar] [CrossRef]
- Walsh, M.D.; Jayasekara, H.; Huang, A.; Winship, I.M.; Buchanan, D.D. Clinico-pathological predictors of mismatch repair deficiency in sebaceous neoplasia: A large case series from a single Australian private pathology service. Australas J. Derm. 2019, 60, 126–133. [Google Scholar] [CrossRef]
- Stewart, W.M.; Lauret, P.; Hemet, J.; Thomine, E.; Gueville, R.M. Multiple kerato-acanthomas and visceral carcinomas: Torre’s syndrome. Ann. Derm. Venereol 1977, 104, 622–626. [Google Scholar] [PubMed]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056.e1010. [Google Scholar] [CrossRef]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Austin, F.; Richard, H.; Idowu, M.; Williamson, V.; Sabato, F.; Ferreira-Gonzalez, A.; Turner, S.A. Lynch syndrome-associated ultra-hypermutated pediatric glioblastoma mimicking a constitutional mismatch repair deficiency syndrome. Cold Spring Harb. Mol. Case Stud. 2019, 5. [Google Scholar] [CrossRef]
- Barresi, V.; Simbolo, M.; Mafficini, A.; Piredda, M.L.; Caffo, M.; Cardali, S.M.; Germano, A.; Cingarlini, S.; Ghimenton, C.; Scarpa, A. Ultra-Mutation in IDH Wild-Type Glioblastomas of Patients Younger than 55 Years is Associated with Defective Mismatch Repair, Microsatellite Instability, and Giant Cell Enrichment. Cancers 2019, 11, 1279. [Google Scholar] [CrossRef]
- Therkildsen, C.; Ladelund, S.; Rambech, E.; Persson, A.; Petersen, A.; Nilbert, M. Glioblastomas, astrocytomas and oligodendrogliomas linked to Lynch syndrome. Eur. J. Neurol. 2015, 22, 717–724. [Google Scholar] [CrossRef]
- Erson-Omay, E.Z.; Caglayan, A.O.; Schultz, N.; Weinhold, N.; Omay, S.B.; Ozduman, K.; Koksal, Y.; Li, J.; Serin Harmanci, A.; Clark, V.; et al. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol. 2015, 17, 1356–1364. [Google Scholar] [CrossRef]
- Shi, Z.F.; Li, K.K.; Kwan, J.S.H.; Yang, R.R.; Aibaidula, A.; Tang, Q.; Bao, Y.; Mao, Y.; Chen, H.; Ng, H.K. Whole-exome sequencing revealed mutational profiles of giant cell glioblastomas. Brain Pathol. 2019, 29, 782–792. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Buhard, O.; Suraweera, N.; Lectard, A.; Duval, A.; Hamelin, R. Quasimonomorphic mononucleotide repeats for high-level microsatellite instability analysis. Dis. Markers 2004, 20, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, N.; Duval, A.; Reperant, M.; Vaury, C.; Furlan, D.; Leroy, K.; Seruca, R.; Iacopetta, B.; Hamelin, R. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology 2002, 123, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Xicola, R.M.; Llor, X.; Pons, E.; Castells, A.; Alenda, C.; Pinol, V.; Andreu, M.; Castellvi-Bel, S.; Paya, A.; Jover, R.; et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J. Natl. Cancer Inst. 2007, 99, 244–252. [Google Scholar] [CrossRef]
- Goel, A.; Nagasaka, T.; Hamelin, R.; Boland, C.R. An optimized pentaplex PCR for detecting DNA mismatch repair-deficient colorectal cancers. PLoS ONE 2010, 5, e9393. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Westers, H.; Sousa, S.; Wu, Y.; Niessen, R.C.; Olderode-Berends, M.; van der Sluis, T.; Reuvekamp, P.T.; Seruca, R.; Kleibeuker, J.H.; et al. Mononucleotide precedes dinucleotide repeat instability during colorectal tumour development in Lynch syndrome patients. J. Pathol. 2009, 219, 96–102. [Google Scholar] [CrossRef]
- You, J.F.; Buhard, O.; Ligtenberg, M.J.; Kets, C.M.; Niessen, R.C.; Hofstra, R.M.; Wagner, A.; Dinjens, W.N.; Colas, C.; Lascols, O.; et al. Tumours with loss of MSH6 expression are MSI-H when screened with a pentaplex of five mononucleotide repeats. Br. J. Cancer 2010, 103, 1840–1845. [Google Scholar] [CrossRef]
- Pagin, A.; Zerimech, F.; Leclerc, J.; Wacrenier, A.; Lejeune, S.; Descarpentries, C.; Escande, F.; Porchet, N.; Buisine, M.P. Evaluation of a new panel of six mononucleotide repeat markers for the detection of DNA mismatch repair-deficient tumours. Br. J. Cancer 2013, 108, 2079–2087. [Google Scholar] [CrossRef]
- Hendriks, Y.M.; Wagner, A.; Morreau, H.; Menko, F.; Stormorken, A.; Quehenberger, F.; Sandkuijl, L.; Moller, P.; Genuardi, M.; Van Houwelingen, H.; et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: Impact on counseling and surveillance. Gastroenterology 2004, 127, 17–25. [Google Scholar] [CrossRef]
- Wang, Q.; Leclerc, J.; Bougeard, G.; Olschwang, S.; Vasseur, S.; Cassinari, K.; Boidin, D.; Lefol, C.; Naibo, P.; Frebourg, T.; et al. Characterisation of heterozygous PMS2 variants in French patients with Lynch syndrome. J. Med. Genet. 2020, 57, 487–499. [Google Scholar] [CrossRef]
- Kuismanen, S.A.; Moisio, A.L.; Schweizer, P.; Truninger, K.; Salovaara, R.; Arola, J.; Butzow, R.; Jiricny, J.; Nystrom-Lahti, M.; Peltomaki, P. Endometrial and colorectal tumors from patients with hereditary nonpolyposis colon cancer display different patterns of microsatellite instability. Am. J. Pathol. 2002, 160, 1953–1958. [Google Scholar] [CrossRef]
- Wong, Y.F.; Cheung, T.H.; Lo, K.W.; Yim, S.F.; Chan, L.K.; Buhard, O.; Duval, A.; Chung, T.K.; Hamelin, R. Detection of microsatellite instability in endometrial cancer: Advantages of a panel of five mononucleotide repeats over the National Cancer Institute panel of markers. Carcinogenesis 2006, 27, 951–955. [Google Scholar] [CrossRef]
- Libera, L.; Sahnane, N.; Carnevali, I.W.; Cimetti, L.; Cerutti, R.; Chiaravalli, A.M.; Riva, C.; Tibiletti, M.G.; Sessa, F.; Furlan, D. Microsatellite analysis of sporadic and hereditary gynaecological cancer in routine diagnostics. J. Clin. Pathol. 2017, 70, 792–797. [Google Scholar] [CrossRef]
- Mongiat-Artus, P.; Miquel, C.; Van der Aa, M.; Buhard, O.; Hamelin, R.; Soliman, H.; Bangma, C.; Janin, A.; Teillac, P.; van der Kwast, T.; et al. Microsatellite instability and mutation analysis of candidate genes in urothelial cell carcinomas of upper urinary tract. Oncogene 2006, 25, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, C.; Eisenberg, R.; Vnencak-Jones, C.L. Differences in Microsatellite Instability Profiles between Endometrioid and Colorectal Cancers: A Potential Cause for False-Negative Results? J. Mol. Diagn. 2017, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Bakry, D.; Aronson, M.; Durno, C.; Rimawi, H.; Farah, R.; Alharbi, Q.K.; Alharbi, M.; Shamvil, A.; Ben-Shachar, S.; Mistry, M.; et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: Report from the constitutional mismatch repair deficiency consortium. Eur. J. Cancer 2014, 50, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lavoine, N.; Colas, C.; Muleris, M.; Bodo, S.; Duval, A.; Entz-Werle, N.; Coulet, F.; Cabaret, O.; Andreiuolo, F.; Charpy, C.; et al. Constitutional mismatch repair deficiency syndrome: Clinical description in a French cohort. J. Med. Genet. 2015, 52, 770–778. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Pawlik, T.M.; Raut, C.P.; Rodriguez-Bigas, M.A. Colorectal carcinogenesis: MSI-H versus MSI-L. Dis. Markers 2004, 20, 199–206. [Google Scholar] [CrossRef]
- Poynter, J.N.; Siegmund, K.D.; Weisenberger, D.J.; Long, T.I.; Thibodeau, S.N.; Lindor, N.; Young, J.; Jenkins, M.A.; Hopper, J.L.; Baron, J.A.; et al. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3208–3215. [Google Scholar] [CrossRef]
- Coelho, H.; Jones-Hughes, T.; Snowsill, T.; Briscoe, S.; Huxley, N.; Frayling, I.M.; Hyde, C. A systematic review of test accuracy studies evaluating molecular micro-satellite instability testing for the detection of individuals with lynch syndrome. BMC Cancer 2017, 17, 836. [Google Scholar] [CrossRef]
- Stelloo, E.; Jansen, A.M.L.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Ruano, D.; Church, D.N.; Morreau, H.; Smit, V.; van Wezel, T.; et al. Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann. Oncol. 2017, 28, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Snowsill, T.; Coelho, H.; Huxley, N.; Jones-Hughes, T.; Briscoe, S.; Frayling, I.M.; Hyde, C. Molecular testing for Lynch syndrome in people with colorectal cancer: Systematic reviews and economic evaluation. Health Technol. Assess. 2017, 21, 1–238. [Google Scholar] [CrossRef]
- Papke, D.J., Jr.; Nowak, J.A.; Yurgelun, M.B.; Frieden, A.; Srivastava, A.; Lindeman, N.I.; Sholl, L.M.; MacConaill, L.E.; Dong, F. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod. Pathol. 2018, 31, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Imai, K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin. Oncol. 2019, 46, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Kautto, E.A.; Bonneville, R.; Miya, J.; Yu, L.; Krook, M.A.; Reeser, J.W.; Roychowdhury, S. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 2017, 8, 7452–7463. [Google Scholar] [CrossRef] [PubMed]
- Mojtahed, A.; Schrijver, I.; Ford, J.M.; Longacre, T.A.; Pai, R.K. A two-antibody mismatch repair protein immunohistochemistry screening approach for colorectal carcinomas, skin sebaceous tumors, and gynecologic tract carcinomas. Mod. Pathol. 2011, 24, 1004–1014. [Google Scholar] [CrossRef]
- O’Regan, T.; Chau, K.; Tatton, M.; Smith, T.; Parry, S.; Bissett, I. Immunochemistry screening for Lynch syndrome in colorectal adenocarcinoma using an initial two antibody panel can replace a four antibody panel. N. Z. Med. J. 2013, 126, 70–77. [Google Scholar] [PubMed]
- Pearlman, R.; Markow, M.; Knight, D.; Chen, W.; Arnold, C.A.; Pritchard, C.C.; Hampel, H.; Frankel, W.L. Two-stain immunohistochemical screening for Lynch syndrome in colorectal cancer may fail to detect mismatch repair deficiency. Mod. Pathol. 2018, 31, 1891–1900. [Google Scholar] [CrossRef]
- Shia, J.; Klimstra, D.S.; Nafa, K.; Offit, K.; Guillem, J.G.; Markowitz, A.J.; Gerald, W.L.; Ellis, N.A. Value of immunohistochemical detection of DNA mismatch repair proteins in predicting germline mutation in hereditary colorectal neoplasms. Am. J. Surg. Pathol. 2005, 29, 96–104. [Google Scholar] [CrossRef]
- Southey, M.C.; Jenkins, M.A.; Mead, L.; Whitty, J.; Trivett, M.; Tesoriero, A.A.; Smith, L.D.; Jennings, K.; Grubb, G.; Royce, S.G.; et al. Use of molecular tumor characteristics to prioritize mismatch repair gene testing in early-onset colorectal cancer. J. Clin. Oncol. 2005, 23, 6524–6532. [Google Scholar] [CrossRef] [PubMed]
- Salahshor, S.; Koelble, K.; Rubio, C.; Lindblom, A. Microsatellite Instability and hMLH1 and hMSH2 expression analysis in familial and sporadic colorectal cancer. Lab. Investig. 2001, 81, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Mangold, E.; Pagenstecher, C.; Friedl, W.; Fischer, H.P.; Merkelbach-Bruse, S.; Ohlendorf, M.; Friedrichs, N.; Aretz, S.; Buettner, R.; Propping, P.; et al. Tumours from MSH2 mutation carriers show loss of MSH2 expression but many tumours from MLH1 mutation carriers exhibit weak positive MLH1 staining. J. Pathol. 2005, 207, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, S.S.; Schmeits, J.; Thomas, G.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D.; Fox, E. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002, 62, 3485–3492. [Google Scholar] [PubMed]
- Chen, W.; Hampel, H.; Pearlman, R.; Jones, D.; Zhao, W.; Alsomali, M.; Knight, D.; Frankel, W.L. Unexpected expression of mismatch repair protein is more commonly seen with pathogenic missense than with other mutations in Lynch syndrome. Hum. Pathol. 2020, 103, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Hechtman, J.F.; Rana, S.; Middha, S.; Stadler, Z.K.; Latham, A.; Benayed, R.; Soslow, R.; Ladanyi, M.; Yaeger, R.; Zehir, A.; et al. Retained mismatch repair protein expression occurs in approximately 6% of microsatellite instability-high cancers and is associated with missense mutations in mismatch repair genes. Mod. Pathol. 2020, 33, 871–879. [Google Scholar] [CrossRef]
- Shia, J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J. Mol. Diagn. 2008, 10, 293–300. [Google Scholar] [CrossRef]
- Van Riel, E.; Ausems, M.G.; Hogervorst, F.B.; Kluijt, I.; van Gijn, M.E.; van Echtelt, J.; Scheidel-Jacobse, K.; Hennekam, E.F.; Stulp, R.P.; Vos, Y.J.; et al. A novel pathogenic MLH1 missense mutation, c.112A>C, p.Asn38His, in six families with Lynch syndrome. Hered. Cancer Clin. Pract. 2010, 8, 7. [Google Scholar] [CrossRef]
- Engel, C.; Forberg, J.; Holinski-Feder, E.; Pagenstecher, C.; Plaschke, J.; Kloor, M.; Poremba, C.; Pox, C.P.; Ruschoff, J.; Keller, G.; et al. Novel strategy for optimal sequential application of clinical criteria, immunohistochemistry and microsatellite analysis in the diagnosis of hereditary nonpolyposis colorectal cancer. Int. J. Cancer 2006, 118, 115–122. [Google Scholar] [CrossRef]
- Watson, N.; Grieu, F.; Morris, M.; Harvey, J.; Stewart, C.; Schofield, L.; Goldblatt, J.; Iacopetta, B. Heterogeneous staining for mismatch repair proteins during population-based prescreening for hereditary nonpolyposis colorectal cancer. J. Mol. Diagn. 2007, 9, 472–478. [Google Scholar] [CrossRef]
- Sarode, V.R.; Robinson, L. Screening for Lynch Syndrome by Immunohistochemistry of Mismatch Repair Proteins: Significance of Indeterminate Result and Correlation With Mutational Studies. Arch. Pathol. Lab. Med. 2019, 143, 1225–1233. [Google Scholar] [CrossRef]
- Bao, F.; Panarelli, N.C.; Rennert, H.; Sherr, D.L.; Yantiss, R.K. Neoadjuvant therapy induces loss of MSH6 expression in colorectal carcinoma. Am. J. Surg. Pathol. 2010, 34, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Kuan, S.F.; Ren, B.; Brand, R.; Dudley, B.; Pai, R.K. Neoadjuvant therapy in microsatellite-stable colorectal carcinoma induces concomitant loss of MSH6 and Ki-67 expression. Hum. Pathol. 2017, 63, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Hissong, E.; Crowe, E.P.; Yantiss, R.K.; Chen, Y.T. Assessing colorectal cancer mismatch repair status in the modern era: A survey of current practices and re-evaluation of the role of microsatellite instability testing. Mod. Pathol. 2018, 31, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Burgart, L.J.; Leontovich, O.; Goldberg, R.M.; Cunningham, J.M.; Sargent, D.J.; Walsh-Vockley, C.; Petersen, G.M.; Walsh, M.D.; Leggett, B.A.; et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J. Clin. Oncol. 2002, 20, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J. Mol. Diagn. 2008, 10, 301–307. [Google Scholar] [CrossRef]
- De Jong, A.E.; Morreau, H.; Van Puijenbroek, M.; Eilers, P.H.; Wijnen, J.; Nagengast, F.M.; Griffioen, G.; Cats, A.; Menko, F.H.; Kleibeuker, J.H.; et al. The role of mismatch repair gene defects in the development of adenomas in patients with HNPCC. Gastroenterology 2004, 126, 42–48. [Google Scholar] [CrossRef]
- Vasen, H.F.; den Hartog Jager, F.C.; Menko, F.H.; Nagengast, F.M. Screening for hereditary non-polyposis colorectal cancer: A study of 22 kindreds in The Netherlands. Am. J. Med. 1989, 86, 278–281. [Google Scholar] [CrossRef]
- Sekine, S.; Mori, T.; Ogawa, R.; Tanaka, M.; Yoshida, H.; Taniguchi, H.; Nakajima, T.; Sugano, K.; Yoshida, T.; Kato, M.; et al. Mismatch repair deficiency commonly precedes adenoma formation in Lynch Syndrome-Associated colorectal tumorigenesis. Mod. Pathol. 2017, 30, 1144–1151. [Google Scholar] [CrossRef]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Blaker, H.; et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef]
- Pino, M.S.; Mino-Kenudson, M.; Wildemore, B.M.; Ganguly, A.; Batten, J.; Sperduti, I.; Iafrate, A.J.; Chung, D.C. Deficient DNA mismatch repair is common in Lynch syndrome-associated colorectal adenomas. J. Mol. Diagn. 2009, 11, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Staffa, L.; Echterdiek, F.; Nelius, N.; Benner, A.; Werft, W.; Lahrmann, B.; Grabe, N.; Schneider, M.; Tariverdian, M.; von Knebel Doeberitz, M.; et al. Mismatch repair-deficient crypt foci in Lynch syndrome—Molecular alterations and association with clinical parameters. PLoS ONE 2015, 10, e0121980. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Huth, C.; Voigt, A.Y.; Benner, A.; Schirmacher, P.; von Knebel Doeberitz, M.; Blaker, H. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: A pathological study. Lancet Oncol. 2012, 13, 598–606. [Google Scholar] [CrossRef]
- Pai, R.K.; Dudley, B.; Karloski, E.; Brand, R.E.; O’Callaghan, N.; Rosty, C.; Buchanan, D.D.; Jenkins, M.A.; Thibodeau, S.N.; French, A.J.; et al. DNA mismatch repair protein deficient non-neoplastic colonic crypts: A novel indicator of Lynch syndrome. Mod. Pathol. 2018, 31, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.E.; Dudley, B.; Karloski, E.; Das, R.; Fuhrer, K.; Pai, R.K. Detection of DNA mismatch repair deficient crypts in random colonoscopic biopsies identifies Lynch syndrome patients. Fam. Cancer 2020, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.D.; Buchanan, D.D.; Pearson, S.A.; Clendenning, M.; Jenkins, M.A.; Win, A.K.; Walters, R.J.; Spring, K.J.; Nagler, B.; Pavluk, E.; et al. Immunohistochemical testing of conventional adenomas for loss of expression of mismatch repair proteins in Lynch syndrome mutation carriers: A case series from the Australasian site of the colon cancer family registry. Mod. Pathol. 2012, 25, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Goel, A.; Hornick, J.L.; Sen, A.; Turgeon, D.K.; Ruffin, M.T.; Marcon, N.E.; Baron, J.A.; Bresalier, R.S.; Syngal, S.; et al. Microsatellite instability and DNA mismatch repair protein deficiency in Lynch syndrome colorectal polyps. Cancer Prev. Res. 2012, 5, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Balmana, J.; Stockwell, D.H.; Steyerberg, E.W.; Stoffel, E.M.; Deffenbaugh, A.M.; Reid, J.E.; Ward, B.; Scholl, T.; Hendrickson, B.; Tazelaar, J.; et al. Prediction of MLH1 and MSH2 mutations in Lynch syndrome. JAMA 2006, 296, 1469–1478. [Google Scholar] [CrossRef][Green Version]
- Chen, S.; Wang, W.; Lee, S.; Nafa, K.; Lee, J.; Romans, K.; Watson, P.; Gruber, S.B.; Euhus, D.; Kinzler, K.W.; et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA 2006, 296, 1479–1487. [Google Scholar] [CrossRef]
- Tresallet, C.; Brouquet, A.; Julie, C.; Beauchet, A.; Vallot, C.; Menegaux, F.; Mitry, E.; Radvanyi, F.; Malafosse, R.; Rougier, P.; et al. Evaluation of predictive models in daily practice for the identification of patients with Lynch syndrome. Int. J. Cancer 2012, 130, 1367–1377. [Google Scholar] [CrossRef]
- Peltomaki, P. Update on Lynch syndrome genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Heinen, C.D.; Juel Rasmussen, L. Determining the functional significance of mismatch repair gene missense variants using biochemical and cellular assays. Hered. Cancer Clin. Pract. 2012, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Gazzoli, I.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002, 62, 3925–3928. [Google Scholar] [PubMed]
- Chan, T.L.; Yuen, S.T.; Kong, C.K.; Chan, Y.W.; Chan, A.S.; Ng, W.F.; Tsui, W.Y.; Lo, M.W.; Tam, W.Y.; Li, V.S.; et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat. Genet. 2006, 38, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P. The role of epigenetics in Lynch syndrome. Fam. Cancer 2013, 12, 189–205. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Smith, C.; Salipante, S.J.; Lee, M.K.; Thornton, A.M.; Nord, A.S.; Gulden, C.; Kupfer, S.S.; Swisher, E.M.; Bennett, R.L.; et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J. Mol. Diagn. 2012, 14, 357–366. [Google Scholar] [CrossRef]
- Clendenning, M.; Hampel, H.; LaJeunesse, J.; Lindblom, A.; Lockman, J.; Nilbert, M.; Senter, L.; Sotamaa, K.; de la Chapelle, A. Long-range PCR facilitates the identification of PMS2-specific mutations. Hum. Mutat. 2006, 27, 490–495. [Google Scholar] [CrossRef]
- Vaughn, C.P.; Robles, J.; Swensen, J.J.; Miller, C.E.; Lyon, E.; Mao, R.; Bayrak-Toydemir, P.; Samowitz, W.S. Clinical analysis of PMS2: Mutation detection and avoidance of pseudogenes. Hum. Mutat. 2010, 31, 588–593. [Google Scholar]
- Vaughn, C.P.; Baker, C.L.; Samowitz, W.S.; Swensen, J.J. The frequency of previously undetectable deletions involving 3’ Exons of the PMS2 gene. Genes Chromosomes Cancer 2013, 52, 107–112. [Google Scholar] [CrossRef]
- Wimmer, K.; Wernstedt, A. PMS2 gene mutational analysis: Direct cDNA sequencing to circumvent pseudogene interference. Methods Mol. Biol. 2014, 1167, 289–302. [Google Scholar]
- Li, J.; Dai, H.; Feng, Y.; Tang, J.; Chen, S.; Tian, X.; Gorman, E.; Schmitt, E.S.; Hansen, T.A.; Wang, J.; et al. A Comprehensive Strategy for Accurate Mutation Detection of the Highly Homologous PMS2. J. Mol. Diagn. 2015, 17, 545–553. [Google Scholar] [CrossRef]
- Gould, G.M.; Grauman, P.V.; Theilmann, M.R.; Spurka, L.; Wang, I.E.; Melroy, L.M.; Chin, R.G.; Hite, D.H.; Chu, C.S.; Maguire, J.R.; et al. Detecting clinically actionable variants in the 3′ exons of PMS2 via a reflex workflow based on equivalent hybrid capture of the gene and its pseudogene. BMC Med. Genet. 2018, 19, 176. [Google Scholar] [CrossRef]
- Antelo, M.; Golubicki, M.; Roca, E.; Mendez, G.; Carballido, M.; Iseas, S.; Cuatrecasas, M.; Moreira, L.; Sanchez, A.; Carballal, S.; et al. Lynch-like syndrome is as frequent as Lynch syndrome in early-onset nonfamilial nonpolyposis colorectal cancer. Int. J. Cancer 2019, 145, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Rumilla, K.; Schowalter, K.V.; Lindor, N.M.; Thomas, B.C.; Mensink, K.A.; Gallinger, S.; Holter, S.; Newcomb, P.A.; Potter, J.D.; Jenkins, M.A.; et al. Frequency of deletions of EPCAM (TACSTD1) in MSH2-associated Lynch syndrome cases. J. Mol. Diagn. 2011, 13, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, R.P.; Vissers, L.E.; Venkatachalam, R.; Bodmer, D.; Hoenselaar, E.; Goossens, M.; Haufe, A.; Kamping, E.; Niessen, R.C.; Hogervorst, F.B.; et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum. Mutat. 2011, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P. Finding the needle in a haystack: Identification of cases of Lynch syndrome with MLH1 epimutation. Fam. Cancer 2016, 15, 413–422. [Google Scholar] [CrossRef]
- Leclerc, J.; Flament, C.; Lovecchio, T.; Delattre, L.; Ait Yahya, E.; Baert-Desurmont, S.; Burnichon, N.; Bronner, M.; Cabaret, O.; Lejeune, S.; et al. Diversity of genetic events associated with MLH1 promoter methylation in Lynch syndrome families with heritable constitutional epimutation. Genet. Med. 2018, 20, 1589–1599. [Google Scholar] [CrossRef]
- Damaso, E.; Canet-Hermida, J.; Vargas-Parra, G.; Velasco, A.; Marin, F.; Darder, E.; Del Valle, J.; Fernandez, A.; Izquierdo, A.; Mateu, G.; et al. Highly sensitive MLH1 methylation analysis in blood identifies a cancer patient with low-level mosaic MLH1 epimutation. Clin. Epigenetics 2019, 11, 171. [Google Scholar] [CrossRef]
- Arnold, A.M.; Morak, M.; Benet-Pages, A.; Laner, A.; Frishman, D.; Holinski-Feder, E. Targeted deep-intronic sequencing in a cohort of unexplained cases of suspected Lynch syndrome. Eur. J. Hum. Genet. 2020, 28, 597–608. [Google Scholar] [CrossRef]
- Clendenning, M.; Buchanan, D.D.; Walsh, M.D.; Nagler, B.; Rosty, C.; Thompson, B.; Spurdle, A.B.; Hopper, J.L.; Jenkins, M.A.; Young, J.P. Mutation deep within an intron of MSH2 causes Lynch syndrome. Fam. Cancer 2011, 10, 297–301. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Casadei, S.; Gulsuner, S.; Shirts, B.H.; Mandell, J.B.; Kortbawi, H.M.; Norquist, B.S.; Swisher, E.M.; Lee, M.K.; Goldberg, Y.; O′Connor, R.; et al. Characterization of splice-altering mutations in inherited predisposition to cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 26798–26807. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.; Koehler, U.; Schackert, H.K.; Steinke, V.; Royer-Pokora, B.; Schulmann, K.; Kloor, M.; Hochter, W.; Weingart, J.; Keiling, C.; et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J. Med. Genet. 2011, 48, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; van der Klift, H.; Franken, P.; Wijnen, J.; Breukel, C.; Bezrookove, V.; Smits, R.; Kinarsky, Y.; Barrows, A.; Franklin, B.; et al. A 10-Mb paracentric inversion of chromosome arm 2p inactivates MSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosomes Cancer 2002, 35, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M. The 10-Mb paracentric inversion of chromosome arm 2p in activating MSH2 and causing hereditary nonpolyposis colorectal cancer: Re-annotation and mutational mechanisms. Genes Chromosomes Cancer 2008, 47, 543–545. [Google Scholar] [CrossRef]
- Liu, Q.; Hesson, L.B.; Nunez, A.C.; Packham, D.; Williams, R.; Ward, R.L.; Sloane, M.A. A cryptic paracentric inversion of MSH2 exons 2-6 causes Lynch syndrome. Carcinogenesis 2016, 37, 10–17. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Rafnar, T.; Frankel, W.L.; Einarsdottir, S.; Sigurdsson, A.; Hampel, H.; Snaebjornsson, P.; Masson, G.; Weng, D.; Arngrimsson, R.; et al. Comprehensive population-wide analysis of Lynch syndrome in Iceland reveals founder mutations in MSH6 and PMS2. Nat. Commun. 2017, 8, 14755. [Google Scholar] [CrossRef]
- Van der Klift, H.M.; Tops, C.M.; Hes, F.J.; Devilee, P.; Wijnen, J.T. Insertion of an SVA element, a nonautonomous retrotransposon, in PMS2 intron 7 as a novel cause of Lynch syndrome. Hum. Mutat. 2012, 33, 1051–1055. [Google Scholar] [CrossRef]
- Morak, M.; Steinke-Lange, V.; Massdorf, T.; Benet-Pages, A.; Locher, M.; Laner, A.; Kayser, K.; Aretz, S.; Holinski-Feder, E. Prevalence of CNV-neutral structural genomic rearrangements in MLH1, MSH2, and PMS2 not detectable in routine NGS diagnostics. Fam. Cancer 2020, 19, 161–167. [Google Scholar] [CrossRef]
- Morak, M.; Schaefer, K.; Steinke-Lange, V.; Koehler, U.; Keinath, S.; Massdorf, T.; Mauracher, B.; Rahner, N.; Bailey, J.; Kling, C.; et al. Full-length transcript amplification and sequencing as universal method to test mRNA integrity and biallelic expression in mismatch repair genes. Eur. J. Hum. Genet. 2019, 27, 1808–1820. [Google Scholar] [CrossRef]
- Rhees, J.; Arnold, M.; Boland, C.R. Inversion of exons 1-7 of the MSH2 gene is a frequent cause of unexplained Lynch syndrome in one local population. Fam. Cancer 2014, 13, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Sutter, C.; Wentzensen, N.; Cremer, F.W.; Buckowitz, A.; Keller, M.; von Knebel Doeberitz, M.; Gebert, J. A large MSH2 Alu insertion mutation causes HNPCC in a German kindred. Hum. Genet. 2004, 115, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Solassol, J.; Larrieux, M.; Leclerc, J.; Ducros, V.; Corsini, C.; Chiesa, J.; Pujol, P.; Rey, J.M. Alu element insertion in the MLH1 exon 6 coding sequence as a mutation predisposing to Lynch syndrome. Hum. Mutat. 2019, 40, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Pastrello, C.; Fornasarig, M.; Pin, E.; Berto, E.; Pivetta, B.; Viel, A. Somatic mosaicism in a patient with Lynch syndrome. Am. J. Med. Genet. A 2009, 149A, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Sourrouille, I.; Coulet, F.; Lefevre, J.H.; Colas, C.; Eyries, M.; Svrcek, M.; Bardier-Dupas, A.; Parc, Y.; Soubrier, F. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam. Cancer 2013, 12, 27–33. [Google Scholar] [CrossRef]
- Geurts-Giele, W.R.; Rosenberg, E.H.; Rens, A.V.; Leerdam, M.E.V.; Dinjens, W.N.; Bleeker, F.E. Somatic mosaicism by a de novo MLH1 mutation as a cause of Lynch syndrome. Mol. Genet. Genom. Med. 2019, 7, e00699. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.M.L.; Goel, A. Mosaicism in Patients With Colorectal Cancer or Polyposis Syndromes: A Systematic Review. Clin. Gastroenterol. Hepatol. 2020, 18, 1949–1960. [Google Scholar] [CrossRef]
- Sutcliffe, E.G.; Bartenbaker Thompson, A.; Stettner, A.R.; Marshall, M.L.; Roberts, M.E.; Susswein, L.R.; Wang, Y.; Klein, R.T.; Hruska, K.S.; Solomon, B.D. Multi-gene panel testing confirms phenotypic variability in MUTYH-Associated Polyposis. Fam. Cancer 2019, 18, 203–209. [Google Scholar] [CrossRef]
- Morak, M.; Heidenreich, B.; Keller, G.; Hampel, H.; Laner, A.; de la Chapelle, A.; Holinski-Feder, E. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur. J. Hum. Genet. 2014, 22, 1334–1337. [Google Scholar] [CrossRef]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- Lefevre, J.H.; Colas, C.; Coulet, F.; Bonilla, C.; Mourra, N.; Flejou, J.F.; Tiret, E.; Bodmer, W.; Soubrier, F.; Parc, Y. MYH biallelic mutation can inactivate the two genetic pathways of colorectal cancer by APC or MLH1 transversions. Fam. Cancer 2010, 9, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; van den Akker, B.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2015, 23, 1080–1084. [Google Scholar] [CrossRef]
- Adam, R.; Spier, I.; Zhao, B.; Kloth, M.; Marquez, J.; Hinrichsen, I.; Kirfel, J.; Tafazzoli, A.; Horpaopan, S.; Uhlhaas, S.; et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am. J. Hum. Genet. 2016, 99, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Damaso, E.; Gonzalez-Acosta, M.; Vargas-Parra, G.; Navarro, M.; Balmana, J.; Ramon, Y.C.T.; Tuset, N.; Thompson, B.A.; Marin, F.; Fernandez, A.; et al. Comprehensive Constitutional Genetic and Epigenetic Characterization of Lynch-Like Individuals. Cancers 2020, 12, 1799. [Google Scholar] [CrossRef]
- Xavier, A.; Olsen, M.F.; Lavik, L.A.; Johansen, J.; Singh, A.K.; Sjursen, W.; Scott, R.J.; Talseth-Palmer, B.A. Comprehensive mismatch repair gene panel identifies variants in patients with Lynch-like syndrome. Mol. Genet. Genom. Med. 2019, 7, e850. [Google Scholar] [CrossRef]
- Vargas-Parra, G.M.; Gonzalez-Acosta, M.; Thompson, B.A.; Gomez, C.; Fernandez, A.; Damaso, E.; Pons, T.; Morak, M.; Del Valle, J.; Iglesias, S.; et al. Elucidating the molecular basis of MSH2-deficient tumors by combined germline and somatic analysis. Int. J. Cancer 2017, 141, 1365–1380. [Google Scholar] [CrossRef]
- Xicola, R.M.; Clark, J.R.; Carroll, T.; Alvikas, J.; Marwaha, P.; Regan, M.R.; Lopez-Giraldez, F.; Choi, J.; Emmadi, R.; Alagiozian-Angelova, V.; et al. Implication of DNA repair genes in Lynch-like syndrome. Fam. Cancer 2019, 18, 331–342. [Google Scholar] [CrossRef]
- Golubicki, M.; Bonjoch, L.; Acuna-Ochoa, J.G.; Diaz-Gay, M.; Munoz, J.; Cuatrecasas, M.; Ocana, T.; Iseas, S.; Mendez, G.; Cisterna, D.; et al. Germline biallelic Mcm8 variants are associated with early-onset Lynch-like syndrome. JCI Insight 2020, 5, e140698. [Google Scholar] [CrossRef]
- Vos, J.R.; Fakkert, I.E.; Spruijt, L.; Willems, R.W.; Langenveld, S.; Mensenkamp, A.R.; Leter, E.M.; Nagtegaal, I.D.; Ligtenberg, M.J.L.; Hoogerbrugge, N. Evaluation of yield and experiences of age-related molecular investigation for heritable and nonheritable causes of mismatch repair deficient colorectal cancer to identify Lynch syndrome. Int. J. Cancer 2020, 147, 2150–2158. [Google Scholar] [CrossRef]
- Jansen, A.M.; van Wezel, T.; van den Akker, B.E.; Ventayol Garcia, M.; Ruano, D.; Tops, C.M.; Wagner, A.; Letteboer, T.G.; Gomez-Garcia, E.B.; Devilee, P.; et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur. J. Hum. Genet. 2016, 24, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Porkka, N.; Lahtinen, L.; Ahtiainen, M.; Bohm, J.P.; Kuopio, T.; Eldfors, S.; Mecklin, J.P.; Seppala, T.T.; Peltomaki, P. Epidemiological, clinical and molecular characterization of Lynch-like syndrome: A population-based study. Int. J. Cancer 2019, 145, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Mensenkamp, A.R.; Vogelaar, I.P.; van Zelst-Stams, W.A.; Goossens, M.; Ouchene, H.; Hendriks-Cornelissen, S.J.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology 2014, 146, 643–646.e8. [Google Scholar] [CrossRef]
- Geurts-Giele, W.R.; Leenen, C.H.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.; Kuipers, E.J.; Goverde, A.; van den Ouweland, A.M.; van Lier, M.G.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014, 147, 1308–1316.e1. [Google Scholar] [CrossRef]
- Billingsley, C.C.; Cohn, D.E.; Mutch, D.G.; Stephens, J.A.; Suarez, A.A.; Goodfellow, P.J. Polymerase varepsilon (POLE) mutations in endometrial cancer: Clinical outcomes and implications for Lynch syndrome testing. Cancer 2015, 121, 386–394. [Google Scholar] [CrossRef]
- Joly, M.O.; Attignon, V.; Saurin, J.C.; Desseigne, F.; Leroux, D.; Martin-Denavit, T.; Giraud, S.; Bonnet-Dupeyron, M.N.; Faivre, L.; Auclair, J.; et al. Somatic MMR gene mutations as a cause for MSI-H sebaceous neoplasms in Muir-Torre syndrome-like patients. Hum. Mutat. 2015, 36, 292–295. [Google Scholar] [CrossRef]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Rigter, L.S.; Snaebjornsson, P.; Rosenberg, E.H.; Atmodimedjo, P.N.; Aleman, B.M.; Ten Hoeve, J.; Geurts-Giele, W.R.; van Ravesteyn, T.W.; Hoeksel, J.; Meijer, G.A.; et al. Double somatic mutations in mismatch repair genes are frequent in colorectal cancer after Hodgkin′s lymphoma treatment. Gut 2018, 67, 447–455. [Google Scholar] [CrossRef]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef]
- Ortega, J.; Li, J.Y.; Lee, S.; Tong, D.; Gu, L.; Li, G.M. Phosphorylation of PCNA by EGFR inhibits mismatch repair and promotes misincorporation during DNA synthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 5667–5672. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, D.D.; Rosty, C.; Clendenning, M.; Spurdle, A.B.; Win, A.K. Clinical problems of colorectal cancer and endometrial cancer cases with unknown cause of tumor mismatch repair deficiency (suspected Lynch syndrome). Appl. Clin. Genet. 2014, 7, 183–193. [Google Scholar] [PubMed]
- Steinke, V.; Holzapfel, S.; Loeffler, M.; Holinski-Feder, E.; Morak, M.; Schackert, H.K.; Gorgens, H.; Pox, C.; Royer-Pokora, B.; von Knebel-Doeberitz, M.; et al. Evaluating the performance of clinical criteria for predicting mismatch repair gene mutations in Lynch syndrome: A comprehensive analysis of 3671 families. Int. J. Cancer 2014, 135, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, D.D.; Clendenning, M.; Rosty, C.; Eriksen, S.V.; Walsh, M.D.; Walters, R.J.; Thibodeau, S.N.; Stewart, J.; Preston, S.; Win, A.K.; et al. Tumor testing to identify lynch syndrome in two Australian colorectal cancer cohorts. J. Gastroenterol. Hepatol. 2017, 32, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.M.; Sloan, E.A.; Thomas, M.; Modesitt, S.C.; Stoler, M.H.; Atkins, K.A.; Moskaluk, C.A. Clinicopathologic Comparison of Lynch Syndrome-associated and “Lynch-like” Endometrial Carcinomas Identified on Universal Screening Using Mismatch Repair Protein Immunohistochemistry. Am. J. Surg. Pathol. 2016, 40, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Park, C.K.; Chang, D.K.; Kim, J.W.; Son, H.J.; Cho, Y.B.; Yun, S.H.; Kim, H.C.; Kwon, M.; Kim, K.M. Lynch-like syndrome: Characterization and comparison with EPCAM deletion carriers. Int. J. Cancer 2015, 136, 1568–1578. [Google Scholar] [CrossRef]
- Rodriguez-Soler, M.; Perez-Carbonell, L.; Guarinos, C.; Zapater, P.; Castillejo, A.; Barbera, V.M.; Juarez, M.; Bessa, X.; Xicola, R.M.; Clofent, J.; et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology 2013, 144, 926–932.e1. [Google Scholar] [CrossRef]
- Win, A.K.; Buchanan, D.D.; Rosty, C.; MacInnis, R.J.; Dowty, J.G.; Dite, G.S.; Giles, G.G.; Southey, M.C.; Young, J.P.; Clendenning, M.; et al. Role of tumour molecular and pathology features to estimate colorectal cancer risk for first-degree relatives. Gut 2015, 64, 101–110. [Google Scholar] [CrossRef]
- Dempsey, K.M.; Broaddus, R.; You, Y.N.; Noblin, S.J.; Mork, M.; Fellman, B.; Urbauer, D.; Daniels, M.; Lu, K. Is it all Lynch syndrome?: An assessment of family history in individuals with mismatch repair-deficient tumors. Genet. Med. 2015, 17, 476–484. [Google Scholar] [CrossRef][Green Version]
- Kato, A.; Sato, N.; Sugawara, T.; Takahashi, K.; Kito, M.; Makino, K.; Sato, T.; Shimizu, D.; Shirasawa, H.; Miura, H.; et al. Isolated Loss of PMS2 Immunohistochemical Expression is Frequently Caused by Heterogenous MLH1 Promoter Hypermethylation in Lynch Syndrome Screening for Endometrial Cancer Patients. Am. J. Surg. Pathol. 2016, 40, 770–776. [Google Scholar] [CrossRef]
- Newton, K.; Jorgensen, N.M.; Wallace, A.J.; Buchanan, D.D.; Lalloo, F.; McMahon, R.F.; Hill, J.; Evans, D.G. Tumour MLH1 promoter region methylation testing is an effective prescreen for Lynch Syndrome (HNPCC). J. Med. Genet. 2014, 51, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.T.; Buchanan, D.D.; Thompson, B.; Young, J.P.; Spurdle, A.B. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: A literature review assessing utility of tumour features for MMR variant classification. J. Med. Genet. 2012, 49, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Schorge, J.O.; Rodabaugh, K.J.; Daniels, M.S.; Sun, C.C.; Soliman, P.T.; White, K.G.; Luthra, R.; Gershenson, D.M.; Broaddus, R.R. Prospective determination of prevalence of lynch syndrome in young women with endometrial cancer. J. Clin. Oncol. 2007, 25, 5158–5164. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Riegert-Johnson, D.L.; Thomas, B.C.; Rumilla, K.M.; Thomas, C.S.; Heckman, M.G.; Purcell, J.U.; Hanson, N.B.; Leppig, K.A.; Lim, J.; et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome. Genet. Med. 2014, 16, 711–716. [Google Scholar] [CrossRef]
- Shannon, C.; Kirk, J.; Barnetson, R.; Evans, J.; Schnitzler, M.; Quinn, M.; Hacker, N.; Crandon, A.; Harnett, P. Incidence of microsatellite instability in synchronous tumors of the ovary and endometrium. Clin. Cancer Res. 2003, 9, 1387–1392. [Google Scholar]
- Soliman, P.T.; Broaddus, R.R.; Schmeler, K.M.; Daniels, M.S.; Gonzalez, D.; Slomovitz, B.M.; Gershenson, D.M.; Lu, K.H. Women with synchronous primary cancers of the endometrium and ovary: Do they have Lynch syndrome? J. Clin. Oncol. 2005, 23, 9344–9350. [Google Scholar] [CrossRef]
- Wagner, A.; Hendriks, Y.; Meijers-Heijboer, E.J.; de Leeuw, W.J.; Morreau, H.; Hofstra, R.; Tops, C.; Bik, E.; Brocker-Vriends, A.H.; van Der Meer, C.; et al. Atypical HNPCC owing to MSH6 germline mutations: Analysis of a large Dutch pedigree. J. Med. Genet. 2001, 38, 318–322. [Google Scholar] [CrossRef]
- Pal, T.; Permuth-Wey, J.; Sellers, T.A. A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer 2008, 113, 733–742. [Google Scholar] [CrossRef]
- Murphy, M.A.; Wentzensen, N. Frequency of mismatch repair deficiency in ovarian cancer: A systematic review This article is a US Government work and, as such, is in the public domain of the United States of America. Int. J. Cancer 2011, 129, 1914–1922. [Google Scholar] [CrossRef]
- Joost, P.; Veurink, N.; Holck, S.; Klarskov, L.; Bojesen, A.; Harbo, M.; Baldetorp, B.; Rambech, E.; Nilbert, M. Heterogenous mismatch-repair status in colorectal cancer. Diagn. Pathol. 2014, 9, 126. [Google Scholar] [CrossRef]
- Pai, R.K.; Plesec, T.P.; Abdul-Karim, F.W.; Yang, B.; Marquard, J.; Shadrach, B.; Roma, A.R. Abrupt loss of MLH1 and PMS2 expression in endometrial carcinoma: Molecular and morphologic analysis of 6 cases. Am. J. Surg. Pathol. 2015, 39, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Chen, A.; Hong, J.; Chae, H.S.; Kim, Y.S. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999, 59, 2029–2033. [Google Scholar] [PubMed]
- Capel, E.; Flejou, J.F.; Hamelin, R. Assessment of MLH1 promoter methylation in relation to gene expression requires specific analysis. Oncogene 2007, 26, 7596–7600. [Google Scholar] [CrossRef]
- Ruemmele, P.; Dietmaier, W.; Terracciano, L.; Tornillo, L.; Bataille, F.; Kaiser, A.; Wuensch, P.H.; Heinmoeller, E.; Homayounfar, K.; Luettges, J.; et al. Histopathologic features and microsatellite instability of cancers of the papilla of vater and their precursor lesions. Am. J. Surg. Pathol. 2009, 33, 691–704. [Google Scholar] [CrossRef]
- Bennett, J.A.; Pesci, A.; Morales-Oyarvide, V.; Da Silva, A.; Nardi, V.; Oliva, E. Incidence of Mismatch Repair Protein Deficiency and Associated Clinicopathologic Features in a Cohort of 104 Ovarian Endometrioid Carcinomas. Am. J. Surg. Pathol. 2019, 43, 235–243. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Rad, R.; Cadinanos, J.; Rad, L.; Varela, I.; Strong, A.; Kriegl, L.; Constantino-Casas, F.; Eser, S.; Hieber, M.; Seidler, B.; et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell 2013, 24, 15–29. [Google Scholar] [CrossRef]
- Sakamoto, N.; Feng, Y.; Stolfi, C.; Kurosu, Y.; Green, M.; Lin, J.; Green, M.E.; Sentani, K.; Yasui, W.; McMahon, M.; et al. BRAF(V600E) cooperates with CDX2 inactivation to promote serrated colorectal tumorigenesis. Elife 2017, 6, e20331. [Google Scholar] [CrossRef]
- Bond, C.E.; Liu, C.; Kawamata, F.; McKeone, D.M.; Fernando, W.; Jamieson, S.; Pearson, S.A.; Kane, A.; Woods, S.L.; Lannagan, T.R.M.; et al. Oncogenic BRAF mutation induces DNA methylation changes in a murine model for human serrated colorectal neoplasia. Epigenetics 2018, 13, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cunningham, J.M.; Winters, J.L.; Guenther, J.C.; French, A.J.; Boardman, L.A.; Burgart, L.J.; McDonnell, S.K.; Schaid, D.J.; Thibodeau, S.N. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003, 63, 5209–5212. [Google Scholar] [PubMed]
- Bouzourene, H.; Hutter, P.; Losi, L.; Martin, P.; Benhattar, J. Selection of patients with germline MLH1 mutated Lynch syndrome by determination of MLH1 methylation and BRAF mutation. Fam. Cancer 2010, 9, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Laiho, P.; Ollikainen, M.; Pinto, M.; Wang, L.; French, A.J.; Westra, J.; Frebourg, T.; Espin, E.; Armengol, M.; et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J. Med. Genet. 2004, 41, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Loughrey, M.B.; Waring, P.M.; Tan, A.; Trivett, M.; Kovalenko, S.; Beshay, V.; Young, M.A.; McArthur, G.; Boussioutas, A.; Dobrovic, A. Incorporation of somatic BRAF mutation testing into an algorithm for the investigation of hereditary non-polyposis colorectal cancer. Fam. Cancer 2007, 6, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Bessa, X.; Balleste, B.; Andreu, M.; Castells, A.; Bellosillo, B.; Balaguer, F.; Castellvi-Bel, S.; Paya, A.; Jover, R.; Alenda, C.; et al. A prospective, multicenter, population-based study of BRAF mutational analysis for Lynch syndrome screening. Clin. Gastroenterol. Hepatol. 2008, 6, 206–214. [Google Scholar] [CrossRef]
- Moreira, L.; Munoz, J.; Cuatrecasas, M.; Quintanilla, I.; Leoz, M.L.; Carballal, S.; Ocana, T.; Lopez-Ceron, M.; Pellise, M.; Castellvi-Bel, S.; et al. Prevalence of somatic mutl homolog 1 promoter hypermethylation in Lynch syndrome colorectal cancer. Cancer 2015, 121, 1395–1404. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Mutch, D.G.; Powell, M.A.; Mallon, M.A.; Goodfellow, P.J. RAS/RAF mutation and defective DNA mismatch repair in endometrial cancers. Am. J. Obs. Gynecol. 2004, 190, 935–942. [Google Scholar] [CrossRef]
- Salvesen, H.B.; Kumar, R.; Stefansson, I.; Angelini, S.; MacDonald, N.; Smeds, J.; Jacobs, I.J.; Hemminki, K.; Das, S.; Akslen, L.A. Low frequency of BRAF and CDKN2A mutations in endometrial cancer. Int. J. Cancer 2005, 115, 930–934. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Yanokura, M.; Banno, K.; Kobayashi, Y.; Kuwabara, Y.; Kobayashi, M.; Nomura, H.; Hirasawa, A.; Susumu, N.; Aoki, D. Analysis of a correlation between the BRAF V600E mutation and abnormal DNA mismatch repair in patients with sporadic endometrial cancer. Int. J. Oncol. 2009, 34, 1541–1547. [Google Scholar]
- Peterson, L.M.; Kipp, B.R.; Halling, K.C.; Kerr, S.E.; Smith, D.I.; Distad, T.J.; Clayton, A.C.; Medeiros, F. Molecular characterization of endometrial cancer: A correlative study assessing microsatellite instability, MLH1 hypermethylation, DNA mismatch repair protein expression, and PTEN, PIK3CA, KRAS, and BRAF mutation analysis. Int. J. Gynecol. Pathol. 2012, 31, 195–205. [Google Scholar] [CrossRef]
- Feng, Y.Z.; Shiozawa, T.; Miyamoto, T.; Kashima, H.; Kurai, M.; Suzuki, A.; Konishi, I. BRAF mutation in endometrial carcinoma and hyperplasia: Correlation with KRAS and p53 mutations and mismatch repair protein expression. Clin. Cancer Res. 2005, 11, 6133–6138. [Google Scholar] [CrossRef]
- Schrock, A.B.; Devoe, C.E.; McWilliams, R.; Sun, J.; Aparicio, T.; Stephens, P.J.; Ross, J.S.; Wilson, R.; Miller, V.A.; Ali, S.M.; et al. Genomic Profiling of Small-Bowel Adenocarcinoma. JAMA Oncol. 2017, 3, 1546–1553. [Google Scholar] [CrossRef]
- Cornejo, K.M.; Hutchinson, L.; Deng, A.; Tomaszewicz, K.; Welch, M.; Lyle, S.; Dresser, K.; Cosar, E.F. BRAF/KRAS gene sequencing of sebaceous neoplasms after mismatch repair protein analysis. Hum. Pathol. 2014, 45, 1213–1220. [Google Scholar] [CrossRef]
- Capper, D.; Voigt, A.; Bozukova, G.; Ahadova, A.; Kickingereder, P.; von Deimling, A.; von Knebel Doeberitz, M.; Kloor, M. BRAF V600E-specific immunohistochemistry for the exclusion of Lynch syndrome in MSI-H colorectal cancer. Int. J. Cancer 2013, 133, 1624–1630. [Google Scholar] [CrossRef]
- Thiel, A.; Heinonen, M.; Kantonen, J.; Gylling, A.; Lahtinen, L.; Korhonen, M.; Kytola, S.; Mecklin, J.P.; Orpana, A.; Peltomaki, P.; et al. BRAF mutation in sporadic colorectal cancer and Lynch syndrome. Virchows Arch. 2013, 463, 613–621. [Google Scholar] [CrossRef]
- Toon, C.W.; Walsh, M.D.; Chou, A.; Capper, D.; Clarkson, A.; Sioson, L.; Clarke, S.; Mead, S.; Walters, R.J.; Clendenning, M.; et al. BRAFV600E immunohistochemistry facilitates universal screening of colorectal cancers for Lynch syndrome. Am. J. Surg. Pathol. 2013, 37, 1592–1602. [Google Scholar] [CrossRef]
- Lasota, J.; Kowalik, A.; Wasag, B.; Wang, Z.F.; Felisiak-Golabek, A.; Coates, T.; Kopczynski, J.; Gozdz, S.; Miettinen, M. Detection of the BRAF V600E mutation in colon carcinoma: Critical evaluation of the imunohistochemical approach. Am. J. Surg. Pathol. 2014, 38, 1235–1241. [Google Scholar] [CrossRef]
- Adackapara, C.A.; Sholl, L.M.; Barletta, J.A.; Hornick, J.L. Immunohistochemistry using the BRAF V600E mutation-specific monoclonal antibody VE1 is not a useful surrogate for genotyping in colorectal adenocarcinoma. Histopathology 2013, 63, 187–193. [Google Scholar] [CrossRef]
- Perez-Carbonell, L.; Alenda, C.; Paya, A.; Castillejo, A.; Barbera, V.M.; Guillen, C.; Rojas, E.; Acame, N.; Gutierrez-Avino, F.J.; Castells, A.; et al. Methylation analysis of MLH1 improves the selection of patients for genetic testing in Lynch syndrome. J. Mol. Diagn. 2010, 12, 498–504. [Google Scholar] [CrossRef]
- Gausachs, M.; Mur, P.; Corral, J.; Pineda, M.; Gonzalez, S.; Benito, L.; Menendez, M.; Espinas, J.A.; Brunet, J.; Iniesta, M.D.; et al. MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: A cost-effectiveness study. Eur. J. Hum. Genet. 2012, 20, 762–768. [Google Scholar] [CrossRef]
- Adar, T.; Rodgers, L.H.; Shannon, K.M.; Yoshida, M.; Ma, T.; Mattia, A.; Lauwers, G.Y.; Iafrate, A.J.; Chung, D.C. A tailored approach to BRAF and MLH1 methylation testing in a universal screening program for Lynch syndrome. Mod. Pathol. 2017, 30, 440–447. [Google Scholar] [CrossRef]
- Rahner, N.; Friedrichs, N.; Steinke, V.; Aretz, S.; Friedl, W.; Buettner, R.; Mangold, E.; Propping, P.; Walldorf, C. Coexisting somatic promoter hypermethylation and pathogenic MLH1 germline mutation in Lynch syndrome. J. Pathol. 2008, 214, 10–16. [Google Scholar] [CrossRef]
- Yokoyama, T.; Takehara, K.; Sugimoto, N.; Kaneko, K.; Fujimoto, E.; Okazawa-Sakai, M.; Okame, S.; Shiroyama, Y.; Teramoto, N.; Ohsumi, S.; et al. Lynch syndrome-associated endometrial carcinoma with MLH1 germline mutation and MLH1 promoter hypermethylation: A case report and literature review. BMC Cancer 2018, 18, 576. [Google Scholar] [CrossRef]
- Bettstetter, M.; Dechant, S.; Ruemmele, P.; Grabowski, M.; Keller, G.; Holinski-Feder, E.; Hartmann, A.; Hofstaedter, F.; Dietmaier, W. Distinction of hereditary nonpolyposis colorectal cancer and sporadic microsatellite-unstable colorectal cancer through quantification of MLH1 methylation by real-time PCR. Clin. Cancer Res. 2007, 13, 3221–3228. [Google Scholar] [CrossRef]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar]
- Ahuja, N.; Mohan, A.L.; Li, Q.; Stolker, J.M.; Herman, J.G.; Hamilton, S.R.; Baylin, S.B.; Issa, J.P. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res. 1997, 57, 3370–3374. [Google Scholar]
- Paya, A.; Alenda, C.; Perez-Carbonell, L.; Rojas, E.; Soto, J.L.; Guillen, C.; Castillejo, A.; Barbera, V.M.; Carrato, A.; Castells, A.; et al. Utility of p16 immunohistochemistry for the identification of Lynch syndrome. Clin. Cancer Res. 2009, 15, 3156–3162. [Google Scholar] [CrossRef]
- Boissiere-Michot, F.; Frugier, H.; Ho-Pun-Cheung, A.; Lopez-Crapez, E.; Duffour, J.; Bibeau, F. Immunohistochemical staining for p16 and BRAFV600E is useful to distinguish between sporadic and hereditary (Lynch syndrome-related) microsatellite instable colorectal carcinomas. Virchows Arch. 2016, 469, 135–144. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.J.; Rhee, Y.Y.; Bae, J.M.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Gastric-type expression signature in serrated pathway-associated colorectal tumors. Hum. Pathol. 2015, 46, 643–656. [Google Scholar] [CrossRef]
- Pai, R.K.; Shadrach, B.L.; Carver, P.; Heald, B.; Moline, J.; Church, J.; Kalady, M.F.; Burke, C.A.; Plesec, T.P.; Lai, K.K.; et al. Immunohistochemistry for annexin A10 can distinguish sporadic from Lynch syndrome-associated microsatellite-unstable colorectal carcinoma. Am. J. Surg. Pathol. 2014, 38, 518–525. [Google Scholar] [CrossRef]
- Cohen, S.A.; Turner, E.H.; Beightol, M.B.; Jacobson, A.; Gooley, T.A.; Salipante, S.J.; Haraldsdottir, S.; Smith, C.; Scroggins, S.; Tait, J.F.; et al. Frequent PIK3CA Mutations in Colorectal and Endometrial Tumors with 2 or More Somatic Mutations in Mismatch Repair Genes. Gastroenterology 2016, 151, 440–447.e1. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef]
- Georgeson, P.; Walsh, M.D.; Clendenning, M.; Daneshvar, S.; Pope, B.J.; Mahmood, K.; Joo, J.E.; Jayasekara, H.; Jenkins, M.A.; Winship, I.M.; et al. Tumor mutational signatures in sebaceous skin lesions from individuals with Lynch syndrome. Mol. Genet. Genom. Med. 2019, 7, e00781. [Google Scholar] [CrossRef]
- Hampel, H.; Pearlman, R.; Beightol, M.; Zhao, W.; Jones, D.; Frankel, W.L.; Goodfellow, P.J.; Yilmaz, A.; Miller, K.; Bacher, J.; et al. Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol. 2018, 4, 806–813. [Google Scholar] [CrossRef]



{kind=link}
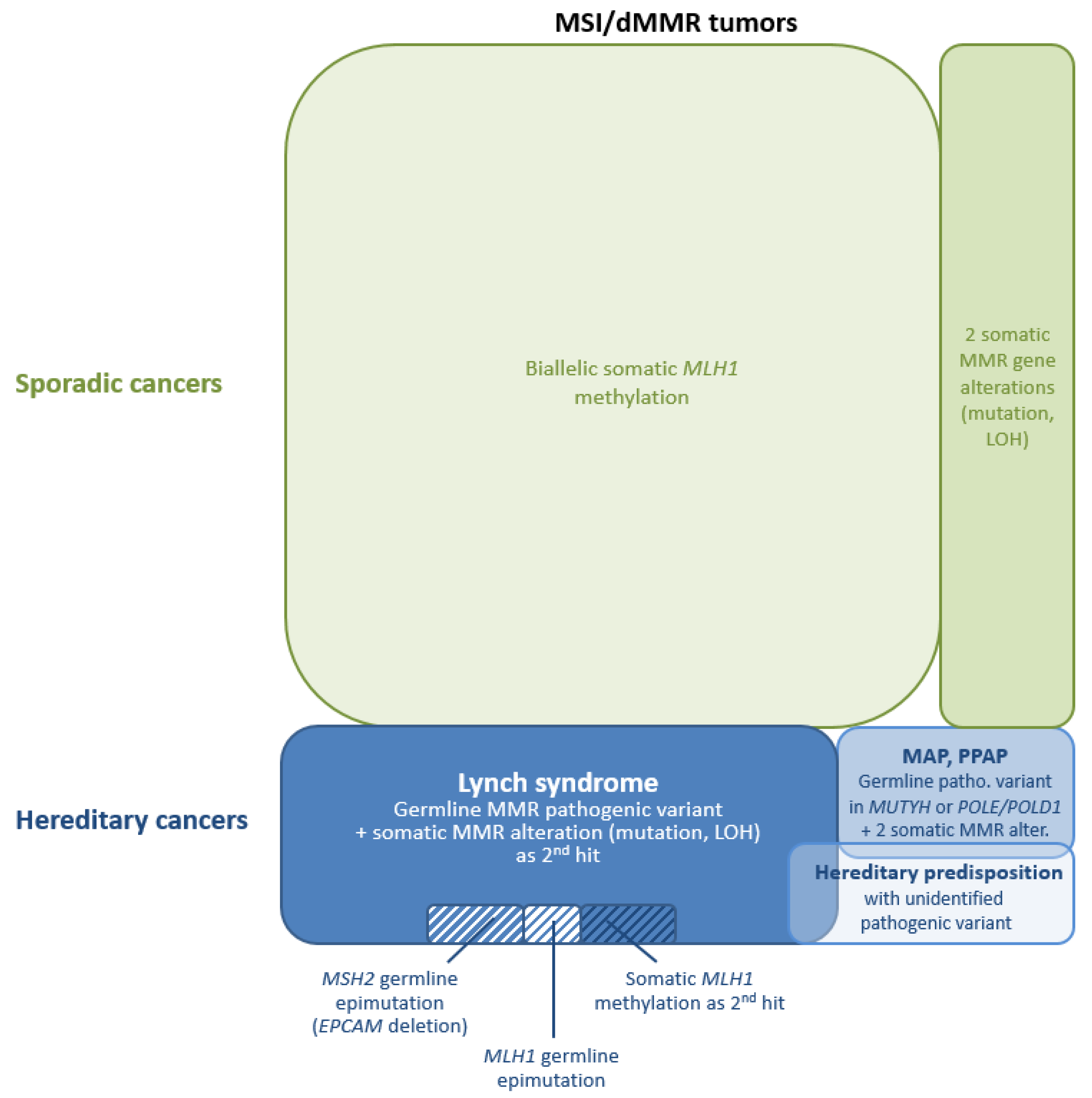{kind=link}
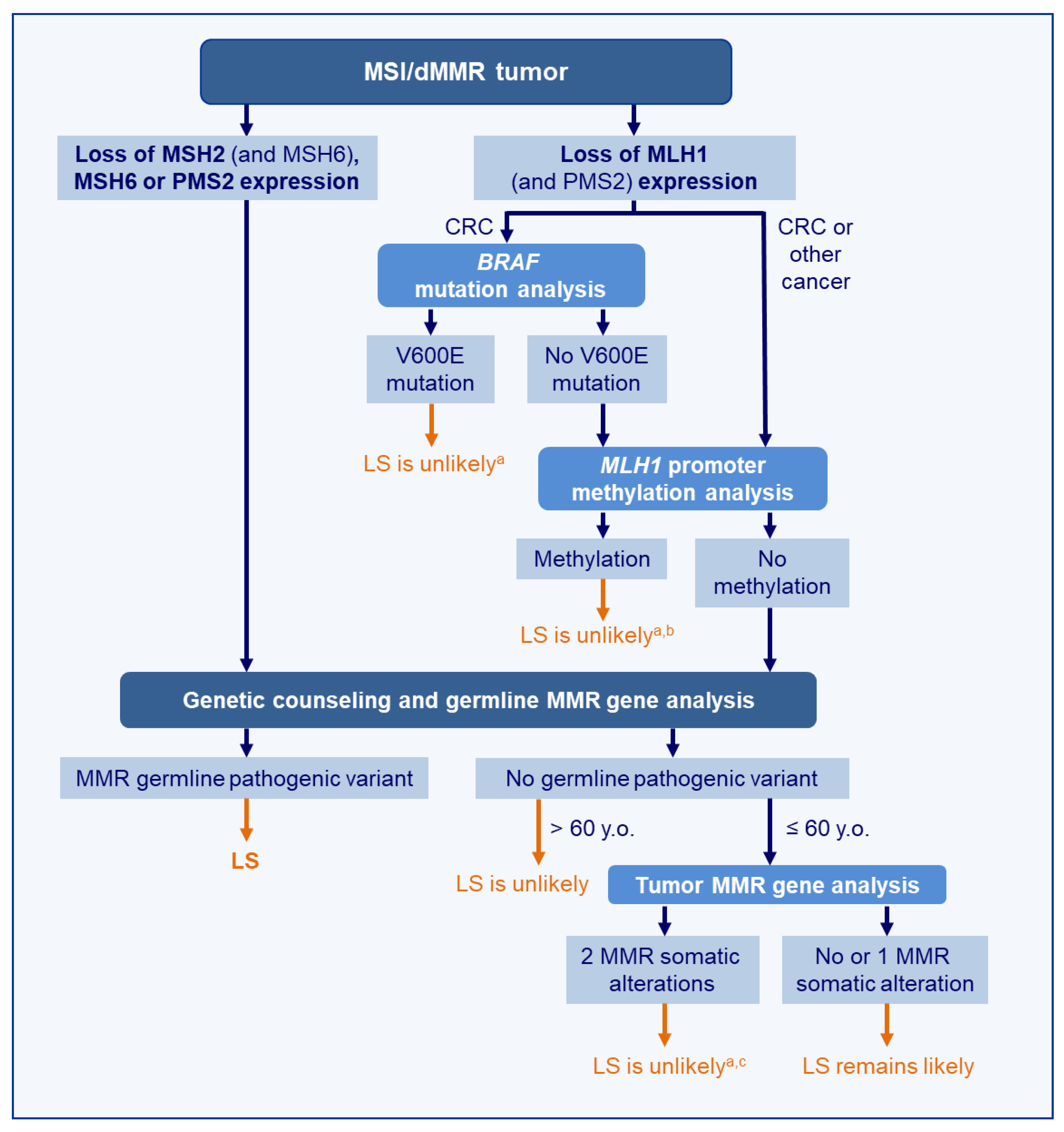{kind=link}
| Amsterdam II Criteria |
| (1) Three or more relatives with histologically verified LS-associated cancer (colorectal cancer, cancer of the endometrium, small bowel, ureter, or renal pelvis), 1 of which is a first-degree relative of the others. (2) Cancer involving at least 2 generations. (3) One or more cancer cases diagnosed before the age of 50. (4) Familial adenomatous polyposis should be excluded. |
| Revised Bethesda Criteria 1 |
| (1) CRC or endometrial cancer diagnosed at age younger than 50. (2) Presence of synchronous or metachronous CRC or other LS-associated tumors, regardless of age. (3) CRC with MSI-high pathologic-associated features (Crohn-like lymphocytic reaction, mucinous/signet cell differentiation, or medullary growth pattern) diagnosed in an individual younger than 60. (4) Patient with CRC or LS-associated tumor diagnosed below 50 in at least 1 first-degree relative. (5) Patient with CRC or LS-associated tumor diagnosed at any age in 2 first-degree or second-degree relatives. |
| Clinical Criteria/Molecular Test | Data Evocative of LS | Limitations |
|---|---|---|
| Clinical presentation | Personal and/or family history of (multiple) LS-related cancers [239] Age of cancer onset ≤ 60 y.o.: | A number of LS patient develop their cancer > 60 y.o.:
|
| Histopathological characteristics | CRC: associated conventional adenomas; MMR-deficient crypts in peritumoral mucosa: Se 30–70%, Sp ≥ 99% [163,164,165] EC: increased number of CD8+ immune cells [51] | |
| IHC for MMR proteins | Loss of MSH2/MSH6, isolated loss of MSH6 or isolated loss of PMS2 | Some may be due to somatic inactivation (up to 60% of sebaceous tumors with loss of MSH2/MSH6 [93,95]; up to 50% of isolated loss of PMS2 may be explained by MLH1 hypermethylation [240]) |
| BRAF mutation analysis (V600E) | Absence of BRAF V600E mutation CRC: Se 98.5%; Sp 66% [241] | BRAF mutation in 1.4% of CRC from LS patients [241] No utility in non-CRC |
| MLH1 promoter methylation analysis | Absence of methylation CRC: Se 94%; Sp 88% [241] | Methylation in up to 6% of CRC from LS patients [242] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leclerc, J.; Vermaut, C.; Buisine, M.-P. Diagnosis of Lynch Syndrome and Strategies to Distinguish Lynch-Related Tumors from Sporadic MSI/dMMR Tumors. Cancers 2021, 13, 467. https://doi.org/10.3390/cancers13030467
Leclerc J, Vermaut C, Buisine M-P. Diagnosis of Lynch Syndrome and Strategies to Distinguish Lynch-Related Tumors from Sporadic MSI/dMMR Tumors. Cancers. 2021; 13(3):467. https://doi.org/10.3390/cancers13030467
Chicago/Turabian StyleLeclerc, Julie, Catherine Vermaut, and Marie-Pierre Buisine. 2021. "Diagnosis of Lynch Syndrome and Strategies to Distinguish Lynch-Related Tumors from Sporadic MSI/dMMR Tumors" Cancers 13, no. 3: 467. https://doi.org/10.3390/cancers13030467
APA StyleLeclerc, J., Vermaut, C., & Buisine, M.-P. (2021). Diagnosis of Lynch Syndrome and Strategies to Distinguish Lynch-Related Tumors from Sporadic MSI/dMMR Tumors. Cancers, 13(3), 467. https://doi.org/10.3390/cancers13030467