
An Easily Expandable Multi-Drug LC-MS Assay for the Simultaneous Quantification of 57 Oral Antitumor Drugs in Human Plasma

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Chromatography and Mass Spectrometer Equipment
2.3. Chromatographic Conditions
2.4. MS Conditions
2.5. Sample Preparation
2.6. Patient Plasma Collections
2.7. Data Analysis
2.8. Method Validation
2.9. Calibration Curves, Linearity, and Sensitivity
2.10. Accuracy and Precision
2.11. Stability
2.12. Recovery and Matrix Effect
2.13. Selectivity
2.14. Carryover
2.15. Dilution Integrity
3. Results
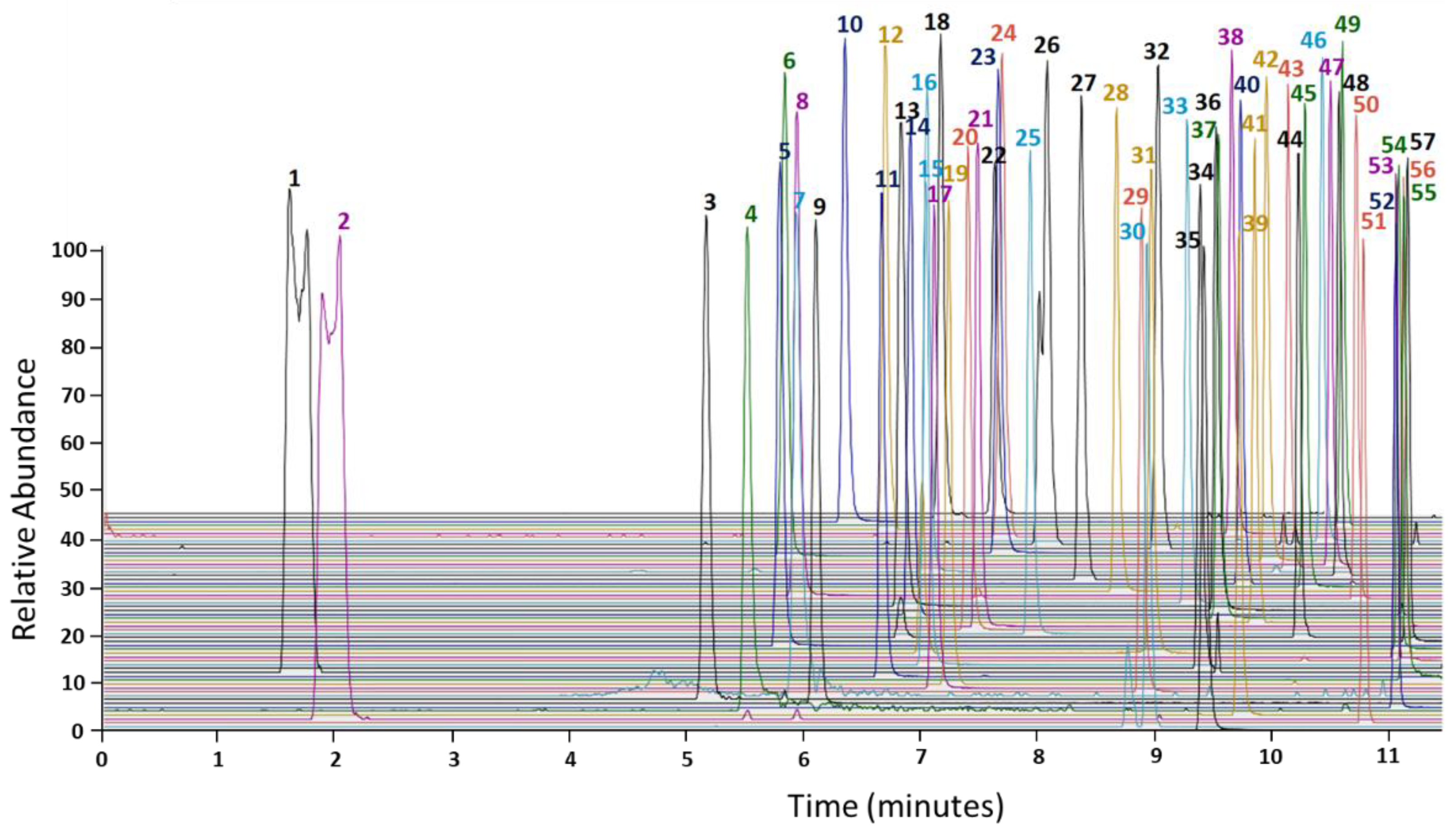
3.1. Calibration Curves, Linearity, Carryover, Selectivity, and Sensitivity
3.2. Accuracy and Precision
3.3. Recovery and Matrix Effect
3.4. Stability
3.5. Dilution Integrity
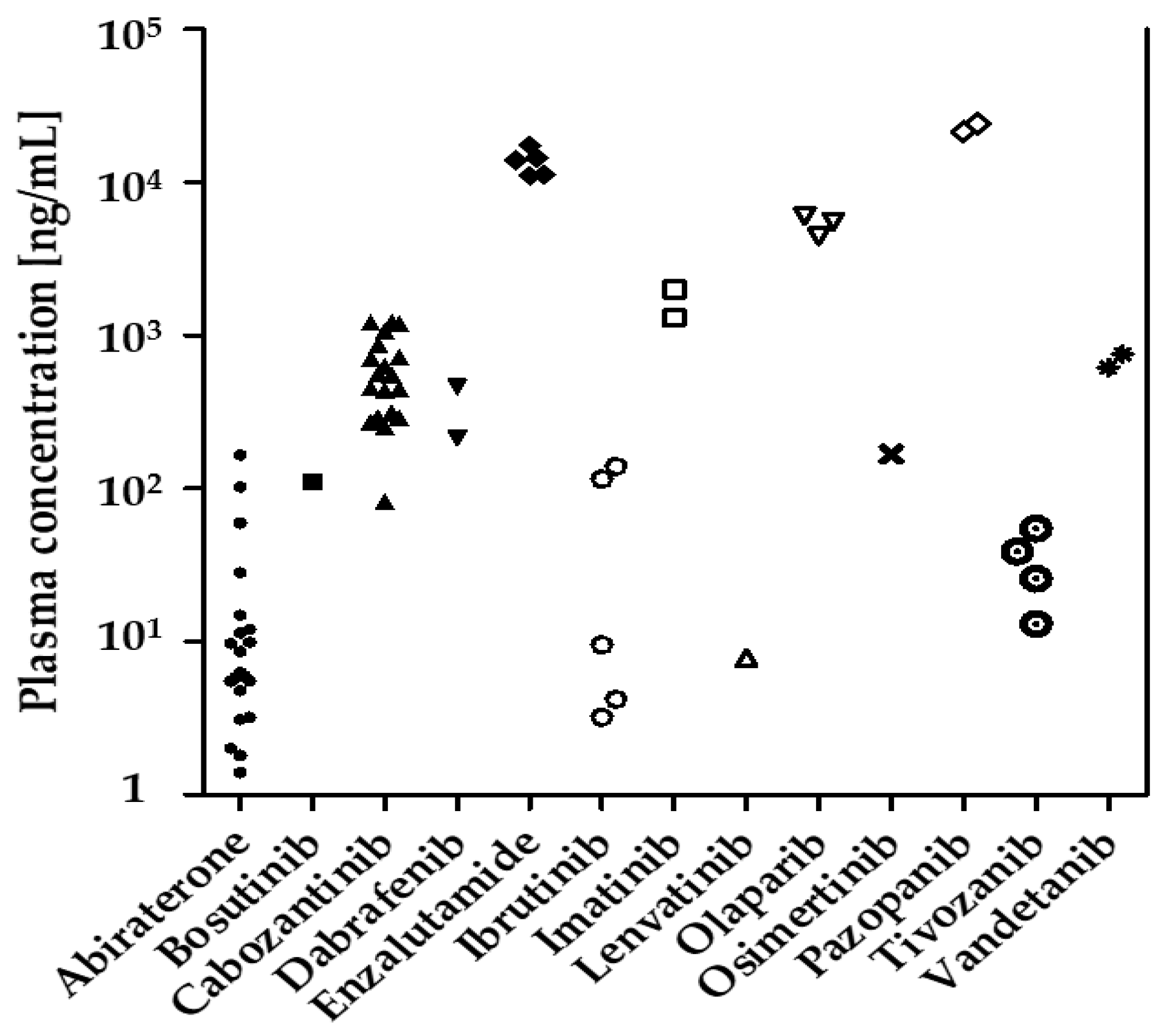
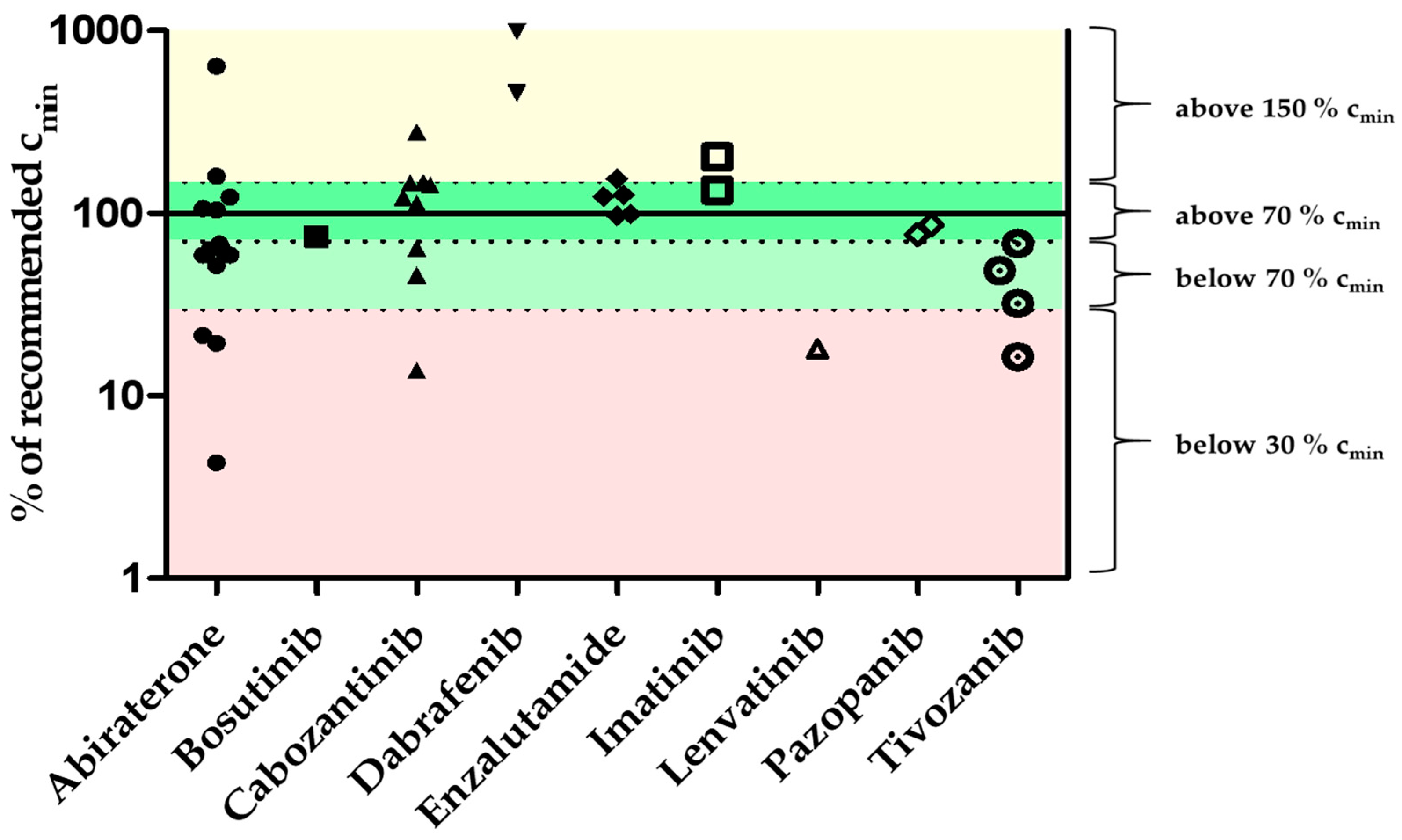
3.6. Patient Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weingart, S.N.; Brown, E.; Bach, P.B.; Eng, K.; Johnson, S.A.; Kuzel, T.M.; Langbaum, T.S.; Leedy, R.D.; Muller, R.J.; Newcomer, L.N.; et al. NCCN task force report: Oral chemotherapy. J. Natl. Compr. Cancer Netw. 2008, 6 (Suppl. S3), S1–S14. [Google Scholar] [CrossRef]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquali, S.; Hadjinicolaou, A.V.; Chiarion Sileni, V.; Rossi, C.R.; Mocellin, S. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database Syst. Rev. 2018, 2, CD011123. [Google Scholar] [CrossRef]
- Retmana, I.A.; Beijnen, J.H.; Sparidans, R.W. Chromatographic bioanalytical assays for targeted covalent kinase inhibitors and their metabolites. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2021, 1162, 122466. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Schoell, A.; Groenland, S.L.; Scherf-Clavel, O.; van Dyk, M.; Huisinga, W.; Michelet, R.; Jaehde, U.; Steeghs, N.; Huitema, A.D.R.; Kloft, C. Therapeutic drug monitoring of oral targeted antineoplastic drugs. Eur. J. Clin. Pharmacol. 2021, 77, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Schlichtig, K.; Dürr, P.; Dörje, F.; Fromm, M.F. New oral anti-cancer drugs and medication safety. Dtsch. Arztebl. Int. 2019, 116, 775–782. [Google Scholar] [CrossRef]
- Hömke, R.; Neue Krebsmedikamente im EU-Zulassungsverfahren Sowie Zugelassene Krebsmedikamente, Die Noch Nicht auf den Deutschen Markt Gebracht Wurden. vfa. Die Forschenden Pharma-Unternehmen. Available online: https://www.vfa.de/de/presse/pressemitteilungen/krebsmedikamente-in-zulassung.pdf (accessed on 24 September 2021).
- Dürr, P.; Schlichtig, K.; Kelz, C.; Deutsch, B.; Maas, R.; Eckart, M.J.; Wilke, J.; Wagner, H.; Wolff, K.; Preuß, C.; et al. The randomized AMBORA trial: Impact of pharmacological/pharmaceutical care on medication safety and patient-reported outcomes during treatment with new oral anticancer agents. J. Clin. Oncol. 2021, 39, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, R.B.; Yu, H.; Schellens, J.H.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin. Pharmacol. Ther. 2017, 102, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Groenland, S.L.; Verheijen, R.B.; Joerger, M.; Mathijssen, R.H.J.; Sparreboom, A.; Beijnen, J.H.; Beumer, J.H.; Steeghs, N.; Huitema, A.D.R. Precision dosing of targeted therapies is ready for prime time. Clin. Cancer Res. 2021, 27, 6644–6652. [Google Scholar] [CrossRef] [PubMed]
- Marin, D.; Bazeos, A.; Mahon, F.X.; Eliasson, L.; Milojkovic, D.; Bua, M.; Apperley, J.F.; Szydlo, R.; Desai, R.; Kozlowski, K.; et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J. Clin. Oncol. 2010, 28, 2381–2388. [Google Scholar] [CrossRef] [PubMed]
- Noens, L.; van Lierde, M.A.; De Bock, R.; Verhoef, G.; Zachée, P.; Berneman, Z.; Martiat, P.; Mineur, P.; Van Eygen, K.; MacDonald, K.; et al. Prevalence, determinants, and outcomes of nonadherence to imatinib therapy in patients with chronic myeloid leukemia: The ADAGIO study. Blood 2009, 113, 5401–5411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.M.; Baran, A.M.; Casulo, C.; Reagan, P.; Friedberg, J.W.; Helber, M.; Moore, J.; Baloga, E.; Zent, C.S.; Barr, P.M. Ibrutinib dose adherence and therapeutic efficacy in non-hodgkin lymphoma: A single-center experience. Clin. Lymphoma Myeloma Leuk. 2019, 19, 41–47. [Google Scholar] [CrossRef]
- Groenland, S.L.; van Eerden, R.A.G.; Verheijen, R.B.; Koolen, S.L.W.; Moes, D.J.A.R.; Desar, I.M.E.; Reyners, A.K.L.; Gelderblom, H.J.; van Erp, N.P.; Mathijssen, R.H.J.; et al. Therapeutic drug monitoring of oral anticancer drugs: The dutch pharmacology oncology group–therapeutic drug monitoring protocol for a prospective study. Ther. Drug Monit. 2019, 41, 561–567. [Google Scholar] [CrossRef]
- Gardner, E.R.; Burger, H.; van Schaik, R.H.; van Oosterom, A.T.; de Bruijn, E.A.; Guetens, G.; Prenen, H.; de Jong, F.A.; Baker, S.D.; Bates, S.E.; et al. Association of enzyme and transporter genotypes with the pharmacokinetics of imatinib. Clin. Pharmacol. Ther. 2006, 80, 192–201. [Google Scholar] [CrossRef]
- Greer, J.A.; Amoyal, N.; Nisotel, L.; Fishbein, J.N.; MacDonald, J.; Stagl, J.; Lennes, I.; Temel, J.S.; Safren, S.A.; Pirl, W.F. A systematic review of adherence to oral antineoplastic therapies. Oncologist 2016, 21, 354–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Brar, S.S.; Lesko, L.J. To take or not to take with meals? Unraveling issues related to food effects labeling for oral antineoplastic drugs. Clin. Pharmacol. Drug Dev. 2018, 7, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.M.; Reddy, N.J.; Cohen, R.B.; Lewis, N.L.; Whitehead, B.; Mackay, K.; Stead, A.; Beelen, A.P.; Lewis, L.D. Effects of food on the relative bioavailability of lapatinib in cancer patients. J. Clin. Oncol. 2009, 27, 1191–1196. [Google Scholar] [CrossRef]
- Kletzl, H.; Giraudon, M.; Ducray, P.S.; Abt, M.; Hamilton, M.; Lum, B.L. Effect of gastric pH on erlotinib pharmacokinetics in healthy individuals: Omeprazole and ranitidine. Anticancer Drugs 2015, 26, 565–572. [Google Scholar] [CrossRef]
- Widmer, N.; Bardin, C.; Chatelut, E.; Paci, A.; Beijnen, J.; Levêque, D.; Veal, G.; Astier, A. Review of therapeutic drug monitoring of anticancer drugs part two-targeted therapies. Eur. J. Cancer 2014, 50, 2020–2036. [Google Scholar] [CrossRef]
- Molenaar-Kuijsten, L.; Van Balen, D.E.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. A review of CYP3A drug-drug interaction studies: Practical guidelines for patients using targeted oral anticancer drugs. Front. Pharmacol. 2021, 12, 670862. [Google Scholar] [CrossRef] [PubMed]
- Schlichtig, K.; Dürr, P.; Dörje, F.; Fromm, M.F. Medication errors during treatment with new oral anticancer agents: Consequences for clinical practice based on the AMBORA study. Clin. Pharmacol. Ther. 2021, 110, 1075–1086. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Igarashi, R.; Suzuki-Honma, N.; Fujiyama, N.; Narita, S.; Inoue, T.; Saito, M.; Akihama, S.; Tsuruta, H.; Miura, M.; et al. Association of pharmacokinetics of axitinib with treatment outcome and adverse events in advanced renal cell carcinoma patients. J. Clin. Oncol. 2015, 33, 506. [Google Scholar] [CrossRef]
- Kurata, Y.; Miyauchi, N.; Suno, M.; Ito, T.; Sendo, T.; Kiura, K. Correlation of plasma crizotinib trough concentration with adverse events in patients with anaplastic lymphoma kinase positive non-small-cell lung cancer. J. Pharm. Health Care Sci. 2015, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheijen, R.B.; Bins, S.; Mathijssen, R.H.; Lolkema, M.P.; van Doorn, L.; Schellens, J.H.; Beijnen, J.H.; Langenberg, M.H.; Huitema, A.D.; Steeghs, N.; et al. Individualized pazopanib dosing: A prospective feasibility study in cancer patients. Clin. Cancer Res. 2016, 22, 5738–5746. [Google Scholar] [CrossRef] [Green Version]
- ON-TARGET. Available online: https://www.bcp.fu-berlin.de/pharmazie/faecher/klinische_pharmazie/arbeitsgruppe_kloft/on-target-studie/index.html (accessed on 7 October 2021).
- Lankheet, N.A.G.; Knapen, L.M.; Schellens, J.H.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. Plasma concentrations of tyrosine kinase inhibitors imatinib, erlotinib, and sunitinib in routine clinical outpatient cancer care. Ther. Drug Monit. 2014, 36, 326–334. [Google Scholar] [CrossRef]
- Merienne, C.; Rousset, M.; Ducint, D.; Castaing, N.; Titier, K.; Molimard, M.; Bouchet, S. High throughput routine determination of 17 tyrosine kinase inhibitors by LC-MS/MS. J. Pharm. Biomed. Anal. 2018, 150, 112–120. [Google Scholar] [CrossRef]
- Aghai, F.; Zimmermann, S.; Kurlbaum, M.; Jung, P.; Pelzer, T.; Klinker, H.; Isberner, N.; Scherf-Clavel, O. Development and validation of a sensitive liquid chromatography tandem mass spectrometry assay for the simultaneous determination of ten kinase inhibitors in human serum and plasma. Anal. Bioanal. Chem. 2021, 413, 599–612. [Google Scholar] [CrossRef]
- Reis, R.; Labat, L.; Allard, M.; Boudou-Rouquette, P.; Chapron, J.; Bellesoeur, A.; Thomas-Schoemann, A.; Arrondeau, J.; Giraud, F.; Alexandre, J.; et al. Liquid chromatography-tandem mass spectrometric assay for therapeutic drug monitoring of the EGFR inhibitors afatinib, erlotinib and osimertinib, the ALK inhibitor crizotinib and the VEGFR inhibitor nintedanib in human plasma from non-small cell lung cancer patients. J. Pharm. Biomed. Anal. 2018, 158, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Herbrink, M.; de Vries, N.; Rosing, H.; Huitema, A.D.R.; Nuijen, B.; Schellens, J.H.M.; Beijnen, J.H. Development and validation of a liquid chromatography-tandem mass spectrometry analytical method for the therapeutic drug monitoring of eight novel anticancer drugs. Biomed. Chromatogr. 2018, 32, e4147. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.M.; de Vries, N.; Venekamp, N.; Rosing, H.; Huitema, A.D.R.; Beijnen, J.H. Development and validation of a liquid chromatography-tandem mass spectrometry assay for nine oral anticancer drugs in human plasma. J. Pharm. Biomed. Anal. 2019, 174, 561–566. [Google Scholar] [CrossRef]
- Koller, D.; Vaitsekhovich, V.; Mba, C.; Steegmann, J.L.; Zubiaur, P.; Abad-Santos, F.; Wojnicz, A. Effective quantification of 11 tyrosine kinase inhibitors and caffeine in human plasma by validated LC-MS/MS method with potent phospholipids clean-up procedure. Application to therapeutic drug monitoring. Talanta 2020, 208, 120450. [Google Scholar] [CrossRef]
- Andriamanana, I.; Gana, I.; Duretz, B.; Hulin, A. Simultaneous analysis of anticancer agents bortezomib, imatinib, nilotinib, dasatinib, erlotinib, lapatinib, sorafenib, sunitinib and vandetanib in human plasma using LC/MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 926, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, E.; Mercier, T.; Wagner, A.D.; Homicsko, K.; Michielin, O.; Ellefsen-Lavoie, K.; Cagnon, L.; Diezi, M.; Buclin, T.; Widmer, N.; et al. Quantification of the next-generation oral anti-tumor drugs dabrafenib, trametinib, vemurafenib, cobimetinib, pazopanib, regorafenib and two metabolites in human plasma by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1083, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Ezzeldin, E.; Iqbal, M.; Herqash, R.N.; ElNahhas, T. Simultaneous quantitative determination of seven novel tyrosine kinase inhibitors in plasma by a validated UPLC-MS/MS method and its application to human microsomal metabolic stability study. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2020, 1136, 121851. [Google Scholar] [CrossRef]
- Krens, S.D.; van der Meulen, E.; Jansman, F.G.A.; Burger, D.M.; van Erp, N.P. Quantification of cobimetinib, cabozantinib, dabrafenib, niraparib, olaparib, vemurafenib, regorafenib and its metabolite regorafenib M2 in human plasma by UPLC-MS/MS. Biomed. Chromatogr. 2020, 34, e4758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, M.; Titier, K.; Bouchet, S.; Dutriaux, C.; Pham-Ledard, A.; Prey, S.; Canal-Raffin, M.; Molimard, M. An UPLC-MS/MS method for the quantification of BRAF inhibitors (vemurafenib, dabrafenib) and MEK inhibitors (cobimetinib, trametinib, binimetinib) in human plasma. Application to treated melanoma patients. Clin. Chim. Acta 2017, 470, 8–13. [Google Scholar] [CrossRef]
- Ni, M.-W.; Zhou, J.; Li, H.; Chen, W.; Mou, H.-Z.; Zheng, Z.-G. Simultaneous determination of six tyrosine kinase inhibitors in human plasma using HPLC-Q-Orbitrap mass spectrometry. Bioanalysis 2017, 9, 925–935. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). Bioanalytical Method Validation Guidance for Industry. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 24 September 2021).
- Veerman, G.D.M.; Lam, M.H.; Mathijssen, R.H.J.; Koolen, S.L.W.; de Bruijn, P. Quantification of afatinib, alectinib, crizotinib and osimertinib in human plasma by liquid chromatography/triple-quadrupole mass spectrometry; focusing on the stability of osimertinib. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1113, 37–44. [Google Scholar] [CrossRef]
- Sparidans, R.W.; Iusuf, D.; Schinkel, A.H.; Schellens, J.H.; Beijnen, J.H. Liquid chromatography-tandem mass spectrometric assay for the light sensitive tyrosine kinase inhibitor axitinib in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 4090–4096. [Google Scholar] [CrossRef]
- Nijenhuis, C.M.; Haverkate, H.; Rosing, H.; Schellens, J.H.; Beijnen, J.H. Simultaneous quantification of dabrafenib and trametinib in human plasma using high-performance liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2016, 125, 270–279. [Google Scholar] [CrossRef]
- van Nuland, M.; Groenland, S.L.; Bergman, A.M.; Steeghs, N.; Rosing, H.; Venekamp, N.; Huitema, A.D.R.; Beijnen, J.H. Exposure-response analyses of abiraterone and its metabolites in real-world patients with metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2020, 23, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chia, Y.L.; Nedelman, J.; Schran, H.; Mahon, F.X.; Molimard, M. A therapeutic drug monitoring algorithm for refining the imatinib trough level obtained at different sampling times. Ther. Drug Monit. 2009, 31, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.M.; Dorlo, T.P.C.; Beijnen, J.H.; Huitema, A.D.R. Evaluation of extrapolation methods to predict trough concentrations to guide therapeutic drug monitoring of oral anticancer drugs. Ther. Drug Monit. 2020, 42, 532–539. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oral Antitumor Drug | Number of Samples/Patients | Samples at ±4 h Trough Level | Median Concentration of All Samples [% of r.c.] | Min–Max Concentration [% of r.c.] | Interpatient Coefficient of Variation [%] |
|---|---|---|---|---|---|
| Abiraterone | 22/12 | 14 | 229.0 | 4.3–1784.9 | 187.3 |
| Axitinib | 1/1 | 0 | - | 6.0 | - |
| Bosutinib | 1/1 | 1 | - | 74.7 | - |
| Cabozantinib * | 19/11 | 9 | 153.3 | 13.8–207.2 | 58.5 |
| Dabrafenib | 2/1 | 2 | 722.3 | 455.8–988.8 | - |
| Enzalutamide | 5/3 | 5 | 120.1 | 97.1–154.2 | 19.5 |
| Ibrutinib | 4/2 | 0 | 110.1 | 4.8–211.4 | 110.0 |
| Imatinib | 2/2 | 2 | 168.9 | 133.8–204.1 | 29.4 |
| Lenvatinib | 2/1 | 2 | - | n.d.–18.0 | - |
| Olaparib | 4/2 | 0 | 287.6 | n.d.–325.5 | 51.6 |
| Osimertinib | 1/1 | 0 | - | 101.1 | - |
| Pazopanib | 2/1 | 2 | 81.5 | 76.6–86.5 | - |
| Tivozanib | 4/2 | 4 | 41.4 | 16.4–68.3 | 53.8 |
| Vandetanib | 2/1 | 0 | 86.7 | 78.2–95.3 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kehl, N.; Schlichtig, K.; Dürr, P.; Bellut, L.; Dörje, F.; Fietkau, R.; Pavel, M.; Mackensen, A.; Wullich, B.; Maas, R.; et al. An Easily Expandable Multi-Drug LC-MS Assay for the Simultaneous Quantification of 57 Oral Antitumor Drugs in Human Plasma. Cancers 2021, 13, 6329. https://doi.org/10.3390/cancers13246329
Kehl N, Schlichtig K, Dürr P, Bellut L, Dörje F, Fietkau R, Pavel M, Mackensen A, Wullich B, Maas R, et al. An Easily Expandable Multi-Drug LC-MS Assay for the Simultaneous Quantification of 57 Oral Antitumor Drugs in Human Plasma. Cancers. 2021; 13(24):6329. https://doi.org/10.3390/cancers13246329
Chicago/Turabian StyleKehl, Niklas, Katja Schlichtig, Pauline Dürr, Laura Bellut, Frank Dörje, Rainer Fietkau, Marianne Pavel, Andreas Mackensen, Bernd Wullich, Renke Maas, and et al. 2021. "An Easily Expandable Multi-Drug LC-MS Assay for the Simultaneous Quantification of 57 Oral Antitumor Drugs in Human Plasma" Cancers 13, no. 24: 6329. https://doi.org/10.3390/cancers13246329
APA StyleKehl, N., Schlichtig, K., Dürr, P., Bellut, L., Dörje, F., Fietkau, R., Pavel, M., Mackensen, A., Wullich, B., Maas, R., Fromm, M. F., Gessner, A., & Taudte, R. V. (2021). An Easily Expandable Multi-Drug LC-MS Assay for the Simultaneous Quantification of 57 Oral Antitumor Drugs in Human Plasma. Cancers, 13(24), 6329. https://doi.org/10.3390/cancers13246329