
Transcription Regulation and Genome Rewiring Governing Sensitivity and Resistance to FOXM1 Inhibition in Breast Cancer

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
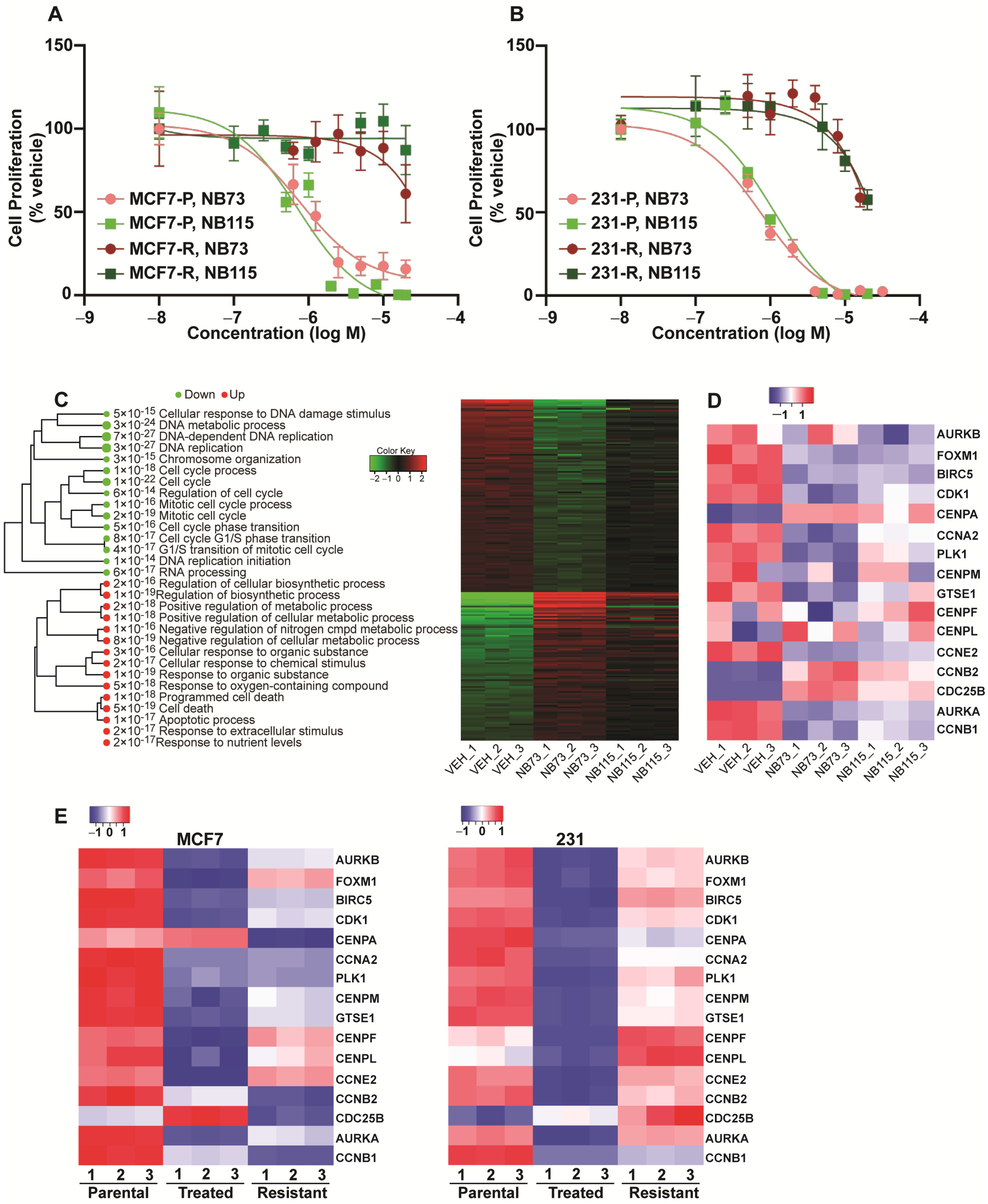
2.1. Transcriptional Effects of FOXM1 Inhibition in Sensitive and Resistant Breast Cancer Cells
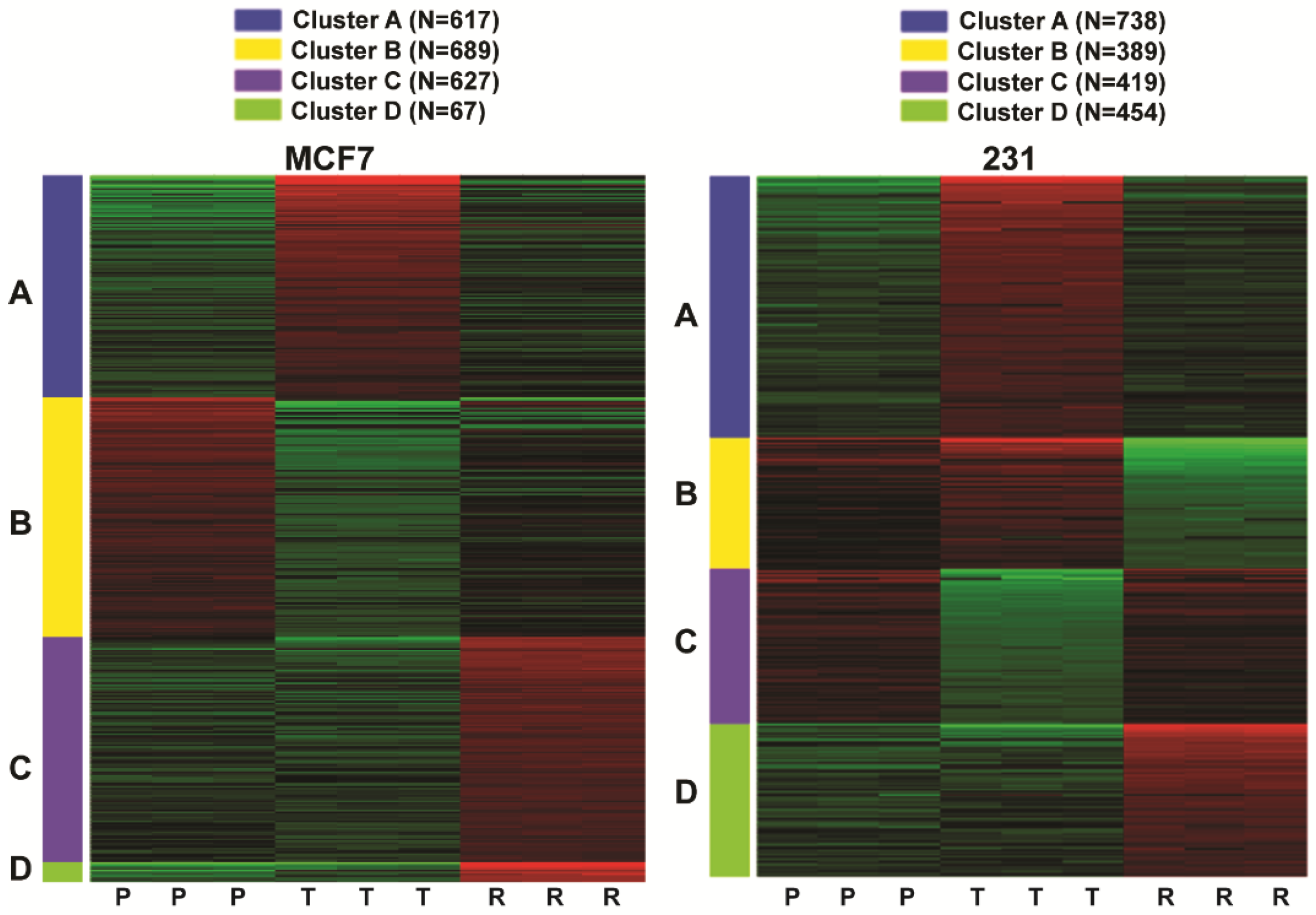
2.2. Comparison of Gene Regulations in Cells Sensitive or Resistant to FOXM1 Inhibitors
2.3. Increased Interferon Inflammatory Gene Expressions and Signaling in ER-Positive Cells with Inhibitor Resistance
2.4. The IRFMS Predicts Poor Clinical Outcome in Patients with ER-Positive Breast Cancers
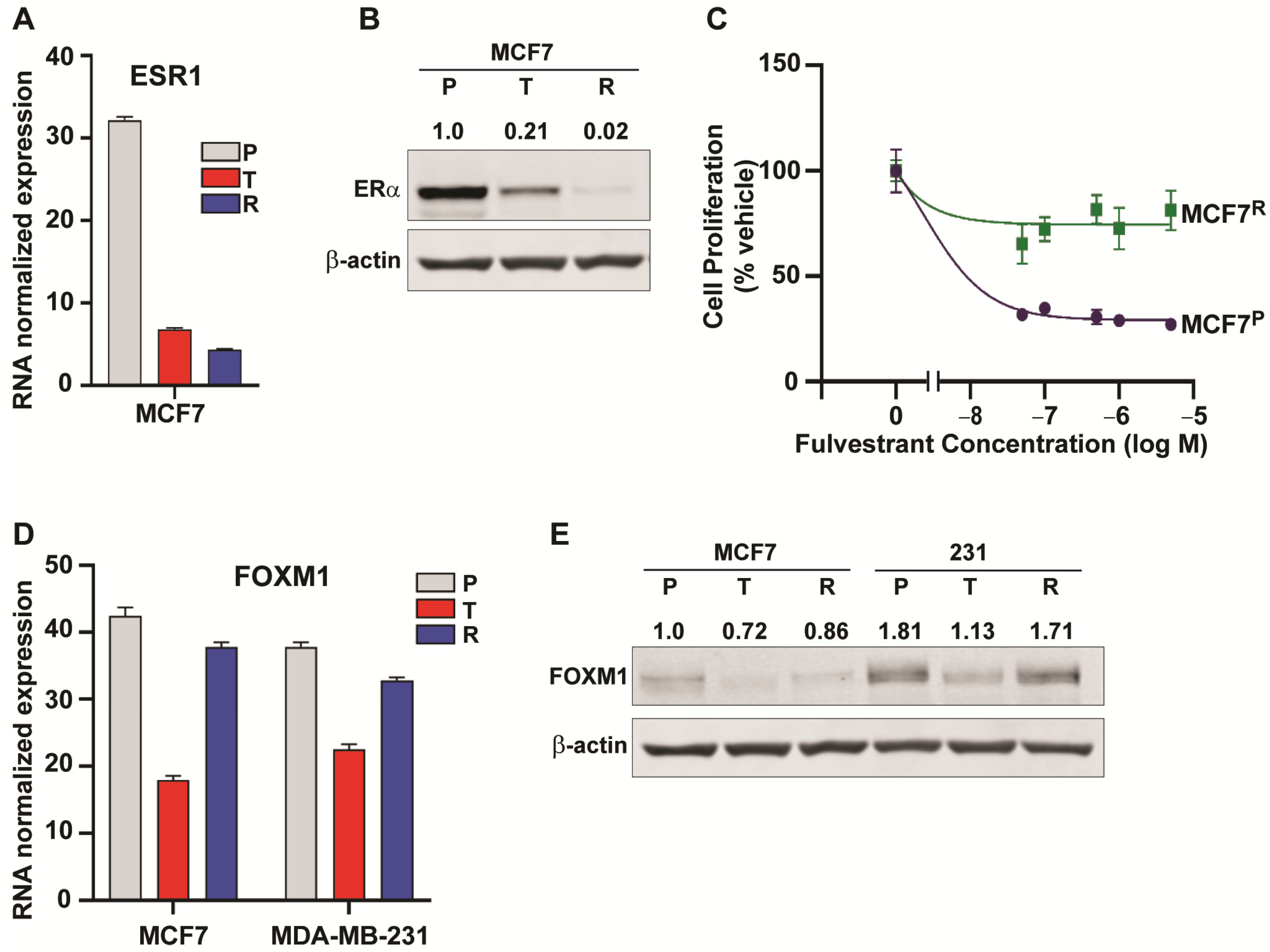
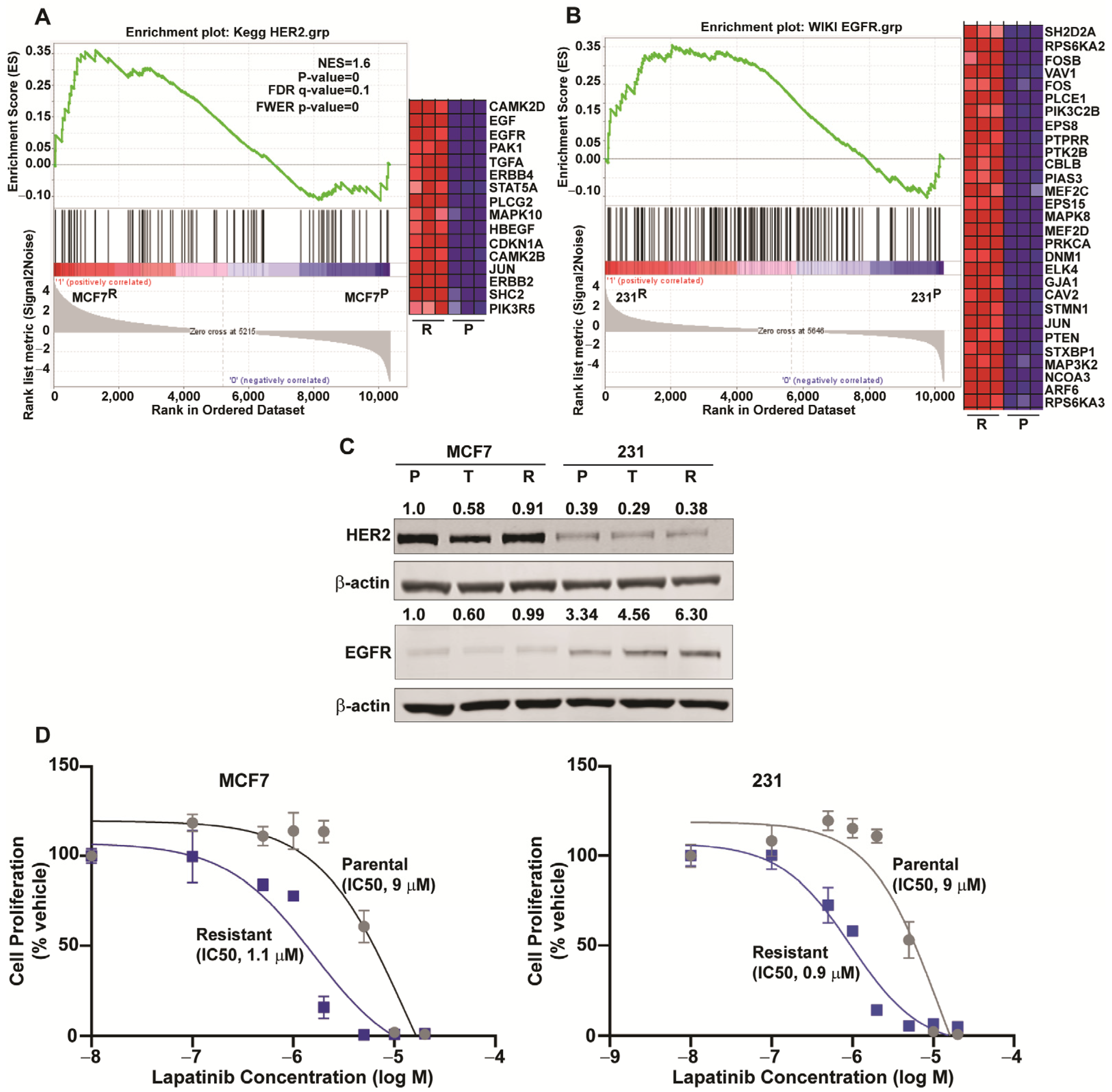
2.5. Downregulation of ESR1 and Upregulation of HER2 and EGFR Pathways in Resistance
2.6. Changes in Genes Associated with the Cell Cycle and Energy Generation in Inhibitor Sensitive and Resistant Breast Cancer Cell States
3. Discussion
3.1. Patterns of Gene and Pathway Alterations from FOXM1 Inhibition and the Development of Resistance
3.2. Common and Contrasting Cellular Alterations Associated with Resistance to FOXM1 Inhibition
4. Material and Methods
4.1. Materials, Cell Culture, and Development of FOXM1 Inhibitor Resistant Cells
4.2. Cell Viability Assay
4.3. Western Blot Analyses
4.4. RNA-Seq Transcriptional Profiling and Gene Ontology and Pathway Signature and Network Analyses
4.5. DNA Sequencing of the FOXM1 Gene
4.6. Our Interferon-Related FOXM1 Inhibitor Resistance Signature (IRFMS), and Clinical Datasets and Kaplan-Meier Survival Analyses
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Regan, R.M.; Nahta, R. Targeting forkhead box M1 transcription factor in breast cancer. Biochem. Pharmacol. 2018, 154, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Song, B.N.; Chu, I.S. A gene expression signature of FOXM1 predicts the prognosis of hepatocellular carcinoma. Exp. Mol. Med. 2018, 50, e418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 2019, 11, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartel, A.L. FOXM1 in Cancer: Interactions and Vulnerabilities. Cancer Res. 2017, 77, 3135–3139. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.F.; Zeng, D.; Liang, W.Q.; Chen, C.F.; Sun, S.M.; Lin, H.Y. FoxM1 is a promising candidate target in the treatment of breast cancer. Oncotarget 2018, 9, 842–852. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, P.; Park, H.J. FoxM1: A master regulator of tumor metastasis. Cancer Res. 2011, 71, 4329–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergamaschi, A.; Madak-Erdogan, Z.; Kim, Y.J.; Choi, Y.L.; Lu, H.; Katzenellenbogen, B.S. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. 2014, 16, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, P.; Wang, A.; Ziegler, Y.; Kim, S.H.; El-Ashry, D.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Suppression of Tumor Growth, Metastasis, and Signaling Pathways by Reducing FOXM1 Activity in Triple Negative Breast Cancer. Cancers 2020, 12, 2677. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, Z.; Ashour, A.; Kahraman, N.; Ozpolat, B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016, 7, 16619–16635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tryfonidis, K.; Zardavas, D.; Katzenellenbogen, B.S.; Piccart, M. Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat Rev. 2016, 50, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, Y.; Laws, M.J.; Sanabria, G.V.; Kim, S.H.; Dey, P.; Smith, B.P.; Gong, P.; Bindman, N.; Zhao, Y.; Carlson, K.; et al. Suppression of FOXM1 activities and breast cancer growth in vitro and in vivo by a new class of compounds. NPJ Breast Cancer 2019, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Angelis, C.; Fu, X.; Cataldo, M.L.; Nardone, A.; Pereira, R.; Veeraraghavan, J.; Nanda, S.; Qin, L.; Sethunath, V.; Wang, T.; et al. Activation of the IFN Signaling Pathway is Associated with Resistance to CDK4/6 Inhibitors and Immune Checkpoint Activation in ER-Positive Breast Cancer. Clin. Cancer Res. 2021, 27, 4870–4882. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Post, A.E.M.; Smid, M.; Nagelkerke, A.; Martens, J.W.M.; Bussink, J.; Sweep, F.; Span, P.N. Interferon-Stimulated Genes Are Involved in Cross-resistance to Radiotherapy in Tamoxifen-Resistant Breast Cancer. Clin. Cancer Res. 2018, 24, 3397–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [Green Version]
- Pidugu, V.K.; Pidugu, H.B.; Wu, M.M.; Liu, C.J.; Lee, T.C. Emerging Functions of Human IFIT Proteins in Cancer. Front. Mol. Biosci. 2019, 6, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal 2020, 18, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Zhou, Y.; Ogawa, M.; Shia, J.; Klimstra, D.S.; Wang, J.Y.; Roehrl, M.H. STAT1 as a potential prognosis marker for poor outcomes of early stage colorectal cancer with microsatellite instability. PLoS ONE 2020, 15, e0229252. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Chiang, L.; He, B.; Zhao, Y.Y.; Winoto, A. FoxM1, a forkhead transcription factor is a master cell cycle regulator for mouse mature T cells but not double positive thymocytes. PLoS ONE 2010, 5, e9229. [Google Scholar] [CrossRef]
- Zona, S.; Bella, L.; Burton, M.J.; Nestal de Moraes, G.; Lam, E.W. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim. Biophys. Acta 2014, 1839, 1316–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laoukili, J.; Kooistra, M.R.; Bras, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Wonsey, D.R.; Follettie, M.T. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005, 65, 5181–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andruska, N.D.; Zheng, X.; Yang, X.; Mao, C.; Cherian, M.M.; Mahapatra, L.; Helferich, W.G.; Shapiro, D.J. Estrogen receptor alpha inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc. Natl. Acad. Sci. USA 2015, 112, 4737–4742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettner, N.M.; Vijayaraghavan, S.; Durak, M.G.; Bui, T.; Kohansal, M.; Ha, M.J.; Liu, B.; Rao, X.; Wang, J.; Yi, M.; et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin. Cancer Res. 2019, 25, 3996–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Shapiro, G.I.; Keyomarsi, K. Selective CDK4/6 Inhibitors: Biologic Outcomes, Determinants of Sensitivity, Mechanisms of Resistance, Combinatorial Approaches, and Pharmacodynamic Biomarkers. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 115–126. [Google Scholar] [CrossRef]
- Pancholi, S.; Ribas, R.; Simigdala, N.; Schuster, E.; Nikitorowicz-Buniak, J.; Ressa, A.; Gao, Q.; Leal, M.F.; Bhamra, A.; Thornhill, A.; et al. Tumour kinome re-wiring governs resistance to palbociclib in oestrogen receptor positive breast cancers, highlighting new therapeutic modalities. Oncogene 2020, 39, 4781–4797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.; Tsiperson, V.; Gottesman, S.R.S.; Somma, J.; Blain, S.W. Dual Inhibition of CDK4 and CDK2 via Targeting p27 Tyrosine Phosphorylation Induces a Potent and Durable Response in Breast Cancer Cells. Mol. Cancer Res. 2018, 16, 361–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Shi, J.; Lan, S.; Gong, X. FOXM1 regulates the proliferation, apoptosis and inflammatory response of keratinocytes through the NF-kappaB signaling pathway. Hum. Exp. Toxicol. 2021, 40, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Hansen, S.K.; Larsen, S.L.; Reiter, B.E.; Sorensen, B.S.; Lykkesfeldt, A.E. T47D breast cancer cells switch from ER/HER to HER/c-Src signaling upon acquiring resistance to the antiestrogen fulvestrant. Cancer Lett. 2014, 344, 90–100. [Google Scholar] [CrossRef]
- Kastrati, I.; Joosten, S.E.P.; Semina, S.E.; Alejo, L.H.; Brovkovych, S.D.; Stender, J.D.; Horlings, H.M.; Kok, M.; Alarid, E.T.; Greene, G.L.; et al. The NF-kappaB Pathway Promotes Tamoxifen Tolerance and Disease Recurrence in Estrogen Receptor-Positive Breast Cancers. Mol. Cancer Res. 2020, 18, 1018–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madak-Erdogan, Z.; Charn, T.H.; Jiang, Y.; Liu, E.T.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Integrative genomics of gene and metabolic regulation by estrogen receptors alpha and beta, and their coregulators. Mol. Syst. Biol. 2013, 9, 676. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Laws, M.J.; Guillen, V.S.; Ziegler, Y.; Min, J.; Sharma, A.; Kim, S.H.; Chu, D.; Park, B.H.; Oesterreich, S.; et al. Structurally Novel Antiestrogens Elicit Differential Responses from Constitutively Active Mutant Estrogen Receptors in Breast Cancer Cells and Tumors. Cancer Res. 2017, 77, 5602–5613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]

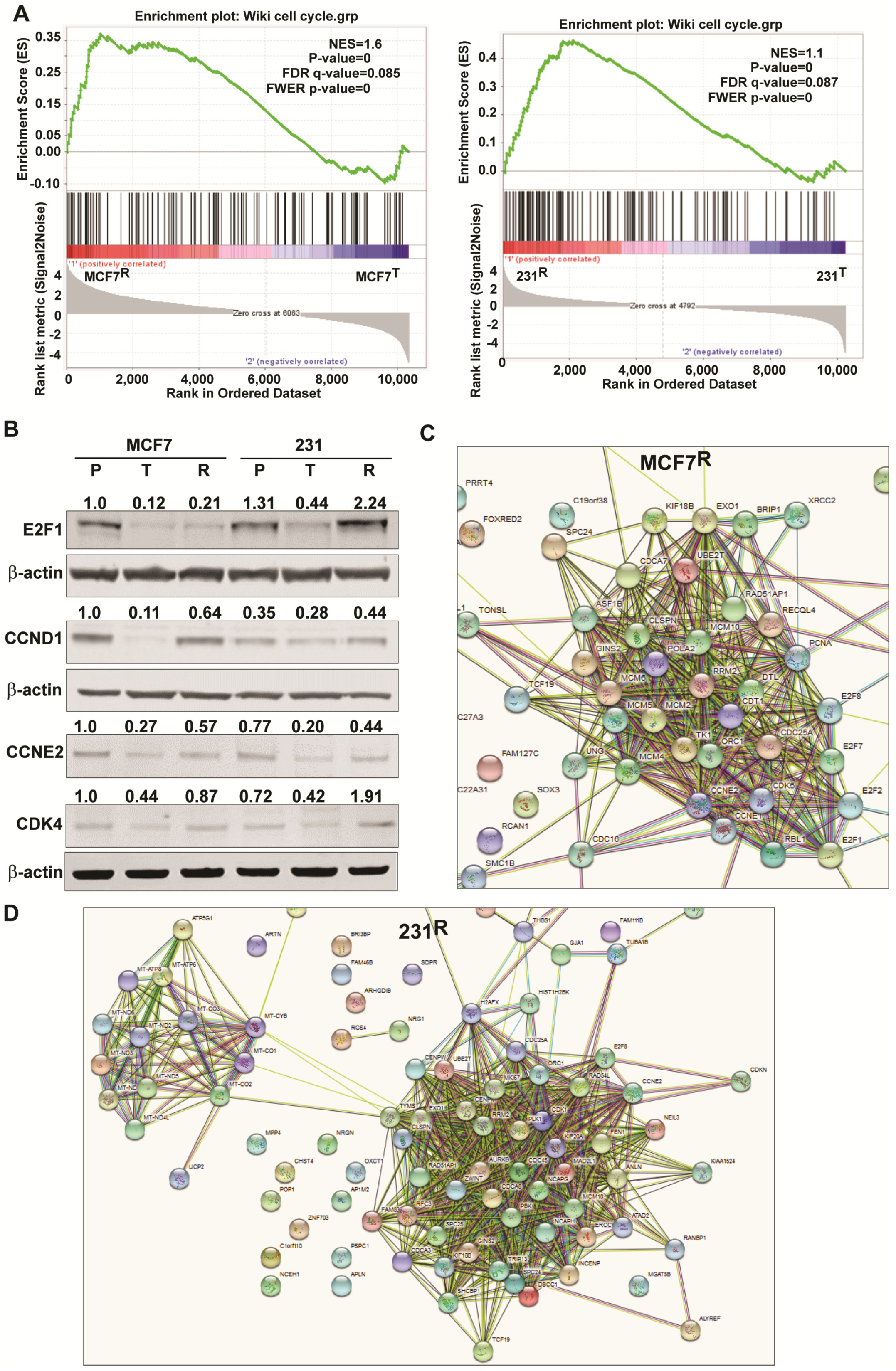
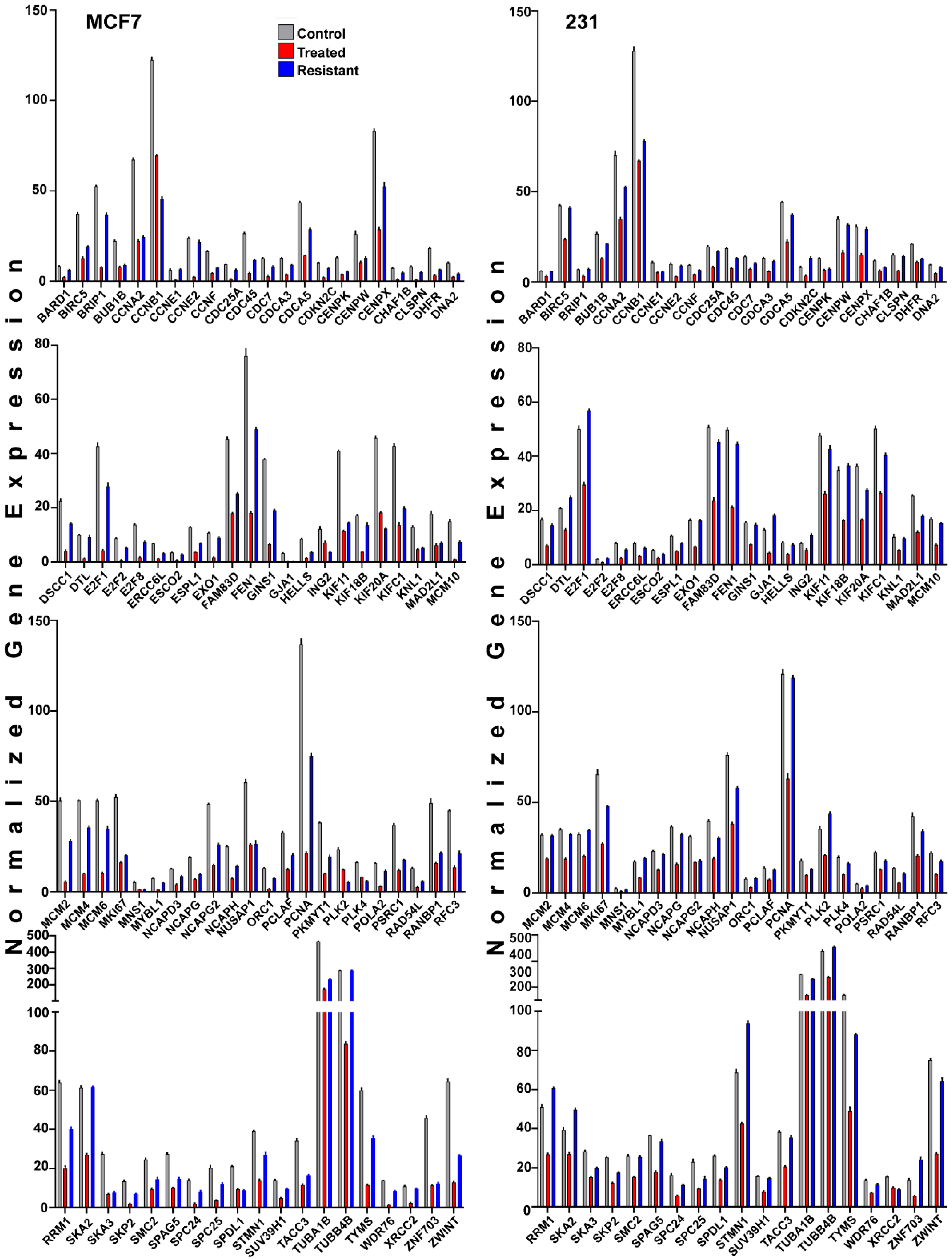

{kind=link}
{kind=link}
{kind=link}
{kind=link}
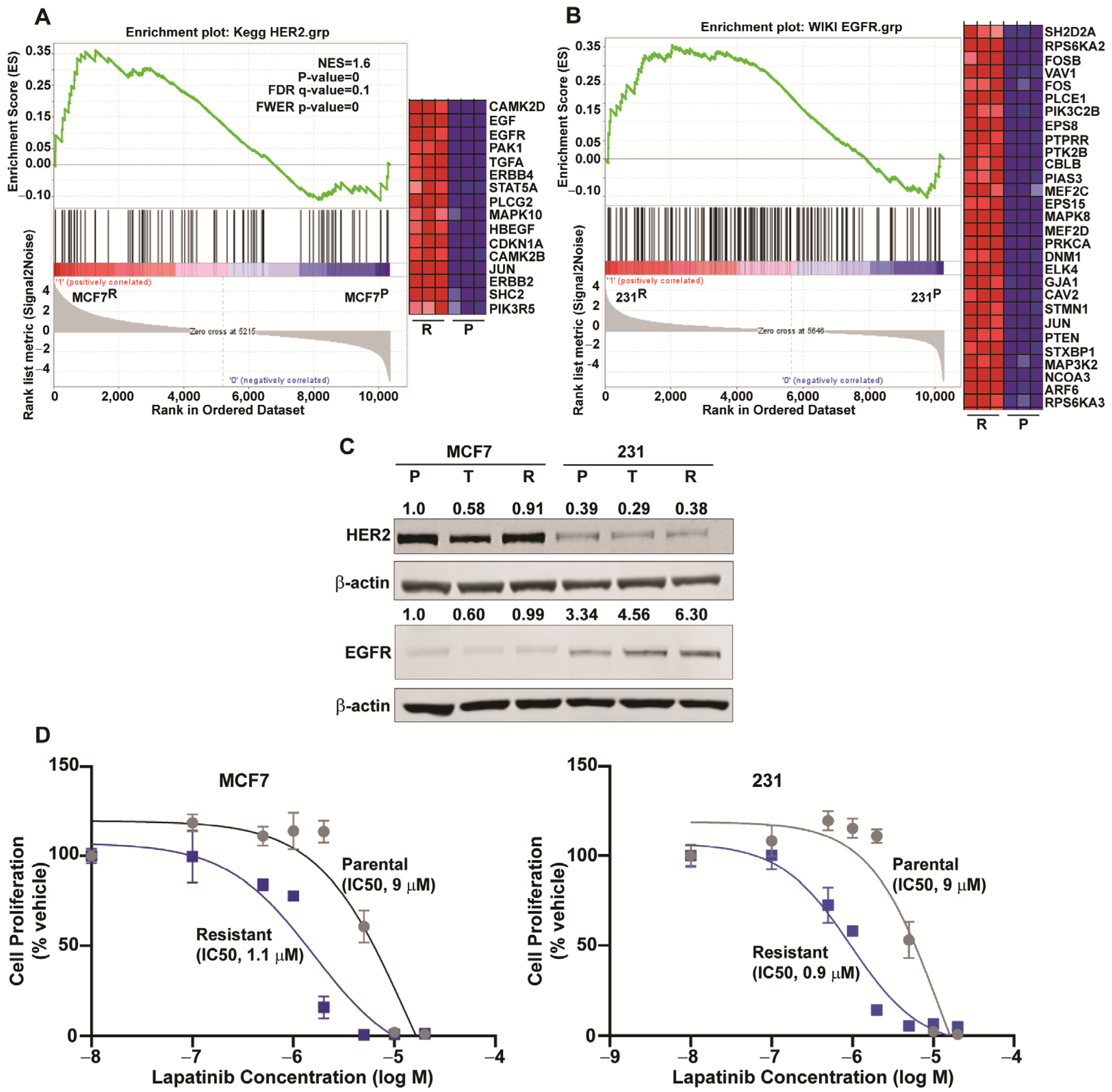{kind=link}
{kind=link}
{kind=link}
| MCF7 | 231 | ||||||
|---|---|---|---|---|---|---|---|
| Cluster | Adj. p-val. | #Genes | Hallmark Pathways | Cluster | Adj. p-val. | #Genes | Hallmark Pathways |
| A | 1.20 × 10−35 | 49 | TNFA signaling via NFκB | A | 1.05 × 10−20 | 38 | TNFA signaling via NFκB |
| A | 5.24 × 10−25 | 27 | Cholesterol homeostasis | A | 6.68 × 10−13 | 29 | Hypoxia |
| A | 1.09 × 10−24 | 39 | MTORC1 signaling | A | 6.68 × 10−13 | 29 | MTORC1 signaling |
| A | 1.41 × 10−21 | 36 | Hypoxia | A | 1.00 × 10−11 | 21 | Unfolded protein response |
| A | 5.29 × 10−21 | 28 | Unfolded protein response | A | 6.31 × 10−10 | 25 | P53 pathway |
| A | 6.97 × 10−16 | 30 | P53 pathway | A | 1.44 × 10−8 | 23 | Interferon gamma response |
| A | 5.44 × 10−11 | 22 | Apoptosis | A | 5.28 × 10−8 | 22 | Heme metabolism |
| A | 2.73 × 10−9 | 16 | Androgen response | A | 1.97 × 10−7 | 19 | Apoptosis |
| A | 1.77 × 10−8 | 21 | Estrogen response early | A | 4.73 × 10−6 | 13 | Interferon alpha response |
| A | 8.68 × 10−8 | 20 | Inflammatory response | A | 4.73 × 10−6 | 19 | IL2 STAT5 signaling |
| B | 1.19 × 10−59 | 72 | E2F Targets | B | 3.59 × 10−30 | 38 | TNFA signaling via NFκB |
| B | 1.50 × 10−29 | 47 | G2M Checkpoint | B | 2.43 × 10−15 | 25 | KRAS signaling up |
| B | 2.79 × 10−24 | 42 | Estrogen response early | B | 1.30 × 10−13 | 23 | Epithelial mesenchymal transition |
| B | 1.40 × 10−18 | 36 | Estrogen response late | B | 1.30 × 10−13 | 23 | Inflammatory response |
| B | 2.53 × 10−6 | 18 | UV response up | B | 7.84 × 10−11 | 20 | Allograft rejection |
| B | 1.51 × 10−5 | 19 | Myc targets v1 | B | 2.15 × 10−8 | 12 | IL6 JAK STAT3 signaling |
| B | 1.80 × 10−5 | 16 | DNA repair | B | 5.97 × 10−6 | 14 | Complement |
| B | 4.47 × 10−5 | 18 | MTORC1 signaling | B | 2.86 × 10−5 | 13 | IL2 STAT5 signaling |
| B | 0.000138 | 17 | Glycolysis | B | 0.000396 | 10 | UV response up |
| B | 0.00068 | 8 | Myc targets v2 | B | 0.000539 | 11 | Interferon gamma response |
| C | 9.01 × 10−12 | 19 | Interferon alpha response | C | 9.55 × 10−68 | 66 | G2M checkpoint |
| C | 3.15 × 10−7 | 20 | Interferon gamma response | C | 3.81 × 10−66 | 65 | E2F targets |
| C | 8.73 × 10−5 | 16 | Adipogenesis | C | 1.36 × 10−17 | 28 | Mitotic spindle |
| C | 8.73 × 10−5 | 16 | Estrogen response early | C | 5.37 × 10−10 | 20 | MTORC1 signaling |
| C | 0.000767 | 14 | Epithelial mesenchymal transition | C | 5.37 × 10−10 | 20 | Myc targets v1 |
| C | 0.000767 | 14 | Glycolysis | C | 2.02 × 10−6 | 9 | Myc targets v2 |
| C | 0.000767 | 14 | P53 pathway | C | 0.000441 | 12 | Estrogen response late |
| C | 0.006029 | 10 | UV response dn | C | 0.000441 | 12 | Glycolysis |
| C | 0.006577 | 12 | Complement | C | 0.000601 | 10 | DNA repair |
| D | 0.009467 | 4 | TNFA signaling via NFκB | D | 3.10 × 10−12 | 21 | UV response dn |
| D | 0.009467 | 4 | KRAS signaling up | D | 7.88 × 10−9 | 20 | Epithelial mesenchymal transition |
| D | 3.45 × 10−8 | 19 | Estrogen response late | ||||
| D | 1.60 × 10−7 | 18 | Estrogen response early | ||||
| D | 4.07 × 10−6 | 16 | Hypoxia | ||||
| D | 3.41 × 10−5 | 9 | Cholesterol homeostasis | ||||
| D | 7.08 × 10−5 | 14 | Myogenesis | ||||
| D | 0.00112 | 12 | KRAS signaling up | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziegler, Y.; Guillen, V.S.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Transcription Regulation and Genome Rewiring Governing Sensitivity and Resistance to FOXM1 Inhibition in Breast Cancer. Cancers 2021, 13, 6282. https://doi.org/10.3390/cancers13246282
Ziegler Y, Guillen VS, Kim SH, Katzenellenbogen JA, Katzenellenbogen BS. Transcription Regulation and Genome Rewiring Governing Sensitivity and Resistance to FOXM1 Inhibition in Breast Cancer. Cancers. 2021; 13(24):6282. https://doi.org/10.3390/cancers13246282
Chicago/Turabian StyleZiegler, Yvonne, Valeria Sanabria Guillen, Sung Hoon Kim, John A. Katzenellenbogen, and Benita S. Katzenellenbogen. 2021. "Transcription Regulation and Genome Rewiring Governing Sensitivity and Resistance to FOXM1 Inhibition in Breast Cancer" Cancers 13, no. 24: 6282. https://doi.org/10.3390/cancers13246282
APA StyleZiegler, Y., Guillen, V. S., Kim, S. H., Katzenellenbogen, J. A., & Katzenellenbogen, B. S. (2021). Transcription Regulation and Genome Rewiring Governing Sensitivity and Resistance to FOXM1 Inhibition in Breast Cancer. Cancers, 13(24), 6282. https://doi.org/10.3390/cancers13246282