
Arginine Depletion in Human Cancers


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
Data Processing
3. Results
3.1. Non-Synonymous Substitution Bias of Arginine Codons
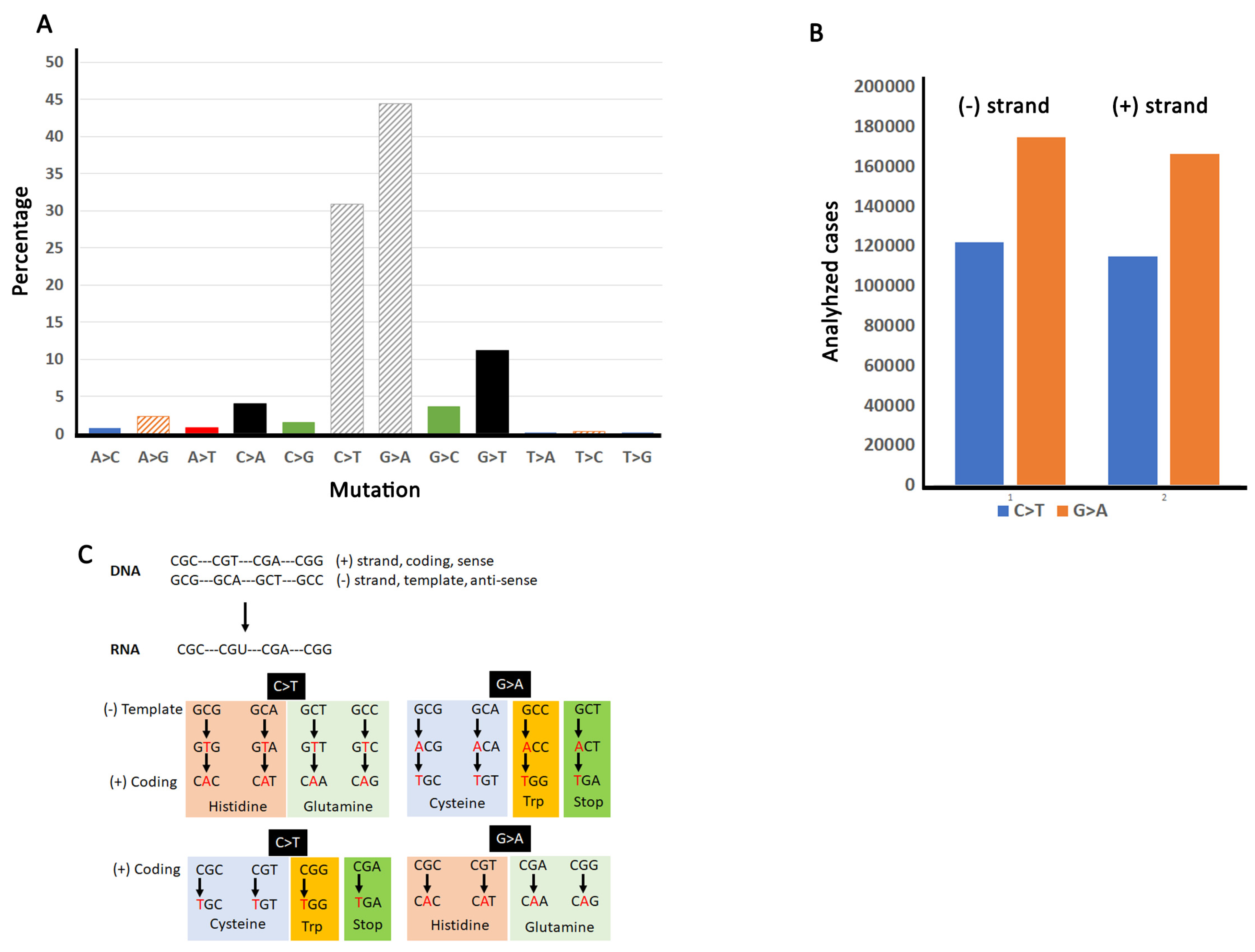
3.2. Arginine Substitutions in Human Cancers Are Driven Mainly by C/G > T/A Transitions
3.3. Purifying Selection at the Amino Acid Level May Be Strongly Biased by Selection at the Nucleotide Level
3.4. Cancer or Gene Specific Arginine Mutation Bias
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Durland, J.; Ahmadian-Moghadam, H. Genetics, Mutagenesis; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Zhang, L.; Vijg, J. Somatic Mutagenesis in Mammals and Its Implications for Human Disease and Aging. Annu. Rev. Genet. 2018, 52, 397–419. [Google Scholar] [CrossRef]
- Lynch, M. Evolution of the mutation rate. Trends Genet. 2010, 26, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N. Human gene mutation in pathology and evolution. J. Inherit. Metab. Dis. 2002, 25, 157–182. [Google Scholar] [CrossRef]
- Bonekamp, F.; Jensen, K.F. The AGG codon is translated slowly in E. coli even at very low expression levels. Nucleic Acids Res. 1988, 16, 3013–3024. [Google Scholar] [CrossRef] [PubMed]
- Chevance, F.F.V.; le Guyon, S.; Hughes, K.T. The Effects of Codon Context on In Vivo Translation Speed. PLoS Genet. 2014, 10, e1004392. [Google Scholar] [CrossRef] [PubMed]
- Charneski, C.A.; Hurst, L.D. Positively Charged Residues Are the Major Determinants of Ribosomal Velocity. PLoS Biol. 2013, 11, e1001508. [Google Scholar] [CrossRef]
- Borders, C., Jr.; Broadwater, J.A.; Bekeny, P.A.; Salmon, J.E.; Lee, A.S.; Eldridge, A.M.; Pett, V.B. A structural role for arginine in proteins: Multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci. 1994, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Mrabet, N.T.; Broeck, A.V.D.; Brande, I.V.D.; Stanssens, P.; Laroche, Y.; Lambeir, A.-M.; Matthijssens, G.; Jenkins, J.; Chiadmi, M. Arginine residues as stabilizing elements in proteins. Biochemistry 1992, 31, 2239–2253. [Google Scholar] [CrossRef]
- Hwang, J.W.; Cho, Y.; Bae, G.-U.; Kim, S.-N.; Kim, Y.K. Protein arginine methyltransferases: Promising targets for cancer therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef] [PubMed]
- Diaz, K.; Huang, R. Non-Histone Arginine Methylation by Protein Arginine Methyltransferases. Curr. Protein Pept. Sci. 2020, 21, 699–712. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Ling, F.; Tang, Y.; Li, M.; Li, Q.-S.; Li, X.; Yang, L.; Zhao, W.; Jin, C.-C.; Zeng, Z.; Liu, C.; et al. Mono-ADP-ribosylation of histone 3 at arginine-117 promotes proliferation through its interaction with P300. Oncotarget 2017, 8, 72773–72787. [Google Scholar] [CrossRef]
- Smith, B.C.; Denu, J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta (BBA)—Bioenerg. 2009, 1789, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Allahverdi, A.; Zahiri, J. Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders. J. Biosci. 2020, 45, 1–29. [Google Scholar] [CrossRef]
- Novoa, E.M.; Jungreis, I.; Jaillon, O.; Kellis, M. Elucidation of Codon Usage Signatures across the Domains of Life. Mol. Biol. Evol. 2019, 36, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, D.; Gold, B. Origins of nonsense mutations in human tumor suppressor genes. Mutat. Res. Mol. Mech. Mutagen. 2021, 823, 111761. [Google Scholar] [CrossRef]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef] [PubMed]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Anoosha, P.; Sakthivel, R.; Gromiha, M.M. Exploring preferred amino acid mutations in cancer genes: Applications to identify potential drug targets. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 155–165. [Google Scholar] [CrossRef]
- Tsuber, V.; Kadamov, Y.; Brautigam, L.; Berglund, U.W.; Helleday, T. Mutations in Cancer Cause Gain of Cysteine, Histidine, and Tryptophan at the Expense of a Net Loss of Arginine on the Proteome Level. Biomolecules 2017, 7, 49. [Google Scholar] [CrossRef]
- Collins, D.W.; Jukes, T.H. Rates of Transition and Transversion in Coding Sequences since the Human-Rodent Divergence. Genomics 1994, 20, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Gold, B. Somatic mutations in cancer: Stochastic versus predictable. Mutat. Res. Toxicol. Environ. Mutagen. 2017, 814, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2010, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Supek, F. The Code of Silence: Widespread Associations between Synonymous Codon Biases and Gene Function. J. Mol. Evol. 2015, 82, 65–73. [Google Scholar] [CrossRef]
- Liu, Y. A code within the genetic code: Codon usage regulates co-translational protein folding. Cell Commun. Signal. 2020, 18, 145. [Google Scholar] [CrossRef]
- Gingold, H.; Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011, 7, 481. [Google Scholar] [CrossRef]
- Angov, E. Codon usage: Nature’s roadmap to expression and folding of proteins. Biotechnol. J. 2011, 6, 650–659. [Google Scholar] [CrossRef]
- Gaither, J.B.S.; Lammi, G.E.; Li, J.L.; Gordon, D.M.; Kuck, H.C.; Kelly, B.J.; Fitch, J.R.; White, P. Synonymous variants that disrupt messenger RNA structure are significantly constrained in the human population. GigaScience 2021, 10, giab023. [Google Scholar] [CrossRef]
- Knapp, K.M.; Fellows, B.; Aggarwal, S.; Dalal, A.; Bicknell, L.S. A synonymous variant in a non-canonical exon of CDC45 disrupts splicing in two affected sibs with Meier-Gorlin syndrome with craniosynostosis. Eur. J. Med Genet. 2021, 64, 104182. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.E.; Pinkard, O.; Martin, S.; Sweet, T.J.; Hanson, G.; Coller, J. Codon and amino acid content are associated with mRNA stability in mammalian cells. PLoS ONE 2020, 15, e0228730. [Google Scholar] [CrossRef]
- Narula, A.; Ellis, J.; Taliaferro, J.M.; Rissland, O.S. Coding regions affect mRNA stability in human cells. RNA 2019, 25, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Gillen, S.L.; Waldron, J.A.; Bushell, M. Codon optimality in cancer. Oncogene 2021, 40, 6309–6320. [Google Scholar] [CrossRef]
- Bernardi, G. The vertebrate genome: Isochores and evolution. Mol. Biol. Evol. 1993, 10, 186–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bernardi, G. Isochores and the evolutionary genomics of vertebrates. Gene 2000, 241, 3–17. [Google Scholar] [CrossRef]
- Knight, R.D.; Freeland, S.J.; Landweber, L.F. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001, 2, 1–13. [Google Scholar]
- Kimura, M. Evolutionary Rate at the Molecular Level. Nat. Cell Biol. 1968, 217, 624–626. [Google Scholar] [CrossRef]
- Ohta, T.; Gillespie, J.H. Development of Neutral and Nearly Neutral Theories. Theor. Popul. Biol. 1996, 49, 128–142. [Google Scholar] [CrossRef]
- Chen, J.; Furano, A.V. Breaking bad: The mutagenic effect of DNA repair. DNA Repair 2015, 32, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D.M.; Holmes, E.C. Molecular Evolution: A Phylogenetic Approach; Blackwell Science: Oxford, UK, 1998. [Google Scholar]
- Li, G.C.; Forster-Benson, E.T.C.; Sanders, C.R. Genetic intolerance analysis as a tool for protein science. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183058. [Google Scholar] [CrossRef] [PubMed]
- Freese, E. The Difference between Spontaneous and Base-Analogue Induced Mutations of Phage T4. Proc. Natl. Acad. Sci. USA 1959, 45, 622–633. [Google Scholar] [CrossRef]
- Fitch, W.M. Evidence suggesting a non-random character to nucleotide replacements in naturally occurring mutations. J. Mol. Biol. 1967, 26, 499–507. [Google Scholar] [CrossRef]
- Vogel, F. Non-randomness of base replacement in point mutation. J. Mol. Evol. 1972, 1, 334–367. [Google Scholar] [CrossRef]
- Vogel, F.; Kopun, M. Higher frequencies of transitions among point mutations. J. Mol. Evol. 1977, 9, 159–180. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Moore, L.; Cagan, A.; Coorens, T.H.H.; Neville, M.D.C.; Sanghvi, R.; Sanders, M.A.; Oliver, T.R.W.; Leongamornlert, D.; Ellis, P.; Noorani, A.; et al. The mutational landscape of human somatic and germline cells. Nat. Cell Biol. 2021, 597, 381–386. [Google Scholar] [CrossRef]
- Abascal, F.; Harvey, L.M.R.; Mitchell, E.; Lawson, A.R.J.; Lensing, S.V.; Ellis, P.; Russell, A.J.C.; Alcantara, R.E.; Baez-Ortega, A.; Wang, Y.; et al. Somatic mutation landscapes at single-molecule resolution. Nat. Cell Biol. 2021, 593, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.; Sale, J.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Shen, J.C.; Rideout, W.M., 3rd; Jones, P.A. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994, 22, 972–976. [Google Scholar] [CrossRef]
- McElhinny, N.S.A.; Stith, C.M.; Burgers, P.M.; Kunkel, T.A. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase delta. J. Biol. Chem. 2007, 282, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Szpiech, Z.A.; Strauli, N.; White, K.; Ruiz, D.G.; Jacobson, M.; Barber, D.L.; Hernandez, R.D. Prominent features of the amino acid mutation landscape in cancer. PLoS ONE 2017, 12, e0183273. [Google Scholar] [CrossRef]
- Tan, H.; Bao, J.; Zhou, X. Genome-wide mutational spectra analysis reveals significant cancer-specific heterogeneity. Sci. Rep. 2015, 5, 12566. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Grillo-Hill, B.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef]
- Ganini, C.; Amelio, I.; Bertolo, R.; Bove, P.; Buonomo, O.C.; Candi, E.; Cipriani, C.; di Daniele, N.; Juhl, H.; Mauriello, A.; et al. Global mapping of cancers: The Cancer Genome Atlas and beyond. Mol. Oncol. 2021, 15, 2823–2840. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Pappula, A.L.; Rasheed, S.; Mirzaei, G.; Petreaca, R.C.; Bouley, R.A. A Genome-Wide Profiling of Glioma Patients with an IDH1 Mutation Using the Catalogue of Somatic Mutations in Cancer Database. Cancers 2021, 13, 4299. [Google Scholar] [CrossRef] [PubMed]
- Sanson, M.; Marie, Y.; Paris, S.; Idbaih, A.; Laffaire, J.; Ducray, F.; el Hallani, S.; Boisselier, B.; Mokhtari, K.; Hoang-Xuan, K.; et al. Isocitrate Dehydrogenase 1 Codon 132 Mutation Is an Important Prognostic Biomarker in Gliomas. J. Clin. Oncol. 2009, 27, 4150–4154. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Liu, Y.; Lang, F.; Chou, F.-J.; Zaghloul, K.A.; Yang, C. Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications. Biomedicines 2020, 8, 294. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1 R132 mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef]
- Ohba, S.; Mukherjee, J.; Johannessen, T.-C.; Mancini, A.; Chow, T.T.; Wood, M.; Jones, L.; Mazor, T.; Marshall, R.E.; Viswanath, P.; et al. Mutant IDH1 Expression Drives TERT Promoter Reactivation as Part of the Cellular Transformation Process. Cancer Res. 2016, 76, 6680–6689. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; el-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef]
- Weaver, A.; Bossaer, J.B. Fibroblast growth factor receptor (FGFR) inhibitors: A review of a novel therapeutic class. J. Oncol. Pharm. Pr. 2021, 27, 702–710. [Google Scholar] [CrossRef]
- Bastians, H.; Ponstingl, H. The novel human protein serine/threonine phosphatase 6 is a functional homologue of budding yeast Sit4p and fission yeast ppe1, which are involved in cell cycle regulation. J. Cell Sci. 1996, 109, 2865–2874. [Google Scholar] [CrossRef]
- Cho, E.; Lou, H.J.; Kuruvilla, L.; Calderwood, D.A.; Turk, B.E. PPP6C negatively regulates oncogenic ERK signaling through dephosphorylation of MEK. Cell Rep. 2021, 34, 108928. [Google Scholar] [CrossRef]
- Ohama, T. The multiple functions of protein phosphatase 6. Biochim. Biophys. Acta (BBA)—Bioenerg. 2019, 1866, 74–82. [Google Scholar] [CrossRef]
- Hurlin, P.J.; Huang, J. The MAX-interacting transcription factor network. Semin. Cancer Biol. 2006, 16, 265–274. [Google Scholar] [CrossRef] [PubMed]
- McAnulty, J.; DiFeo, A. The Molecular ‘Myc-anisms’ behind Myc-Driven Tumorigenesis and the Relevant Myc-Directed Therapeutics. Int. J. Mol. Sci. 2020, 21, 9486. [Google Scholar] [CrossRef]
- Augert, A.; Mathsyaraja, H.; Ibrahim, A.H.; Freie, B.; Geuenich, M.J.; Cheng, P.-F.; Alibeckoff, S.P.; Wu, N.; Hiatt, J.B.; Basom, R.; et al. MAX Functions as a Tumor Suppressor and Rewires Metabolism in Small Cell Lung Cancer. Cancer Cell 2020, 38, 97–114.e7. [Google Scholar] [CrossRef]
- Gaffal, E. Research in practice: Therapeutic targeting of oncogenic GNAQ mutations in uveal melanoma. J. Dtsch. Dermatol. Ges. 2020, 18, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Cai, L.C.; Wang, X.Y.; Cheng, S.Y.; Zhang, D.M.; Jian, W.G.; Wang, T.D.; Yang, J.K.; Yang, K.B.; Zhang, C. BMP8A promotes survival and drug resistance via Nrf2/TRIM24 signaling pathway in clear cell renal cell carcinoma. Cancer Sci. 2020, 111, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Han, Y.K.; Park, G.-Y.; Kim, J.S.; Lee, C.G. Bub1 is required for maintaining cancer stem cells in breast cancer cell lines. Sci. Rep. 2015, 5, 15993. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| 2 Gene | Most Frequent Substituted Residue | Most Frequent Substituted Nucleotide | 3 Driver Gene | ||||
|---|---|---|---|---|---|---|---|
| Cysteine | Glutamine | Histidine | Tryptophan | C > T | G > A | Yes/No | |
| IDH1 | 76% | 76.36% | YES | ||||
| TCP10L2 | 61% | 68.97% | NO | ||||
| NEK9 | 74% | 86.90% | NO | ||||
| TXK | 60% | 83.13% | NO | ||||
| CYP2D6 | 69% | 68.66% | NO | ||||
| NCF1 | 76% | 76.47% | NO | ||||
| OR4C3 | 70% | 76.60% | NO | ||||
| KRTAP4-8 | 85% | 86.96% | NO | ||||
| BMP8A | 78% | 77.78% | NO | ||||
| FGFR3 | 80% | 88.59% | YES | ||||
| RFPL3 | 66% | 71.26% | NO | ||||
| PPP6C | 63% | 66.67% | YES | ||||
| HASPIN | 73% | 76.54% | NO | ||||
| DTX2 | 63% | 66.10% | NO | ||||
| PRSS1 | 77% | 79.25% | NO | ||||
| POTEB2 | 73% | 73.08% | NO | ||||
| CAMKK2 | 64% | 74.00% | NO | ||||
| PARN | 69% | 73.81% | NO | ||||
| OR9G1 | 73% | 73.17% | NO | ||||
| NPIPA5 | 80% | 80.00% | NO | ||||
| PRB2 | 94% | 94.94% | NO | ||||
| BUB1B | 70% | 77.05% | NO | ||||
| AC004223.3 | 88% | 92.56% | NO | ||||
| RAD51D | 89% | 93.04% | NO | ||||
| GNL3 | 93% | 93.58% | NO | ||||
| RNASEL | 69% | 79.31% | NO | ||||
| MAX | 68% | 75.86% | YES | ||||
| GNAQ | 64% | 76.47% | YES | ||||
| IRF5 | 62% | 82.72% | NO | ||||
| CS | 80% | 81.36% | NO | ||||
| PRB1 | 91% | 93.48% | NO | ||||
| FAM120B | 70% | 76.74% | NO | ||||
| CLEC4M | 76% | 87.80% | NO | ||||
| OR1L6 | 68% | 75.00% | NO | ||||
| IST1 | 75% | 82.50% | NO | ||||
| PDCL3 | 61% | 71.74% | NO | ||||
| SPAG11B | 71% | 76.19% | NO | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelakurti, D.D.; Rossetti, T.; Husbands, A.Y.; Petreaca, R.C. Arginine Depletion in Human Cancers. Cancers 2021, 13, 6274. https://doi.org/10.3390/cancers13246274
Nelakurti DD, Rossetti T, Husbands AY, Petreaca RC. Arginine Depletion in Human Cancers. Cancers. 2021; 13(24):6274. https://doi.org/10.3390/cancers13246274
Chicago/Turabian StyleNelakurti, Devi D., Tiffany Rossetti, Aman Y. Husbands, and Ruben C. Petreaca. 2021. "Arginine Depletion in Human Cancers" Cancers 13, no. 24: 6274. https://doi.org/10.3390/cancers13246274
APA StyleNelakurti, D. D., Rossetti, T., Husbands, A. Y., & Petreaca, R. C. (2021). Arginine Depletion in Human Cancers. Cancers, 13(24), 6274. https://doi.org/10.3390/cancers13246274