
Emerging Role of Autophagy in the Development and Progression of Oral Squamous Cell Carcinoma




{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
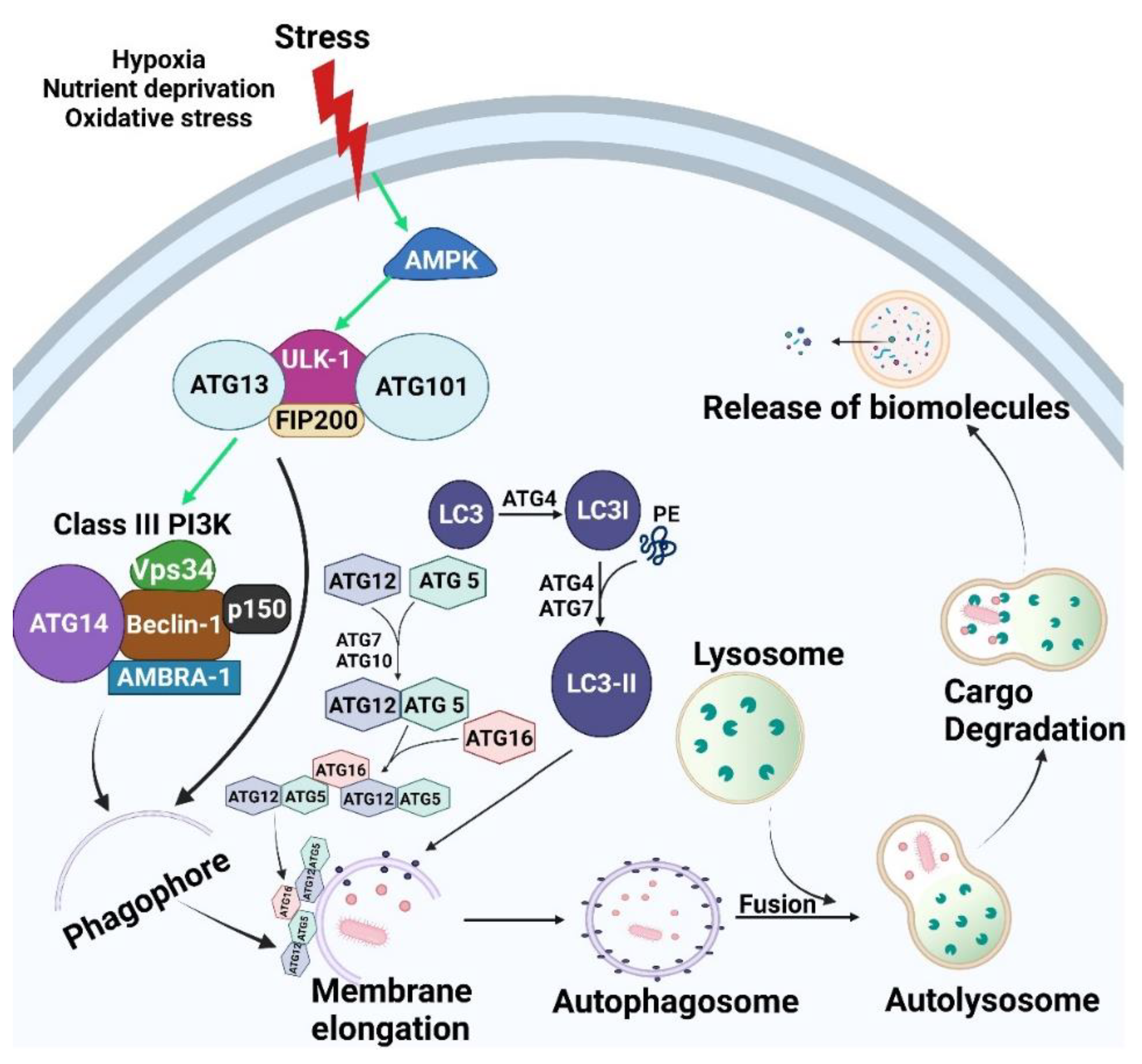
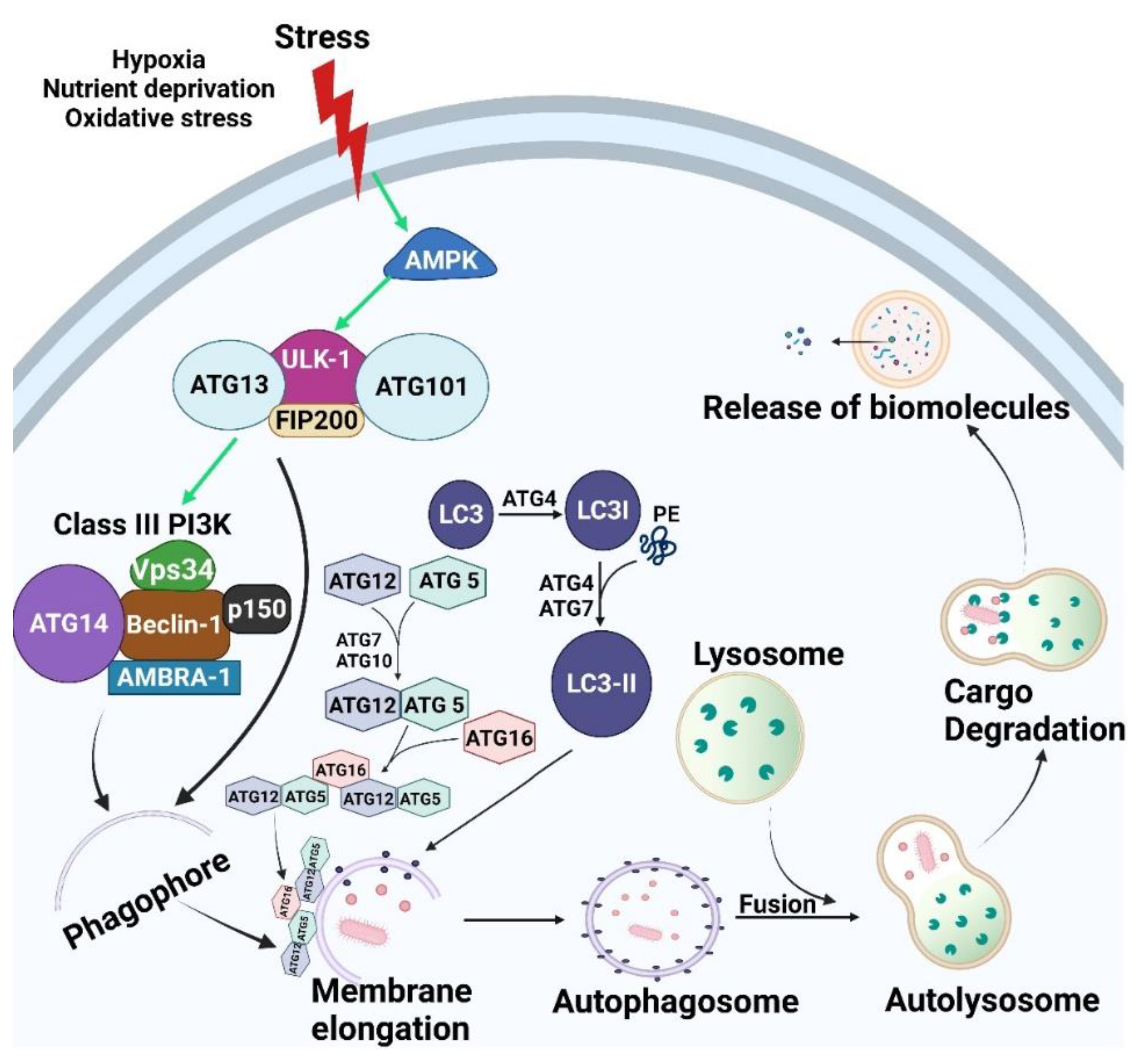
2. Autophagy
2.1. Core Autophagic Machinery
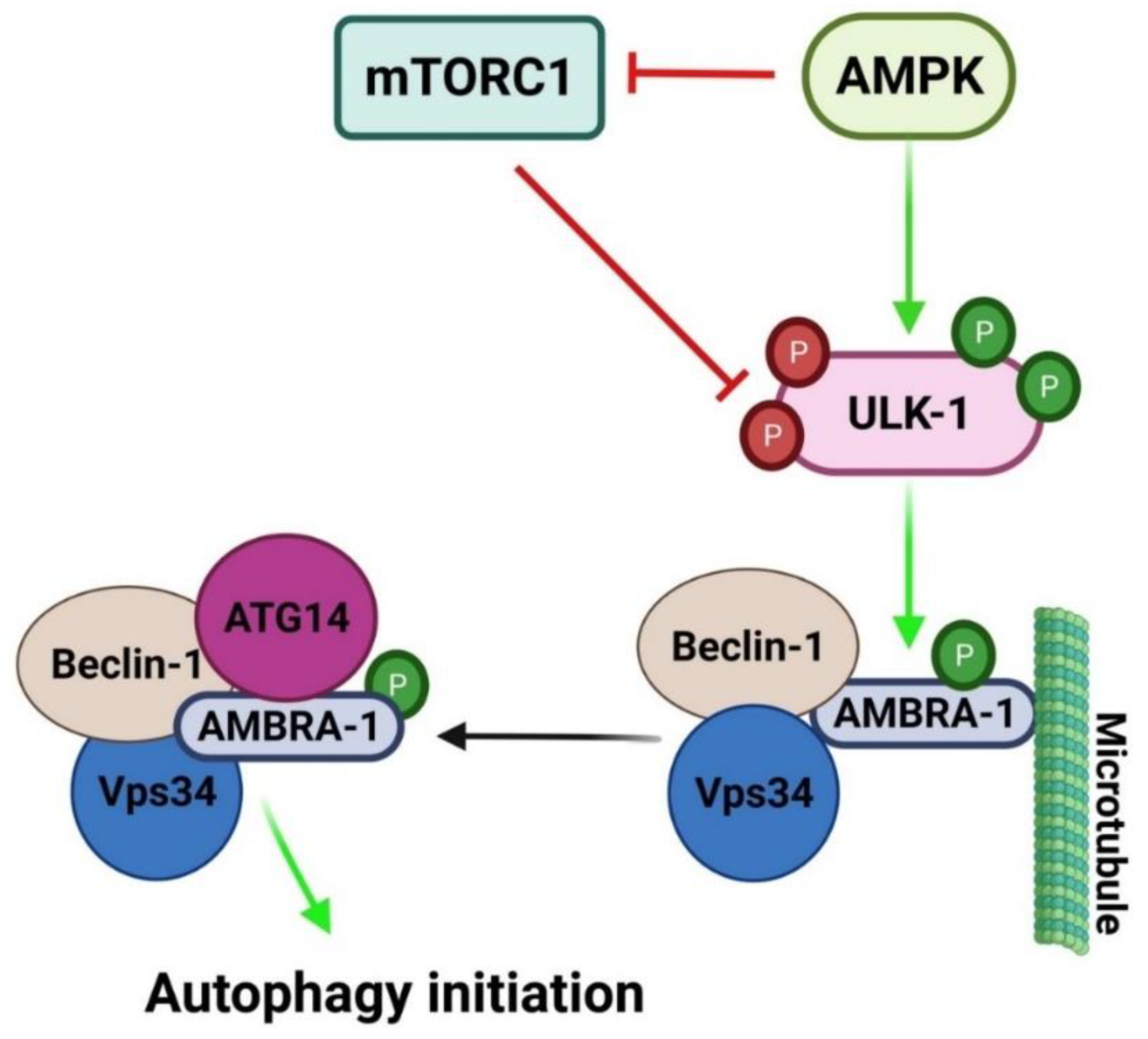
2.1.1. Initiation/Induction and Nucleation
2.1.2. Elongation
2.1.3. Maturation/Degradation
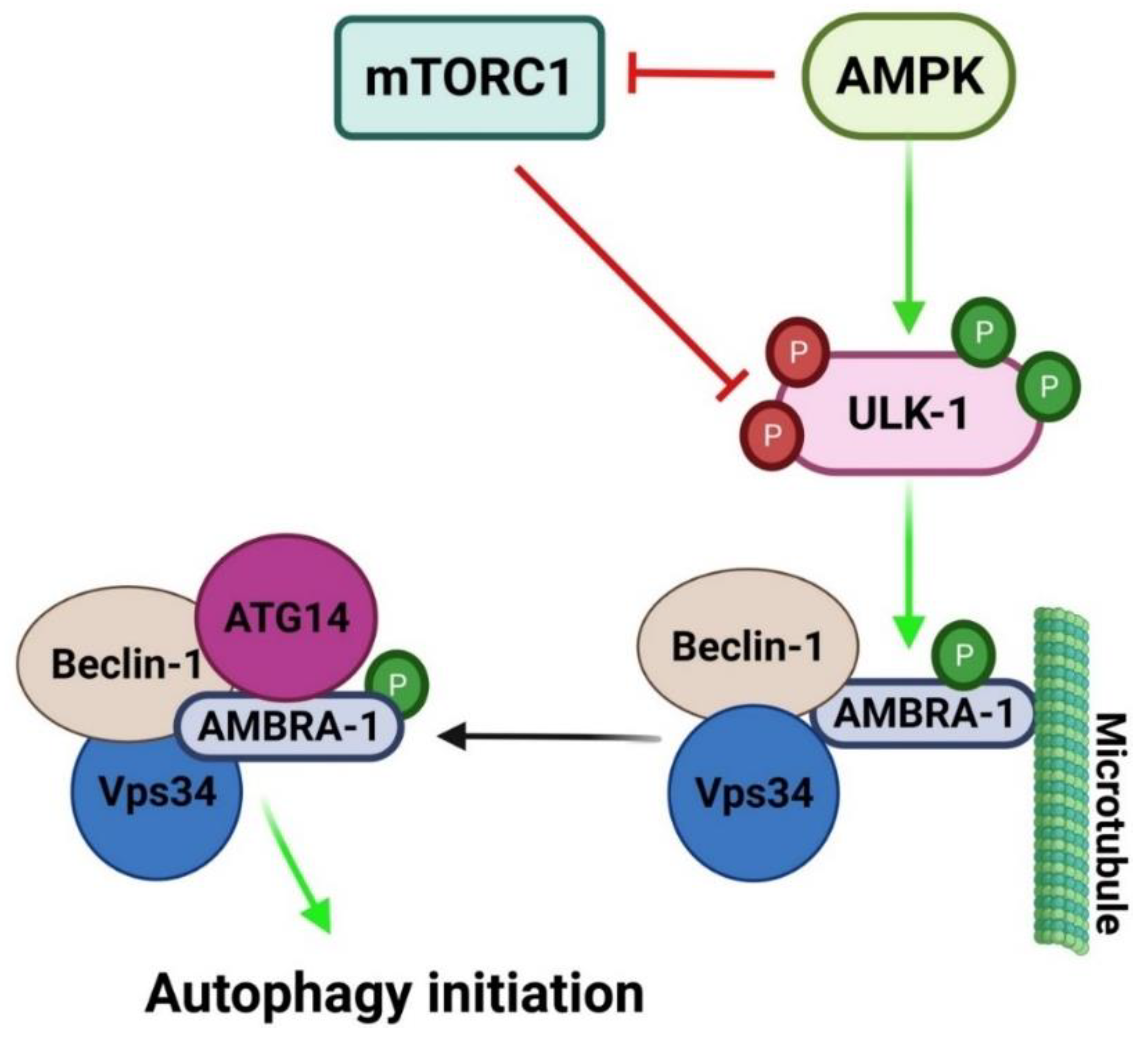
2.2. Mechanisms Regulating Autophagy
3. Crosstalk between Autophagy and Apoptosis
4. Autophagy and Ferroptosis
5. Autophagy and Cancer
5.1. Regulation of Autophagy by Oncogenes and Tumor Suppressors
5.2. Autophagy as a Tumor Suppression Pathway
5.3. Autophagy as a Tumor Promoting Mechanism
5.3.1. Self-Sufficiency of Growth Signals—Sustained Proliferation
5.3.2. Evasion of Apoptosis
5.3.3. Angiogenesis
5.3.4. Invasion and Metastasis
5.3.5. Anoikis Resistance
5.3.6. Avoidance of Immune Destruction
5.3.7. Metabolic Reprogramming
6. Autophagy in Oral Cancer
6.1. Oral Squamous Cell Carcinoma (OSCC)
6.2. Stressful OSCC Microenvironment
6.2.1. Cancer Associated Fibroblast (CAFs)
6.2.2. Tumor Associated Macrophages (TAMs)
6.2.3. Hypoxia and Angiogenesis
6.2.4. ECM
6.3. Role of Autophagy in OSCC Progression
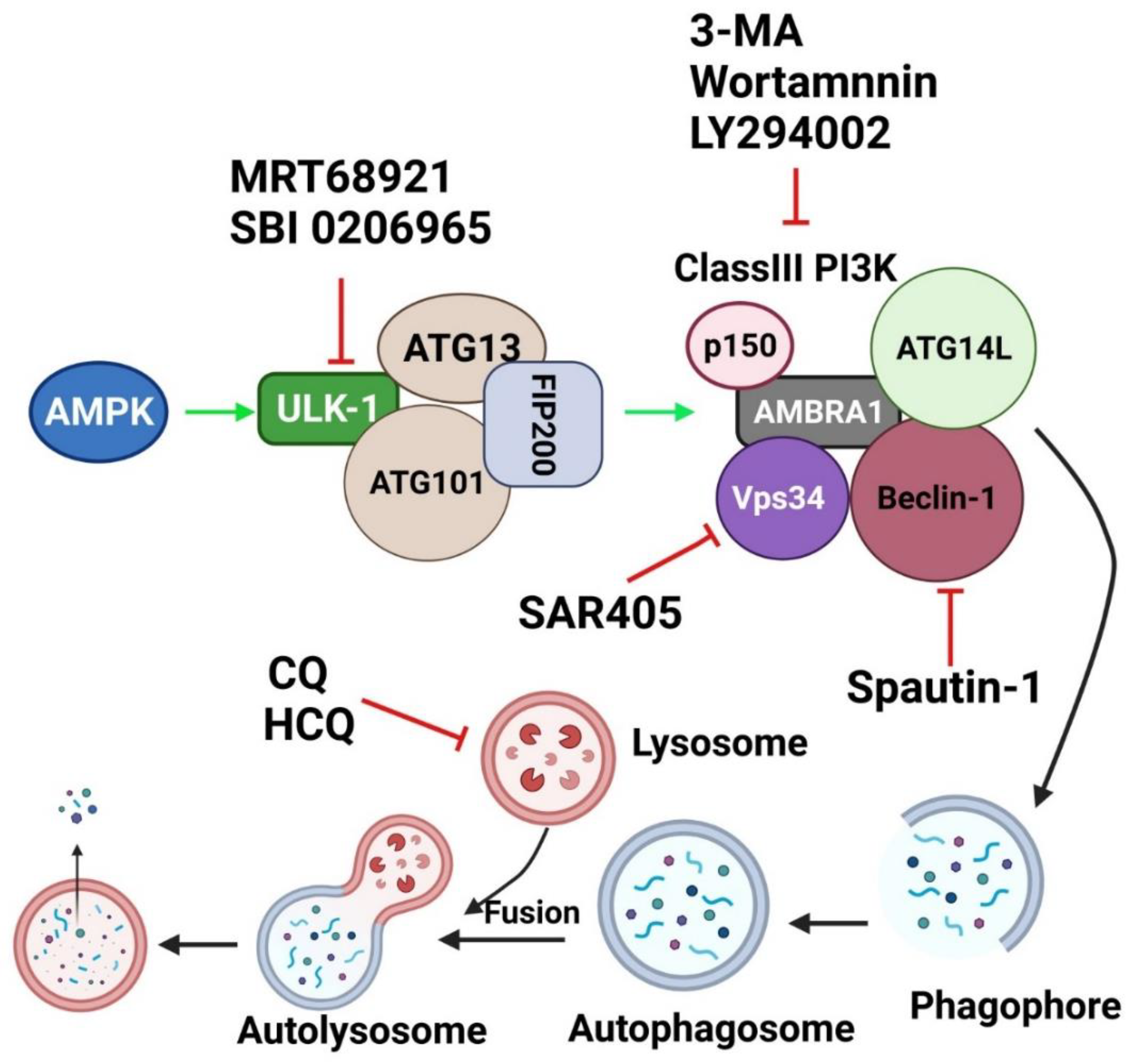
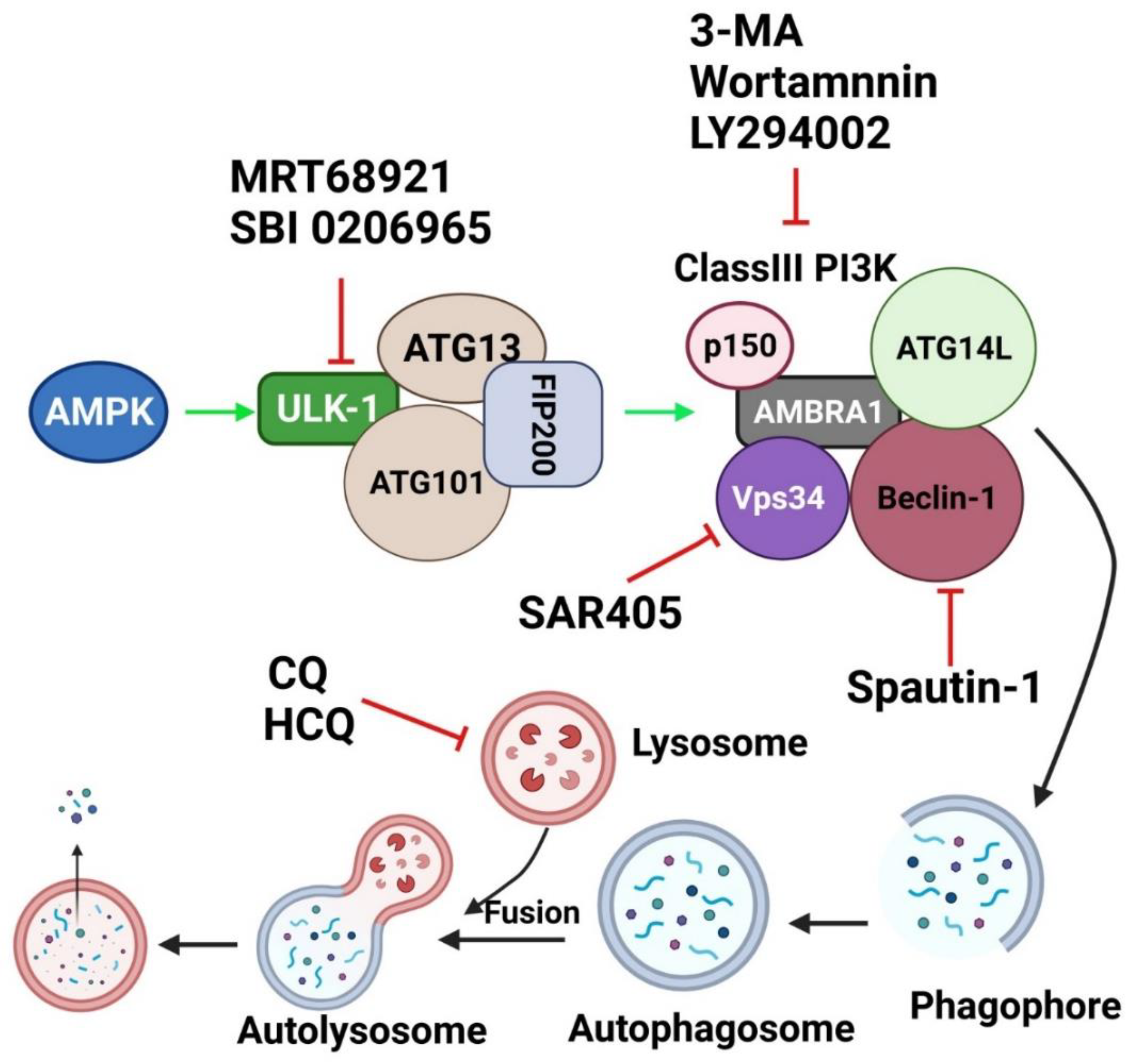
7. Targeting Autophagy as a Therapeutic Approach
7.1. Chloroquine and Its Derivatives
7.2. Specific Autophagy Inhibitors Directed toward Major Molecules in Autophagic Machinery
7.2.1. PI3K Inhibitors
7.2.2. ULK-1/2 Inhibitors
8. Autophagy Inhibitors in OSCC
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bishop, J.A.; Sciubba, J.J.; Westra, H.W. Squamous cell carcinoma of the oral cavity and oropharynx. Surg. Pathol. Clin. 2011, 4, 1127–1151. [Google Scholar] [CrossRef]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef]
- Chatzistefanou, I.; Lubek, J.; Markou, K.; Ord, R.A. The role of neck dissection and postoperative adjuvant radiotherapy in cN0 patients with PNI-positive squamous cell carcinoma of the oral cavity. Oral Oncol. 2014, 50, 753–758. [Google Scholar] [CrossRef]
- Rao, S.K.; Pavicevic, Z.; Du, Z.; Kim, J.-G.; Fan, M.; Jiao, Y.; Rosebush, M.; Samant, S.; Gu, W.; Pfeffer, L.M.; et al. Pro-inflammatory Genes as Biomarkers and Therapeutic Targets in Oral Squamous Cell Carcinoma. J. Biol. Chem. 2010, 285, 32512–32521. [Google Scholar] [CrossRef] [Green Version]
- Das, C.K.; Banerjee, I.; Mandal, M. Pro-survival autophagy: An emerging candidate of tumor progression through maintaining hallmarks of cancer. Semin. Cancer Biol. 2020, 66, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Suzuki, Y.; Fujimoto, C.; Kanzaki, S. Molecular Mechanisms and Biological Functions of Autophagy for Genetics of Hearing Impairment. Genes 2020, 11, 1331. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Viry, E.; Paggetti, J.; Baginska, J.; Mgrditchian, T.; Berchem, G.; Moussay, E.; Janji, B. Autophagy: An adaptive metabolic response to stress shaping the antitumor immunity. Biochem. Pharmacol. 2014, 92, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Wong, A.S.; Cheung, Z.H.; Ip, N.Y. Molecular machinery of macroautophagy and its deregulation in diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1490–1497. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell Mol. Life. Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.; Thamm, D.; Gustafson, D.L. Autophagy and Cancer Therapy. Mol. Pharmacol. 2014, 85, 830–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Hamacher-Brady, A. Autophagy Regulation and Integration with Cell Signaling. Antioxid. Redox Signal. 2012, 17, 756–765. [Google Scholar] [CrossRef]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human: The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.L.; Ravikumar, B.; Rubinsztein, D.C. Microtubule disruption inhibits autophagosome-lysosome fusion: Implications for studying the roles of aggresomes in polyglutamine diseases. Int. J. Biochem. 2004, 36, 2541–2550. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Birgisdottir, Å.B.; Lamark, T.; Johansen, T. The LIR motif–crucial for selective autophagy. J. Cell. Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, Y. AMPK and Autophagy. In Autophagy: Biology and Diseases; Springer: Singapore, 2019; pp. 85–108. [Google Scholar]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Gordy, C.; He, Y.-W. The crosstalk between autophagy and apoptosis: Where does this lead? Protein Cell 2012, 3, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Kriel, J.; Priya, B.S.; Shivananju, N.S.; Loos, B. Modulating autophagy in cancer therapy: Advancements and challenges for cancer cell death sensitization. Biochem. Pharmacol. 2018, 147, 170–182. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against Fatal Sindbis Virus Encephalitis by Beclin, a Novel Bcl-2-Interacting Protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-Induced Expression of Noxa and Beclin-1 Promotes Autophagic Cell Death and Limits Clonogenic Survival. Mol. Cell 2011, 42, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27 (Suppl. S1), S2–S19. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Ann. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pasqualis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; LaRocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, X.; Yi, C.; He, Y.; Chen, X.; Zhao, W.; Yu, D. Ferroptosis-related gene signature predicts the prognosis in Oral squamous cell carcinoma patients. BMC Cancer 2021, 21, 835. [Google Scholar] [CrossRef]
- Gu, W.; Kim, M.; Wang, L.; Yang, Z.; Nakajima, T.; Tsushima, Y. Multi-omics Analysis of Ferroptosis Regulation Patterns and Characterization of Tumor Microenvironment in Patients with Oral Squamous Cell Carcinoma. Int. J. Biol. Sci. 2021, 17, 3476–3492. [Google Scholar] [CrossRef]
- Sato, K.; Shi, L.; Ito, F.; Ohara, Y.; Motooka, Y.; Tanaka, H.; Mizuno, M.; Hori, M.; Hirayama, T.; Hibi, H.; et al. Non-thermal plasma specifically kills oral squamous cell carcinoma cells in a catalytic Fe (II)-dependent manner. J. Clin. Biochem. Nutr. 2019, 65, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Jiang, X.; Dong, Z.; Hu, S.; Xiao, M. Low-Concentration PTX And RSL3 Inhibits Tumor Cell Growth Synergistically by Inducing Ferroptosis In Mutant p53 Hypopharyngeal Squamous Carcinoma. Cancer Manag. Res. 2019, 11, 9783–9792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Sehm, T.; Fan, Z.; Ghoochani, A.; Rauh, M.; Engelhorn, T.; Minakaki, G.; Dörfler, A.; Klucken, J.; Buchfelder, M.; Eyüpoglu, I.Y.; et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget 2016, 7, 36021. [Google Scholar] [CrossRef]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Martinez, M.R.; Lopez, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, S.; Ryan, K.M. Autophagy: An adaptable modifier of tumourigenesis. Curr. Opin. Genet. Dev. 2010, 20, 57–64. [Google Scholar] [CrossRef]
- Lorin, S.; Hamaï, A.; Mehrpour, M.; Codogno, P. Autophagy Regulation and its Role in Cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell. Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C. The role of p53 in apoptosis. Discov. Med. 2010, 9, 145–152. [Google Scholar]
- Sui, X.; Jin, L.; Huang, X.; Geng, S.; He, C.; Hu, X. p53 signaling and autophagy in cancer: A revolutionary strategy could be developed for cancer treatment. Autophagy 2011, 7, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK β1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Shen, Z.; Shang, L.; Wang, X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011, 18, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.A.; Mariño, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.C.; Chano, T.; et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar] [CrossRef] [Green Version]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.R.J.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.J.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-J.; Woo, S.-J.; Yoon, C.-H.; Lee, J.-S.; An, S.; Choi, Y.-H.; Hwang, S.-G.; Yoon, G.; Lee, S.-J. Involvement of Autophagy in Oncogenic K-Ras-induced Malignant Cell Transformation. J. Biol. Chem. 2011, 286, 12924–12932. [Google Scholar] [CrossRef] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, Å.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Amati, B.; Alevizopoulos, K.; Vlach, J. Myc and the cell cycle. Front Biosci. 1998, 3, D250–D268. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Liu, S.; Zhang, G.; Zhou, C.-G.; Zhu, H.-X.; Zhou, X.-B.; Quan, L.-P.; Bai, J.-F.; Xu, N.-Z. Knockdown of c-Myc expression by RNAi inhibits MCF-7 breast tumor cells growth in vitro and in vivo. Breast Cancer Res. 2005, 7, R220–R228. [Google Scholar] [CrossRef] [Green Version]
- Iversen, P.; Arora, V.; Acker, A.J.; Mason, D.H.; Devi, G.R. Efficacy of antisense morpholino oligomer targeted to c-myc in prostate cancer xenograft murine model and a Phase I safety study in humans. Clin. Cancer Res. 2003, 9, 2510–2519. [Google Scholar]
- Schaub, F.X.; Li, W.; Fallahi, M.; Yang, C.; Schaub, S.K.; Lee, S.; Tzankov, A.; Schmitt, C.; Amelio, A.L.; Cleveland, J.L. Myc-Directed Suppression of Autophagy Provides Therapeutic Vulnerabilities Targeting Amino Acid Homeostasis. Blood 2015, 126, 2450. [Google Scholar] [CrossRef]
- Annunziata, I.; van de Vlekkert, D.; Wolf, E.; Finkelstein, D.; Neale, G.; Machado, E.; Mosca, R.; Campos, Y.; Tillman, H.; Roussel, M.F.; et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat. Commun. 2019, 10, 3623. [Google Scholar] [CrossRef]
- Toh, P.P.C.; Luo, S.; Menzies, F.M.; Raskó, T.; Wanker, E.; Rubinsztein, D.C. Myc inhibition impairs autophagosome formation. Hum. Mol. Genet. 2013, 22, 5237–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 Is a Key Regulator of Nutrient Sensing in the mTORC1 Pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Nezis, I.P.; Stenmark, H. p62 at the Interface of Autophagy, Oxidative Stress Signaling, and Cancer. Antioxid. Redox Signal 2012, 17, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.N.; Gilpin, C.; Levine, B. Autophagy Gene-Dependent Clearance of Apoptotic Cells during Embryonic Development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.-L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer. 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-W.; Su, Y.; Zhu, H.; Cao, J.; Ding, W.-J.; Zhao, Y.-C.; He, Q.-J.; Yang, B. HIF-1α-dependent autophagy protects HeLa cells from fenretinide (4-HPR)-induced apoptosis in hypoxia. Pharmacol. Res. 2010, 62, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; White, E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy 2013, 9, 1636–1638. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Bläuer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Shang, Y.; Chen, S.-Z. Chloroquine potentiates the anti-cancer effect of lidamycin on non-small cell lung cancer cells in vitro. Acta Pharmacol. Sin. 2014, 35, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Ameisen, J.C. On the origin, evolution, and nature of programmed cell death: A timeline of four billion years. Cell Death Differ. 2002, 9, 367–393. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Abedin, M.J.; Wang, D.; McDonnell, M.A.; Lehmann, U.; Kelekar, A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007, 14, 500–510. [Google Scholar] [CrossRef]
- Xie, X.; White, E.P.; Mehnert, J.M. Coordinate Autophagy and mTOR Pathway Inhibition Enhances Cell Death in Melanoma. PLoS ONE 2013, 8, e55096. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xing, D.; Zhou, F.; Chen, Q. Mitochondrial autophagy protects against heat shock-induced apoptosis through reducing cytosolic cytochrome c release and downstream caspase-3 activation. Biochem. Biophys. Res. Commun. 2010, 395, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Das, D.N.; Naik, P.P.; Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Meher, B.R.; Bhutia, S.K. Elimination of dysfunctional mitochondria through mitophagy suppresses benzo[a]pyrene-induced apoptosis. Free Radic. Biol. Med. 2017, 112, 452–463. [Google Scholar] [CrossRef]
- Zhao, Z.; Xia, G.; Li, N.; Su, R.; Chen, X.; Zhong, L. Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. J. Cancer 2018, 9, 3407–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhao, C.; He, R.; Zhou, M.; Liu, Y.; Guo, X.; Wang, M.; Zhu, F.; Qin, R.; Li, X. Danthron suppresses autophagy and sensitizes pancreatic cancer cells to doxorubicin. Toxicol. Vitro 2018, 54, 345–353. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Josset, E.; Burckel, H.; Noël, G.; Bischoff, P. The mTOR inhibitor RAD001 potentiates autophagic cell death induced by temozolomide in a glioblastoma cell line. Anticancer Res. 2013, 33, 1845–1851. [Google Scholar] [PubMed]
- Wang, Y.; Luo, Q.; He, X.; Wei, H.; Wang, T.; Shao, J.; Jiang, X. Emodin Induces Apoptosis of Colon Cancer Cells via Induction of Autophagy in a ROS-Dependent Manner. Oncol. Res. 2018, 26, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Shui, L.; Wang, W.; Xie, M.; Ye, B.; Li, X.; Liu, Y.; Zheng, M. Isoquercitrin induces apoptosis and autophagy in hepatocellular carcinoma cells via AMPK/mTOR/p70S6K signaling pathway. Aging 2020, 12, 24318–24332. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Molecular mechanisms of tumor angiogenesis and tumor progression. J. Neuro-Oncol. 2000, 50, 63–70. [Google Scholar] [CrossRef]
- Du, J.; Teng, R.-J.; Guan, T.; Eis, A.; Kaul, S.; Konduri, G.G.; Shi, Y. Role of autophagy in angiogenesis in aortic endothelial cells. Am. J. Physiol. Physiol. 2012, 302, C383–C391. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Livesey, K.M.; Zeh, I.H.J.; Loze, M.T.; Tang, D. HMGB1: A novel Beclin 1-binding protein active in autophagy. Autophagy 2010, 6, 1209–1211. [Google Scholar] [CrossRef]
- Yang, S.; Xu, L.; Yang, T.; Wang, F. High-mobility group box-1 and its role in angiogenesis. J. Leukoc. Biol. 2014, 95, 563–574. [Google Scholar] [CrossRef]
- Sharifi, M.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Macintosh, R.L.; Timpson, P.; Thorburn, J.; Anderson, K.I.; Thorburn, A.; Ryan, K.M. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012, 11, 2022–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.L.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2012, 32, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial–mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, H.; Fesler, A.; Ba, Y.; Wu, S.; Ju, J. Inhibition of colorectal cancer stem cell survival and invasive potential by hsa-miR-140-5p mediated suppression of Smad2 and autophagy. Oncotarget 2015, 6, 19735–19746. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Yan, L.; Xu, Y. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene 2014, 34, 3315–3324. [Google Scholar] [CrossRef] [Green Version]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK Integrates Autophagy and Oxidative Stress Responses to Promote Survival during Extracellular Matrix Detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-F.; Shi, Y.-H.; Ding, Z.-B.; Ke, A.-W.; Gu, C.-Y.; Hui, B.; Zhou, J.; Qiu, S.-J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Li, T.; Wei, Q.; Zhang, Y.; Jia, X.; Wan, Z.; Han, L. Upregulation of BNIP3 mediated by ERK/HIF-1α pathway induces autophagy and contributes to anoikis resistance of hepatocellular carcinoma cells. Future Oncol. 2014, 10, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; David, J.; Cook-Spaeth, D.; Casey, S.; Cohen, D.; Selvendiran, K.; Bekaii-Saab, T.; Hays, J.L. Autophagy Induction Results in Enhanced Anoikis Resistance in Models of Peritoneal Disease. Mol. Cancer Res. 2016, 15, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-Mesenchymal Transition and Autophagy Induction in Breast Carcinoma Promote Escape from T-cell–Mediated Lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tittarelli, A.; Janji, B.; Van Moer, K.; Noman, M.Z.; Chouaib, S. The Selective Degradation of Synaptic Connexin 43 Protein by Hypoxia-induced Autophagy Impairs Natural Killer Cell-mediated Tumor Cell Killing. J. Biol. Chem. 2015, 290, 23670–23679. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Messai, Y.; Noman, M.Z.; Hasmim, M.; Janji, B.; Tittarelli, A.; Boutet, M.; Baud, V.; Viry, E.; Billot, K.; Nanbakhsh, A.; et al. ITPR1 Protects Renal Cancer Cells against Natural Killer Cells by Inducing Autophagy. Cancer Res. 2014, 74, 6820–6832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V.; Semenza, G.L. Oncogenic Alterations of Metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling Rivals for Control of Cancer Cell Metabolism and Proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; Gottlieb, E.; et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ariyawardana, A.; Johnson, N.W. Trends of lip, oral cavity and oropharyngeal cancers in Australia 1982–2008: Overall good news but with rising rates in the oropharynx. BMC Cancer 2013, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Li, K.-Y.; Yang, W.; Su, Y.-X. Induction chemotherapy for squamous cell carcinomas of the oral cavity: A cumulative meta-analysis. Oral Oncol. 2016, 61, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Busch, C.-J.; Tribius, S.; Schafhausen, P.; Knecht, R. The current role of systemic chemotherapy in the primary treatment of head and neck cancer. Cancer Treat. Rev. 2015, 41, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, M.K.; Li, Y.; Murphy, B.; Hussain, M.H.; DeConti, R.C.; Ensley, J.; Forastiere, A.A. Randomized Phase III Evaluation of Cisplatin Plus Fluorouracil Versus Cisplatin Plus Paclitaxel in Advanced Head and Neck Cancer (E1395): An Intergroup Trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2005, 23, 3562–3567. [Google Scholar] [CrossRef]
- Güneri, P.; Epstein, J.B. Late stage diagnosis of oral cancer: Components and possible solutions. Oral Oncol. 2014, 50, 1131–1136. [Google Scholar] [CrossRef]
- Ronca, R.; Van Ginderachter, J.; Turtoi, A. Paracrine interactions of cancer-associated fibroblasts, macrophages and endothelial cells: Tumor allies and foes. Curr. Opin. Oncol. 2018, 30, 45–53. [Google Scholar] [CrossRef]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta Rev. Cancer 2014, 1845, 182–201. [Google Scholar] [CrossRef] [Green Version]
- Dourado, M.R.; Guerra, E.N.S.; Salo, T.; Lambert, D.W.; Coletta, R.D. Prognostic value of the immunohistochemical detection of cancer-associated fibroblasts in oral cancer: A systematic review and meta-analysis. J. Oral Pathol. Med. 2017, 47, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Kellermann, M.G.; Sobral, L.; da Silva, S.D.; Zecchin, K.G.; Graner, E.; Lopes, M.A.; Kowalski, L.P.; Coletta, R.D. Mutual paracrine effects of oral squamous cell carcinoma cells and normal oral fibroblasts: Induction of fibroblast to myofibroblast transdifferentiation and modulation of tumor cell proliferation. Oral Oncol. 2008, 44, 509–517. [Google Scholar] [CrossRef]
- Wei, L.-Y.; Lee, J.-J.; Yeh, C.-Y.; Yang, C.-J.; Kok, S.-H.; Ko, J.-Y.; Tsai, F.-C.; Chia, J.-S. Reciprocal activation of cancer-associated fibroblasts and oral squamous carcinoma cells through CXCL1. Oral Oncol. 2019, 88, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Moon, S.; Kim, D.K.; Zhang, X.; Kim, J. CXCL1 induces senescence of cancer-associated fibroblasts via autocrine loops in oral squamous cell carcinoma. PLoS ONE 2018, 13, e0188847. [Google Scholar] [CrossRef] [PubMed]
- Bello, I.O.; Vered, M.; Dayan, D.; Dobriyan, A.; Yahalom, R.; Alanen, K.; Nieminen, P.; Kantola, S.; Läärä, E.; Salo, T. Cancer-associated fibroblasts, a parameter of the tumor microenvironment, overcomes carcinoma-associated parameters in the prognosis of patients with mobile tongue cancer. Oral Oncol. 2011, 47, 33–38. [Google Scholar] [CrossRef]
- Vered, M.; Dobriyan, A.; Dayan, D.; Yahalom, R.; Talmi, Y.P.; Bedrin, L.; Barshack, I.; Taicher, S. Tumor-host histopathologic variables, stromal myofibroblasts and risk score, are significantly associated with recurrent disease in tongue cancer. Cancer Sci. 2010, 101, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jing, Y.; Ding, L.; Zhang, X.; Song, Y.; Chen, S.; Zhao, X.; Huang, X.; Pu, Y.; Wang, Z.; et al. Epiregulin reprograms cancer-associated fibroblasts and facilitates oral squamous cell carcinoma invasion via JAK2-STAT3 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 274. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.-W.; Che, Z.M.; Kim, J.; Kim, K.; Kim, K.-Y.; Williams, D.; Kim, J. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: A pivotal role of CCL7. Int. J. Cancer 2009, 127, 332–344. [Google Scholar] [CrossRef]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2016, 8, 8633–8647. [Google Scholar] [CrossRef] [Green Version]
- Fabriek, B.O.; Dijkstra, C.D.; Berg, T.K.V.D. The macrophage scavenger receptor CD163. Immunobiology 2005, 210, 153–160. [Google Scholar] [CrossRef]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Qu, C.; Wang, Q.; Meng, Z.; Wang, P. Cancer-associated fibroblasts in pancreatic cancer: Should they be deleted or reeducated? Integr. Cancer Ther. 2018, 17, 1016–1019. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.L.; Parkinson, E.K.; Yap, L.F.; Paterson, I.C. Autophagy is deregulated in cancer-associated fibroblasts from oral cancer and is stimulated during the induction of fibroblast senescence by TGF-β1. Sci. Rep. 2021, 11, 584. [Google Scholar] [CrossRef] [PubMed]
- Hassona, Y.; Cirillo, N.; Lim, K.P.; Herman, A.; Mellone, M.; Thomas, G.J.; Pitiyage, G.N.; Parkinson, E.; Prime, S.S. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-β. Carcinogenesis 2013, 34, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Koontongkaew, S. The Tumor Microenvironment Contribution to Development, Growth, Invasion and Metastasis of Head and Neck Squamous Cell Carcinomas. J. Cancer 2013, 4, 66–83. [Google Scholar] [CrossRef] [PubMed]
- He, K.-F.; Zhang, L.; Huang, C.-F.; Ma, S.-R.; Wang, Y.-F.; Wang, W.-M.; Zhao, Z.; Liu, B.; Zhao, Y.-F.; Zhang, W.-F.; et al. CD163+ Tumor-Associated Macrophages Correlated with Poor Prognosis and Cancer Stem Cells in Oral Squamous Cell Carcinoma. BioMed Res. Int. 2014, 2014, 838632. [Google Scholar] [CrossRef]
- Gomes, F.G.; Nedel, F.; Alves, A.; Nör, J.E.; Tarquinio, S.B.C. Tumor angiogenesis and lymphangiogenesis: Tumor/endothelial crosstalk and cellular/microenvironmental signaling mechanisms. Life Sci. 2012, 92, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.S.M.R.; Moriyama, M.; Kubota, K.; Ishiguro, N.; Sakamoto, M.; Chinju, A.; Mochizuki, K.; Sakamoto, T.; Kaneko, N.; Munemura, R.; et al. CD206+ tumor-associated macrophages promote proliferation and invasion in oral squamous cell carcinoma via EGF production. Sci. Rep. 2019, 9, 14611. [Google Scholar] [CrossRef]
- Fujii, N.; Shomori, K.; Shiomi, T.; Nakabayashi, M.; Takeda, C.; Ryoke, K.; Ito, H. Cancer-associated fibroblasts and CD163-positive macrophages in oral squamous cell carcinoma: Their clinicopathological and prognostic significance. J. Oral Pathol. Med. 2012, 41, 444–451. [Google Scholar] [CrossRef]
- Chang, C.-P.; Su, Y.-C.; Lee, P.-H.; Lei, H.-Y. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy 2013, 9, 619–621. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-P.; Su, Y.-C.; Hu, C.-W.; Lei, H.-Y. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2012, 20, 515–523. [Google Scholar] [CrossRef]
- Yang, M.; Liu, J.; Shao, J.; Qin, Y.; Ji, Q.; Zhang, X.; Du, J. Cathepsin S-mediated autophagic flux in tumor-associated macrophages accelerate tumor development by promoting M2 polarization. Mol. Cancer 2014, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-Y.; Wang, X.; Sun, S.-Z.; Song, Y.; Yang, M.-X.; Qu, X. [Effect of hypoxia inducible factor-1 alpha on vascular endothelial growth factor expression in human tongue squamous carcinoma cells (Tca8113) under hypoxia]. Chin. J. Stomatol. 2007, 42, 747–749. [Google Scholar]
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical Remodeling of the Microenvironment by Stromal Caveolin-1 Favors Tumor Invasion and Metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, R.J.B.; Robins, M.W. Cancer Biology, 3rd ed.; Pearson Education: London, UK, 2006. [Google Scholar]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Yang, M.-H.; Wu, K.-J. TWIST activation by hypoxia inducible factor-1 (HIF-1): Implications in metastasis and development. Cell Cycle 2008, 7, 2090–2096. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 Is an RB/E2F Target Gene Required for Hypoxia-Induced Autophagy. Mol. Cell. Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Huang, Q.; Wang, Y.; Shen, P.; Guan, F.; Li, J.; Huang, H.; Shi, C. Hypoxia-inducible factor-1alpha regulates autophagy to activate hepatic stellate cells. Biochem. Biophys. Res. Commun. 2014, 454, 328–334. [Google Scholar] [CrossRef]
- Qureshi-Baig, K.; Kuhn, D.; Viry, E.; Pozdeev, V.I.; Schmitz, M.; Rodriguez, F.; Ullmann, P.; Koncina, E.; Nurmik, M.; Frasquilho, S.; et al. Hypoxia-induced autophagy drives colorectal cancer initiation and progression by activating the PRKC/PKC-EZR (ezrin) pathway. Autophagy 2019, 16, 1436–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yin, H.; Zhang, Y.; Li, X.; Tong, H.; Zeng, Y.; Wang, Q.; He, W. Hypoxia-induced autophagy promotes gemcitabine resistance in human bladder cancer cells through hypoxia-inducible factor 1α activation. Int. J. Oncol. 2018, 53, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Tanis, T.; Cincin, Z.B.; Gokcen-Rohlig, B.; Bireller, E.S.; Ulusan, M.; Tanyel, C.R.; Cakmakoglu, B. The role of components of the extracellular matrix and inflammation on oral squamous cell carcinoma metastasis. Arch. Oral Biol. 2014, 59, 1155–1163. [Google Scholar] [CrossRef]
- Levental, K.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Méndez, E.; Houck, J.R.; Doody, D.; Fan, W.; Lohavanichbutr, P.; Rue, T.C.; Yueh, B.; Futran, N.D.; Upton, M.P.; Farwell, D.G.; et al. A Genetic Expression Profile Associated with Oral Cancer Identifies a Group of Patients at High Risk of Poor Survival. Clin. Cancer Res. 2009, 15, 1353–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kainulainen, T.; Grenman, R.; Oikarinen, A.; Greenspan, D.; Sale, T. Distribution and synthesis of type VII collagen in oral squamous cell carcinoma. J. Oral Pathol. Med. 1997, 26, 414–418. [Google Scholar] [CrossRef]
- Harada, T.; Shinohara, M.; Nakamura, S.; Oka, M. An immunohistochemical study of the extracellular matrix in oral squamous cell carcinoma and its association with invasive and metastatic potential. Virchows Arch. 1994, 424, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Randriantsilefisoa, R.; Chen, J.; Cuellar-Camacho, J.L.; Liang, W.; Li, W. Matrix Stiffness Regulates Chemosensitivity, Stemness Characteristics, and Autophagy in Breast Cancer Cells. ACS Appl. Bio Mater. 2020, 3, 4474–4485. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [Green Version]
- De Lima, T.B.; Paz, A.H.R.; Rados, P.V.; Leonardi, R.; Bufo, P.; Pedicillo, M.C.; Santoro, A.; Cagiano, S.; Aquino, G.; Botti, G.; et al. Autophagy analysis in oral carcinogenesis. Pathol Res Pract. 2017, 213, 1072–1077. [Google Scholar] [CrossRef]
- Tang, J.-Y.; Hsi, E.; Huang, Y.-C.; Hsu, N.C.-H.; Chu, P.-Y.; Chai, C.-Y. High LC3 expression correlates with poor survival in patients with oral squamous cell carcinoma. Hum. Pathol. 2013, 44, 2558–2562. [Google Scholar] [CrossRef]
- Tang, J.-Y.; Hsi, E.; Huang, Y.-C.; Hsu, N.C.-H.; Chen, Y.-K.; Chu, P.-Y.; Chai, C.-Y. ATG9A overexpression is associated with disease recurrence and poor survival in patients with oral squamous cell carcinoma. Virchows Arch. Int. J. Pathol. 2013, 463, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-Y.; Hsi, E.; Huang, Y.-C.; Hsu, N.C.-H.; Yang, W.-C.; Chang, H.-W.; Chai, C.-Y.; Chu, P.-Y. Overexpression of Autophagy-Related 16-Like 1 in Patients with Oral Squamous Cell Carcinoma. Pathol. Oncol. Res. 2014, 21, 301–305. [Google Scholar] [CrossRef]
- Liu, J.; Chen, F.; Lung, J.; Lo, C.-H.; Lee, F.-H.; Lu, Y.-C.; Hung, C.-H. Prognostic significance of p62/SQSTM1 subcellular localization and LC3B in oral squamous cell carcinoma. Br. J. Cancer 2014, 111, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Luo, H.; He, Q.; You, Y.; Fan, Y.; Liang, J. Investigation of cancer-associated fibroblasts and p62 expression in oral cancer before and after chemotherapy. J. Cranio-Maxillofac. Surg. 2018, 46, 605–610. [Google Scholar] [CrossRef]
- Park, B.-S.; Choi, N.-E.; Lee, J.H.; Kang, H.-M.; Yu, S.-B.; Kim, H.-J.; Kang, H.-K.; Kim, I.-R. Crosstalk between Fisetin-induced Apoptosis and Autophagy in Human Oral Squamous Cell Carcinoma. J. Cancer 2019, 10, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Chin, H.; Lee, S.; Chiu, C.; Chung, J.; Lin, Z.; Wu, C.; Liu, Y.; Hsiao, Y.; Feng, C.; et al. Ursolic acid induces apoptosis and autophagy in oral cancer cells. Environ. Toxicol. 2019, 34, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Dou, Z.-C.; Ren, W.-H.; Li, S.-M.; Liang, X.; Zhi, K.-Q. CircCDR1as upregulates autophagy under hypoxia to promote tumor cell survival via AKT/ERK½/mTOR signaling pathways in oral squamous cell carcinomas. Cell Death Dis. 2019, 10, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, P.P.; Mukhopadhyay, S.; Praharaj, P.P.; Bhol, C.S.; Panigrahi, D.P.; Mahapatra, K.K.; Patra, S.; Saha, S.; Panda, A.K.; Panda, K.; et al. Secretory clusterin promotes oral cancer cell survival via inhibiting apoptosis by activation of autophagy in AMPK/mTOR/ULK1 dependent pathway. Life Sci. 2020, 264, 118722. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.P.; Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, C.K.; Mishra, R.; Patil, S.; Bhutia, S.K. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD 44, ABCB 1 and ADAM 17 in oral squamous cell carcinoma. Cell Prolif. 2018, 51, e12411. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, C.; Tang, H.; Wang, M.; Weng, J.; Liu, X.; Zhang, R.; Huang, H.; Hou, J. Decrease of autophagy activity promotes malignant progression of tongue squamous cell carcinoma. J. Oral Pathol. Med. 2013, 42, 557–564. [Google Scholar] [CrossRef]
- Kapoor, V.; Paliwal, D.; Singh, S.B.; Mohanti, B.K.; Das, S.N. Deregulation of Beclin 1 in patients with tobacco-related oral squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2012, 422, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Wang, C.; Wang, Y.; Tang, H.; Liang, J.; Liu, X.; Huang, H.; Hou, J. Beclin1 inhibits proliferation, migration and invasion in tongue squamous cell carcinoma cell lines. Oral Oncol. 2014, 50, 983–990. [Google Scholar] [CrossRef]
- Wang, X.; Li, S.; Wu, S.; Xie, L.; Wang, P. Silence of Beclin1 in oral squamous cell carcinoma cells promotes proliferation, inhibits apoptosis, and enhances chemosensitivity. Int. J. Clin. Exp. Pathol. 2017, 10, 8424–8433. [Google Scholar]
- Sahni, S.; Merlot, A.M.; Krishan, S.; Jansson, P.; Richardson, D. Gene of the month: BECN1. J. Clin. Pathol. 2014, 67, 656–660. [Google Scholar] [CrossRef]
- Kong, Q.; Liang, Y.; He, Q.; You, Y.; Wu, L.; Liang, L.; Liang, J. Autophagy inhibits TLR4-mediated invasiveness of oral cancer cells via the NF-κB pathway. Oral Dis. 2020, 26, 1165–1174. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, T.J.; Woo, B.H.; Choi, K.U.; Lee, C.H.; Ryu, M.H.; Park, H.R. Curcumin-induced autophagy contributes to the decreased survival of oral cancer cells. Arch. Oral Biol. 2012, 57, 1018–1025. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, C.; Wang, Q.; Zeng, X.; Ji, P. Tanshinone IIA induces cell death via Beclin-1-dependent autophagy in oral squamous cell carcinoma SCC-9 cell line. Cancer Med. 2018, 7, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-T.; Kuo, C.-L.; Chueh, F.-S.; Liu, K.-C.; Bau, D.-T.; Chung, J.-G. Curcuminoids Induce Reactive Oxygen Species and Autophagy to Enhance Apoptosis in Human Oral Cancer Cells. Am. J. Chin. Med. 2018, 46, 1145–1168. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L. Maturation of Autophagic Vacuoles in Mammalian Cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Autophagosome and Phagosome; Humana Press: Totowa, NJ, USA, 2008; Volume 445, pp. 77–88. [Google Scholar]
- Savarino, A.; Lucia, M.B.; Giordano, F.; Cauda, R. Risks and benefits of chloroquine use in anticancer strategies. Lancet Oncol. 2006, 7, 792–793. [Google Scholar] [CrossRef]
- Homewood, C.; Warhurst, D.; Peters, W.; Baggaley, V.C. Lysosomes, p H and the Anti-malarial Action of Chloroquine. Nature 1972, 235, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M. Rational Use of New and Existing Disease-Modifying Agents in Rheumatoid Arthritis. Ann. Intern. Med. 2001, 134, 695–706. [Google Scholar] [CrossRef]
- Rainsford, K.D.; Parke, A.L.; Clifford-Rashotte, M.; Kean, W.F. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 2015, 23, 231–269. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From Malaria to Autoimmunity. Clin. Rev. Allergy Immunol. 2011, 42, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.D.; Munz, S.J.; Paschal, J.; Cohen, H.B.; Pince, K.J.; Peterson, T. Incidence of hydroxychloroquine retinopathy in 1,207 patients in a large multicenter outpatient practice. Arthritis Rheum. 1997, 40, 1482–1486. [Google Scholar] [CrossRef]
- Zhao, H.; Cai, Y.; Santi, S.; Lafrenie, R.; Lee, H. Chloroquine-Mediated Radiosensitization is due to the Destabilization of the Lysosomal Membrane and Subsequent Induction of Cell Death by Necrosis. Radiat. Res. 2005, 164, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Hosoda, R.; Akiyama, Y.; Sebori, R.; Wanibuchi, M.; Mikami, T.; Sugino, T.; Suzuki, K.; Maruyama, M.; Tsukamoto, M.; et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J. Neuro-Oncol. 2014, 122, 11–20. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [Green Version]
- Grimaldi, A.; Balestrieri, M.L.; D′Onofrio, N.; Di Domenico, G.; Nocera, C.; Lamberti, M.; Tonini, G.; Zoccoli, A.; Santini, D.; Caraglia, M.; et al. The Synergistic Effect of Everolimus and Chloroquine on Endothelial Cell Number Reduction Is Paralleled by Increased Apoptosis and Reduced Autophagy Occurrence. PLoS ONE 2013, 8, e79658. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, G.; Shi, Y.; Su, C.; Chang, J. Co-delivery of Gefitinib and chloroquine by chitosan nanoparticles for overcoming the drug acquired resistance. J. Nanobiotechnol. 2015, 13, 57. [Google Scholar] [CrossRef] [Green Version]
- Bokobza, S.M.; Jiang, Y.; Weber, A.M.; Devery, A.M.; Ryan, A.J. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget 2014, 5, 4765–4778. [Google Scholar] [CrossRef]
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; Arimoto, T.; et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecol. Oncol. 2015, 137, 538–545. [Google Scholar] [CrossRef]
- Wu, Z.; Chang, P.-C.; Yang, J.C.; Chu, C.-Y.; Wang, L.-Y.; Chen, N.-T.; Ma, A.-H.; Desai, S.J.; Lo, S.H.; Evans, C.P.; et al. Autophagy Blockade Sensitizes Prostate Cancer Cells towards Src Family Kinase Inhibitors. Genes Cancer 2010, 1, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A., 2nd; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell Mol. Med. 2010, 14, 2448–2459. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.-M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and Current Strategies for Targeting Autophagy for Cancer Treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceño, E.; López-González, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.-S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [Green Version]
- Loaiza-Bonilla, A.; O’Hara, M.H.; Redlinger, M.; Damjanov, N.; Teitelbaum, U.R.; Rosen, I.V.M.A.; Heitjan, D.F.; Amaravadi, R.K.; O’Dwyeret, P.J. Phase II trial of autophagy inhibition using hydroxychloroquine (HCQ) with FOLFOX/bevacizumab in the first-line treatment of advanced colorectal cancer. Am. Soc. Clin. Oncol. 2015, 35 (Suppl. S15), 3545. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients with Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquier, B. Autophagy inhibitors. Cell Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.; Ma, X.-H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Hu, Q.; Shen, H.-M. Pharmacological inhibitors of autophagy as novel cancer therapeutic agents. Pharmacol. Res. 2016, 105, 164–175. [Google Scholar] [CrossRef]
- Yu, X.; Long, Y.C.; Shen, H.-M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [Green Version]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.-M. Dual Role of 3-Methyladenine in Modulation of Autophagy via Different Temporal Patterns of Inhibition on Class I and III Phosphoinositide 3-Kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [Green Version]
- Caro, L.H.P.; Plomp, P.J.A.M.; Wolvetang, E.J.; Kerkhof, C.; Meijer, A.J. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. JBIC J. Biol. Inorg. Chem. 1988, 175, 325–329. [Google Scholar] [CrossRef]
- Ihle, N.T.; Williams, R.; Chow, S.; Chew, W.; Berggren, M.I.; Paine-Murrieta, G.; Minion, D.J.; Halter, R.J.; Wipf, P.; Abraham, R.; et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol. Cancer Ther. 2004, 3, 763–772. [Google Scholar] [PubMed]
- Wiesinger, D.; Gubler, H.U.; Haefliger, W.; Hauser, D. Antiinflammatory activity of the new mould metabolite 11-desacetoxy-wortmannin and of some of its derivatives. Cell. Mol. Life Sci. 1974, 30, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Shokat, K.M. Chemically targeting the PI3K family. Biochem. Soc. Trans. 2007, 35 Pt 2, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takase, Y.; Saeki, T.; Watanabe, N.; Adachi, H.; Souda, S.; Saito, I. Cyclic GMP Phosphodiesterase Inhibitors. 2. Requirement of 6-Substitution of Quinazoline Derivatives for Potent and Selective Inhibitory Activity. J. Med. Chem. 1994, 37, 2106–2111. [Google Scholar] [CrossRef]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of p53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [Green Version]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.-F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Pasquier, B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.D.; Arteaga, C.L.; Cook, R.S. Dual inhibition of Type I and Type III PI3 kinases increases tumor cell apoptosis in HER2+ breast cancers. Breast Cancer Res. 2015, 17, 148. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Li, Y.; Li, B.; Zhang, M.; Xu, C.; Liu, F.; Bian, L.; Liu, Y.; Yao, Y.; Li, D. Autophagy inhibition enhances celecoxib-induced apoptosis in osteosarcoma. Cell Cycle 2018, 17, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Schlütermann, D.; Skowron, M.A.; Berleth, N.; Böhler, P.; Deitersen, J.; Stuhldreier, F.; Wallot-Hieke, N.; Wu, W.; Peter, C.; Hoffmann, M.J.; et al. Targeting urothelial carcinoma cells by combining cisplatin with a specific inhibitor of the autophagy-inducing class III PtdIns3K complex. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 160.e1–160.e13. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1· ATG13· FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Kim, J.; Shaw, R.J.; Guan, K.-L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [Green Version]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef] [Green Version]
- Dite, T.A.; Langendorf, C.; Hoque, A.; Galic, S.; Rebello, R.J.; Ovens, A.J.; Lindqvist, L.M.; Ngoei, K.R.; Ling, N.; Furic, L.; et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J. Biol. Chem. 2018, 293, 8874–8885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahwazi, D.; Neopane, K.; Markby, G.R.; Kopietz, F.; Ovens, A.J.; Dall, M.; Hassing, A.S.; Gräsle, P.; Alshuweishi, Y.; Treebak, J.T.; et al. Investigation of the specificity and mechanism of action of the ULK1/AMPK inhibitor SBI-0206965. Biochem. J. 2021, 478, 2977–2997. [Google Scholar] [CrossRef]
- Zhou, Z.-H.; Zhao, T.-C.; Liang, S.-Y.; Zhang, Z.-Y.; Zhu, D.-W.; Ju, W.-T.; Zhong, L.-P. A therapeutic approach with combination of interferon-gamma and autophagy inhibitor for oral squamous cell carcinoma. 2021, 11, 1503–1521.
- Ahn, M.-Y.; Ahn, S.-G.; Yoon, J.-H. Apicidin, a histone deaceylase inhibitor, induces both apoptosis and autophagy in human oral squamous carcinoma cells. Oral Oncol. 2011, 47, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, J.; Wu, T.; Wu, J.; Ling, J.; Cheng, B. In vitro and in vivo antitumor effects of chloroquine on oral squamous cell carcinoma. Mol. Med. Rep. 2017, 16, 5779–5786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnano, S.; Barroeta, P.H.; Duffy, R.; O′Sullivan, J.; Zisterer, D.M. Cisplatin induces autophagy-associated apoptosis in human oral squamous cell carcinoma (OSCC) mediated in part through reactive oxygen species. Toxicol. Appl. Pharmacol. 2021, 427, 115646. [Google Scholar] [CrossRef]
- Li, B.; Lu, M.; Jiang, X.-X.; Pan, M.-X.; Mao, J.-W.; Chen, M. Inhibiting reactive oxygen species-dependent autophagy enhanced baicalein-induced apoptosis in oral squamous cell carcinoma. J. Nat. Med. 2017, 71, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-C.; Xin, Z.-Y.; Deborah, B.; Zhang, J.-S.; Yuan, D.-Y.; Xu, K.; Liu, X.-B.; Jiang, H.-Q.; Fan, Q.-C.; Zhang, B.; et al. Inhibition of autophagy augments apoptosis in human oral squamous cell carcinoma under nutrient depletion. J. Oral Pathol. Med. 2014, 44, 361–366. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Wang, W.; Piao, S.; Zhou, J.; Saiyin, W.; Zheng, C.; Sun, H.; Li, Y. Inhibition of autophagy by 3-MA enhances IL-24-induced apoptosis in human oral squamous cell carcinoma cells. J. Exp. Clin. Cancer Res. 2015, 34, 97. [Google Scholar] [CrossRef] [Green Version]
- Saiyin, W.; Wang, D.; Li, L.; Zhu, L.; Liu, B.; Sheng, L.; Li, Y.; Zhu, B.; Mao, L.; Li, G.; et al. Sequential Release of Autophagy Inhibitor and Chemotherapeutic Drug with Polymeric Delivery System for Oral Squamous Cell Carcinoma Therapy. Mol. Pharm. 2014, 11, 1662–1675. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd El-Aziz, Y.S.; Leck, L.Y.W.; Jansson, P.J.; Sahni, S. Emerging Role of Autophagy in the Development and Progression of Oral Squamous Cell Carcinoma. Cancers 2021, 13, 6152. https://doi.org/10.3390/cancers13246152
Abd El-Aziz YS, Leck LYW, Jansson PJ, Sahni S. Emerging Role of Autophagy in the Development and Progression of Oral Squamous Cell Carcinoma. Cancers. 2021; 13(24):6152. https://doi.org/10.3390/cancers13246152
Chicago/Turabian StyleAbd El-Aziz, Yomna S., Lionel Y. W. Leck, Patric J. Jansson, and Sumit Sahni. 2021. "Emerging Role of Autophagy in the Development and Progression of Oral Squamous Cell Carcinoma" Cancers 13, no. 24: 6152. https://doi.org/10.3390/cancers13246152
APA StyleAbd El-Aziz, Y. S., Leck, L. Y. W., Jansson, P. J., & Sahni, S. (2021). Emerging Role of Autophagy in the Development and Progression of Oral Squamous Cell Carcinoma. Cancers, 13(24), 6152. https://doi.org/10.3390/cancers13246152