Simple Summary
Pancreatic ductal adenocarcinoma (PDAC) has an extremely poor prognosis. The lack of early diagnosis and the absence of suitable biomarkers coupled with resistance to available therapeutic options has made PDAC one of the deadliest cancers. Despite advances in diagnostics and therapeutics, the prognosis of PDAC remains dismal. PDAC has a prominent desmoplastic stromal microenvironment that includes a dense extracellular matrix together with a series of activated cell types, hypoxia, and an acidic extracellular pH. This activated desmoplastic stroma compromises treatments yet, despite the recognition of its importance, it has not been comprehensively studied in this role. Moreover, PDAC metabolic reprogramming has also been found to be one of the key factors involved in treatment failure. Here, we critically review the role of the various stromal components in determining resistance to available therapeutics with the hope that its comprehensive understanding, if employed in the appropriate combination therapy, may make this recalcitrant cancer more manageable.
Abstract
Currently, the median overall survival of PDAC patients rarely exceeds 1 year and has an overall 5-year survival rate of about 9%. These numbers are anticipated to worsen in the future due to the lack of understanding of the factors involved in its strong chemoresistance. Chemotherapy remains the only treatment option for most PDAC patients; however, the available therapeutic strategies are insufficient. The factors involved in chemoresistance include the development of a desmoplastic stroma which reprograms cellular metabolism, and both contribute to an impaired response to therapy. PDAC stroma is composed of immune cells, endothelial cells, and cancer-associated fibroblasts embedded in a prominent, dense extracellular matrix associated with areas of hypoxia and acidic extracellular pH. While multiple gene mutations are involved in PDAC initiation, this desmoplastic stroma plays an important role in driving progression, metastasis, and chemoresistance. Elucidating the mechanisms underlying PDAC resistance are a prerequisite for designing novel approaches to increase patient survival. In this review, we provide an overview of the stromal features and how they contribute to the chemoresistance in PDAC treatment. By highlighting new paradigms in the role of the stromal compartment in PDAC therapy, we hope to stimulate new concepts aimed at improving patient outcomes.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related mortality in western countries and is projected to be the second-leading cause of cancer-related death in the United States by 2030 [1]. Despite significant breakthroughs in cancer research, PDAC remains a malignant disease with a high mortality. It is among the most chemoresistant cancers due to the broad heterogeneity of the genetic mutations and the dense stromal environment [2]. Due to the lack of both early diagnostic strategies and premature symptoms before the disease reaches its advanced stage, approximately 85% of tumors are not resectable at the time of diagnosis [3], making the median patient survival only 6–10 months [4]. Although several chemotherapies have reported some benefits, they are not enough to prolong survival and improve a patient’s quality of life [5]. Despite improvements made in the approaches for detecting and managing pancreatic cancer, the five-year survival rate only reached 9% in 2020 [6]. A prominent feature of the PDAC microenvironment is an extensive desmoplasia that consists of a highly fibrotic and stiff extracellular matrix (ECM) principally composed of collagen I, fibronectin and hyaluronan, which are secreted by alpha-muscle actin-positive fibroblasts (also known as myofibroblasts or activated pancreatic stellate cells (PSCs)) [7]. Modifications that the ECM architecture suffers during cancer progression have been deeply explored, since it has been recognized that atypical ECM architecture influences therapeutic outcomes specifically by modulating (i) tumor biomechanics [8]; (ii) cancer cell migration/invasion [9,10,11]; and (iii) drug penetration into the tumor [12]. The existence of this dense tumor microenvironment (TME) may be the main reason that therapies targeting specifically only cancer-associated molecular pathways have not given satisfactory results [13].
The TME was first proposed by Ioannides in 1993, and referred especially to the local environment where tumors occurred and developed [14]. Commonly, the TME of PDAC is characterized by abundant stroma, hypoxia, a deficient blood supply and elevated immunosuppression [15]. Studies have shown that the TME, including cancer-related fibroblasts (CAFs), stellate cells, diverse immune cells and cytokines released by them, are involved in the control of the proliferation, metastasis, chemoresistance and immunotherapy of pancreatic cancer cells [16]. Factors associated with TME such as cell plasticity, heterogeneity of the tumor, composition of the tumor stroma, epithelial-to-mesenchymal transition (EMT), reprogrammed metabolism, acidic extracellular pH (pHe) and hypoxia can heavily impact treatment outcomes. Therefore, finding new therapeutic targets within the TME of PDAC is an encouraging and potential research direction in order to understand the lack of efficacy of current treatments for pancreatic cancer.
Resistance to cancer chemotherapy or chemoresistance is the innate and/or acquired ability of cancer cells to survive and maintain uncontrolled proliferation leading to tumor progression in spite of the tumor’s exposure to cytotoxic compounds [17]. Chemoresistance also causes disease relapse and metastasis, thus representing a crucial challenge that oncology research has sought to understand and overcome in order to improve the clinical outcome of cancer patients [18,19]. Moreover, tumor heterogeneity promotes a specific tumor response to diverse types of chemotherapy, and the recent literature on pancreatic tumors has confirmed that tumors have a complex microenvironment containing many independent components, each of which exert a unique role in conferring chemoresistance [15,20].
Herein, we will describe the main mechanisms of chemoresistance in pancreatic cancer with a main emphasis on the emerging role played by the TME, aiming to provide future directions and to discover novel targets for new therapeutic strategies against PDAC.
2. Desmoplastic Reaction in the Pancreatic Tumor Microenvironment
2.1. Desmoplasia: The Impact on Tumor Development and Progression
Despite the sizeable improvement in recent years, chemotherapy remains markedly incompetent in improving PDAC patient survival [21]. There are many factors that contribute to the failure of chemotherapy in this pathology, including the occurrence of desmoplasia in the PDAC TME [21].
Desmoplasia, also known as the desmoplastic reaction, consists of a dense ECM together with myofibroblast-like cells, including CAFs and is a fundamental feature of the pancreatic cancer TME [22]. Initially, the role of this phenomenon was overlooked; however, many studies have since demonstrated that during PDAC development, the cancer cells expend a large amount of energy to promote the recruitment, proliferation, and activation of fibroblasts. Consequent to their activation, CAFs are able to deposit ECM components and secrete several types of factors that strongly affect the behavior of cancer cells [23,24,25]. Indeed, pharmacologic inhibition of the desmoplastic reaction in combination with chemotherapy showed better results in inhibiting PDAC progression than chemotherapy alone, thus highlighting desmoplasia as a likely therapeutic target in pancreatic cancer [26,27,28,29]. Desmoplasia can be divided histopathologically into two groups: (i) overproduction of ECM proteins, and (ii) extensive proliferation of the PSCs [30,31]. Hence, the resultant abundant and fibrotic stroma tissue is encompassed by both cellular and non-cellular elements. In this section, we will focus on the non-cellular elements.
Among all non-cellular components of desmoplasia, the importance of many ECM proteins, namely collagen types I, III and IV, fibronectin, laminin, hyaluronan, as well as the glycoprotein osteonectin [32,33] should be emphasized. Desmoplastic progression derives from the abnormal activation of several intercellular and intracellular signaling processes, such as the transforming growth factor beta (TGFβ), the basic fibroblast growth factor (bFGF), the connective tissue growth factor (CTGF) and interleukin-1β, which by stimulating ECM production drive desmoplastic progression [34,35,36,37,38]. The ECM components can also be divided into two categories: the fibrous proteins, such as collagens, and the polysaccharide chain glycosaminoglycans (GAGs), such as hyaluronan [39,40,41,42]. In the normal pancreas, GAGs structurally function to sustain compressive forces on the tissue, whereas the fibrous proteins act to support the tensile forces on the tissue [43]. On other hand, the marked overproduction of ECM constituents in PDAC has been suggested to be a failed wound healing, leading to fibrosis [43]. The increased deposition of collagen type I, III, and IV in PDAC tissues [44,45,46] is directly linked to the TGFβ/Smad signaling and is a product of the activity of the fibroblasts [47]. Remarkably, in PDAC, the elevated levels of collagen I reduce tissue elasticity and raise interstitial fluid pressure, causing a reduction in drug perfusion [48].
The protein-free GAG, hyaluronan, is also an important component of the ECM, contributing to tissue rigidity and thereby decreasing elasticity [49] and its accumulation within damaged tissue is a product of increased secretion by CAFs in pancreatic cancer [50]. Moreover, hyaluronan maintains its interaction with water molecules, to preserve tissue hydration in normal pancreas [51]. Nevertheless, in the “unhealthy” pancreas the increased deposition of hyaluronan might result in interstitial edema and, consequently, augmented interstitial fluid pressure that, in turn, leads to decreased fluid conveyance [52]. Accordingly, this enhanced interstitial edema together with the absence of an efficient lymphatic system in tumor tissue, results in a remarkable reduction in the exchange of several substances with the bloodstream, including chemotherapeutics [53]. Therefore, this significant deposition of hyaluronan in pancreatic cancer stroma is one of the main features of pancreatic TME responsible for decreased chemotherapeutic penetration (Figure 1) [54,55].
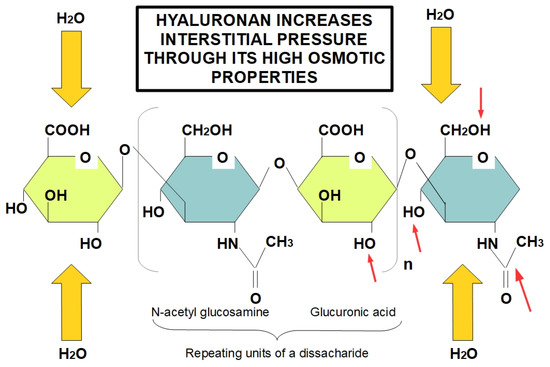
Figure 1.
Role of hyaluronan in increasing tissue interstitial pressure. Hyaluronan forms long chains creating a highly osmotic environment that produces edema and increased interstitial pressure. Despite the fact that the diagram only shows a tetrasacharide, hyaluronan is a very lengthy unbranched chain of repeating disaccharides. Red arrows indicate the hydrophilic parts of glucuronic acid and N-acetyl glucosamine, proving the highly hydrophilic ability of hyaluronan. Increased hyaluronan in tumors is an early event occurring in TME, which leads to increased interstitial pressure due to its hygroscopic properties, causing an obstacle to the adequate delivery of chemotherapeutic drugs.
Lastly, fibronectin is also a crucial ECM component which binds to adhesion receptors in several cell types [56]. Additionally, it supports cell–ECM interactions and is a key factor for wound healing and development and the maintenance of tissue homeostasis [57,58,59,60,61]. While several cell types including tumor cells and endothelial cells are able to produce fibronectin, fibroblasts are the main producers [59]. The elevated expression of fibronectin is displayed by several solid tumors, particularly in pancreatic cancer [62]. Therefore, the interaction of multiple ECM proteins to produce the desmoplastic reaction in PDAC is a feature which clearly contributes to the pathogenesis and ultimately to chemoresistance.
2.2. The Contribution of Desmoplastic Components towards Chemoresistance
Desmoplasia can promote chemoresistance through several mechanisms which can be divided in two main groups: biological and physiological chemoresistance [31]. Biological chemoresistance can arise from different mechanisms. Target cells can (a) acquire resistance to drug uptake, (b) reduce their sensitivity to drugs by increasing the expression of anti-apoptotic proteins and activating protective mechanisms such as autophagy [63], (c) use DNA repair mechanisms to counteract the drug-dependent destruction of the tumor cell DNA [64,65] and (d) recruit more transporters/proteins responsible for drug efflux, thus preventing their action in the cancer cell. In PDAC, both physiological and biological chemoresistance are present and constitute a considerable problem for effective chemotherapy [31]. Moreover, the non-cellular components of the desmoplastic reaction can contribute to biological chemoresistance. Indeed, the binding of hyaluronan to its receptor, CD44, caused a Stat-3-mediated increase in the expression of the multi-drug resistance protein 1 (MDR1) in pancreatic cancer cell lines [66]. Moreover, the interaction of hyaluronan with CD44 is able to activate the phosphatidylinositol-3-kinase (PI3K/AkT) signaling pathway, which is upregulated in several cancers, resulting in the phosphorylation of Bad, and the consequent downregulation of apoptosis (Figure 2) [67,68].
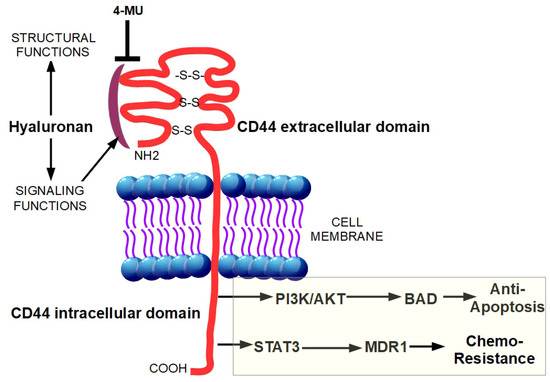
Figure 2.
Binding of hyaluronan to CD44 unleashes a pro-tumoral intracellular signaling. The intracellular signaling functions of hyaluronan are triggered after its binding with CD44. This interaction results in the increased expression of the multi-drug resistance protein 1 (MDR1) through STAT3 activation and in the activation of phosphatidylinositol-3-kinase (PI3K/AkT) signaling pathway, causing phosphorylation of Bad, and the subsequent downregulation of apoptosis. The hyaluronan synthesis inhibitor, 4-methylumbelliferone (4-MU), inhibits cell migration, proliferation, and invasion by blocking the interaction between hyaluronan and CD44.
The ECM components are also involved in physiological chemoresistance and probably to a larger extent than in biological chemoresistance [69]. Physiological chemoresistance is due to poor tissue vascularization in the tumor tissue caused by the overproduction of ECM proteins and the consequent increased interstitial fluid pressure, which together produce a barrier to drug absorption into the target tissue [31]. As mentioned before, collagen type I, III, and IV are highly secreted into the PDAC TME and it was demonstrated, in a PDAC orthotopic xenograft mouse model, that the increased collagen I content decreased the penetration of nanoparticles, resulting in a significant reduction in the response to doxorubicin treatment [70]. Moreover, the collagen-reducing effects of the anti-hypertensive agent, losartan, resulted in a significant augmentation of nanoparticle penetration towards the target cells [70]. However, the decreased collagen I content induced a switch in cancer stem cells (CSCs) from slow growing, avascular-type cells to fast-growing, highly autophagic endothelial-like cells, creating a favorable mechanism for tumor progression and chemoresistance to gemcitabine [63,71]. Altogether, these data suggest that the correct manipulation of ECM collagen composition may be able to enhance the accessibility of drugs to tumor tissues and decrease chemoresistance. However, the role of hyaluronan in physiological chemoresistance is not well established and there is no consensus. While some reports have shown that total tissue hyaluronan content is not correlated with tissue elasticity and hydraulic conductivity [72], another study demonstrated that ECM molecular selectivity is regulated by variation of the hyaluronan content, thus affecting molecule penetration based on charge and size [73]. In line with this second paper, the depletion of hyaluronan in a PC3 xenograft model decreased tumor interstitial fluid pressure and increased vascular area, suggesting that hyaluronan might have an important role in blocking the penetration of chemotherapeutic agents [74]. Regarding PDAC, more studies are needed to disclose whether hyaluronan content may have a role in tissue elasticity and mechanobiology of ECM, and how this can be correlated with drug perfusion and chemoresistance.
2.3. Targeting the Desmoplasia Improves Chemotherapy Outcomes
Several stroma-targeting drugs are currently being tested as new treatment strategies to reduce chemoresistance in PDAC. One of the more appealing targets of the tumor stroma is the potent cytokine TGFβ which regulates developments, differentiation, and homeostasis in mammalian [75]. TGFβ binds to TGFβ receptor 1 or 2 to inhibits cell proliferation, motility, invasion, EMT, and metastasis [76] and its tumor-inhibitory effect is controlled by Smad-dependent TGFβ signaling [77]. Indeed, in human prostate cancer, overexpression of TGFβ1 correlates with collagen I levels, suggesting that TGFβ can be directly linked to the desmoplastic process [35]. Furthermore, knocking down Smad 3 abolishes collagen fibrosis induced by the EMT-regulator Snail, hence corroborating the role of TGFβ as a crucial signaling pathway in the propagation of the desmoplastic reaction [78]. Moreover, Smad4 is frequently mutated in PDAC and this could be one of the reasons for the resistance to the growth inhibitory effects of TGFβ [35]. In this line, some TGFβ receptor 1 inhibitors, such as SB431542 and SB525334, have been developed and tested in combination with gemcitabine, resulting in a higher cytotoxic effect compared to gemcitabine alone due to an increased delivery and penetration of the drugs into the tumor (desmoplastic) tissues [79].
Moreover, the hyaluronan cell surface receptor CD44 has a critical role in pancreatic carcinogenesis [80] such that disruption of the hyaluronan-CD44 complex is a crucial therapeutic target to prevent PDAC drug resistance [80]. Indeed, the compound, 4-methylumbelliferone (4-MU), by inhibiting hyaluronan synthesis and accumulation on cancer cells and their surrounding stroma, blocked cell proliferation, migration, and spreading in several tumor cell types [81,82] and reduced bone metastases in breast cancer (Figure 2) [83]. In both PDAC cell lines and in vivo, 4-MU slowed the development and progression of the disease and also increased tumor response to gemcitabine [84,85,86]. In a similar way, PEGylated human recombinant PH20 hyaluronidase (PEGPH20) acted as a hyaluronan “consumer” enhancing the delivery of chemotherapeutic agents such as doxorubicin and gemcitabine [55,74]. Injection of PEGPH20 into KPC mice tumors rapidly degraded hyaluronan, restored the patency of intra-tumoral vessels, increased vessel diameter and highly reduced the high interstitial fluid pressure within the tumors to normal levels [12]. Importantly, these alterations increased the macromolecules permeability and the combined treatment of gemcitabine with PEGPH20 remarkably improved treatment efficacy by reducing metastases and doubling the median survival time in mice [87].
Further, the enzymatic degradation of ECM components has also been proposed as a possible complementary therapeutic approach against PDAC. Several studies using hyaluronidase showed improved cancer sensitivity to chemotherapeutics and enhanced permeability to drugs in cultured multicellular spheroids [88]. Similarly, collagenases have also displayed beneficial characteristics by increasing the penetration of macromolecules. However, their sensitivity to and stability at physiological pH might be a therapeutic hindrance that has not yet allowed this enzyme to be clinically available [89]. We have proposed a double edged targeting of the hyaluronan-CD44 pathway by combining 4-MU as hyaluronan production inhibitor and bromelain as CD44 inhibitor [90], since this combination has not been experimentally tested yet. Lastly, Hedgehog (Hh) is a signaling pathway that is abnormally activated in most pancreatic cancers leading to cancer initiation, progression and metastatic development [91,92]. More recently, it has been related to the beginning and maintenance of the desmoplastic reaction. Indeed, Hh stimulates the differentiation of myofibroblasts and induces stroma-derived growth promoting molecules [93,94]. There is also some evidence that supports the existence of an interplay between Hh signaling and TGFβ, both being tightly connected with the desmoplastic reaction and involved in fibrosis [94]. Further, it was demonstrated that blocking Hh signaling, both in vitro and in vivo, with the small molecule compound, cyclopamine, markedly improved drug delivery and abrogated pancreatic metastasis [95,96]. Multiple studies have been conducted to verify whether the combined treatment of Hh signaling inhibitors with chemotherapeutic agents can have synergistic anticancer effects [97]. Indeed, the inhibition of the Hh signal with the semisynthetic analogue of cyclopamine, IP-926, reduced the desmoplastic reaction and improved tumor vascularity [98]. A phase Ib trial demonstrated that IPI-926 reduced tumor desmoplasia and increased gemcitabine delivery, such that 31% of patients displayed a partial response and 63% of them showed a reduced expression of the marker Carbohydrate Antigen 19-9 (CA 19-9) in their tumor tissues [99].
In summary, the desmoplastic reaction creates a unique microenvironment, which stimulates tumor growth/progression and forms a “physical barrier” to chemotherapy permeability. Hence, several strategies have arisen to improve chemotherapeutic efficacy by blocking or interfering with the desmoplastic process and enhance tumor penetration, accumulation, and drug distribution. Thus, targeting stroma components of the desmoplastic reaction might be a promising new area of investigation in pancreatic cancer treatment.
Table 1 lists all the drugs that might be employed against the desmoplastic reaction.

Table 1.
List of useful drugs for targeting desmoplastic reaction-mediators.
3. The Role of Extracellular Acidic pH and Hypoxia in the Resistance to Therapy
In recent years, numerous studies on PDAC have focused on the biologic or metabolic TME, which plays a role in tumor malignancy [155]. The TME represents an important factor that allows tumor growth and survival of the most aggressive cells, leading to chemoresistance and metastatic behavior. The pattern of an acidic extracellular environment together with an alkaline cytosol is considered a hallmark of malignant cancers and is referred to as a “reversed pH gradient” [156]. The intracellular alkalinity confers a proliferative advantage for the malignant cells, while a low nutrient supply, extracellular acidic and hypoxic conditions may contribute to the progression from benign to malignant growth and induce the selection of more aggressive tumor cells capable of withstanding this hostile acidic and hypoxic microenvironment [157]. The new blood vessels (angiogenesis) forming in a tumor are not properly formed and are often twisted and abnormal (convoluted). This defective structure leads to a poor ability to deliver oxygen and remove metabolic waste products, resulting in the development of acidic conditions [158]. In the next subsections, the role of both acidic pHe and hypoxia on PDAC chemoresistance will be discussed.
3.1. Extracellular Acidic pH Driving Tumor Progression
The pHe of tumor tissues is often acidic [159] and this phenomenon is the result of multiple factors such as: (i) increased CO2 production, which is converted into carbonic acid by membrane Carbonic Anhydrases IX and XII (CAIX and CAXII) [160]; (ii) increased lactate production as a consequence of high glycolytic flux and aerobic glycolysis [161]; (iii) extrusion of this lactate by monocarboxylate transporters (MCTs) [162] and (iv) active translocation of cellular protons into the ECM mainly by sodium-hydrogen exchanger isoform-1 (NHE1) [163]. Hence, all these mechanisms are altered in tumors and lead to low pHe (~6.7–7.1) compared to normal tissues (7.4) [164], and to an alkaline intracellular milieu [164]. This inverted pH gradient may initiate and then drive the further development of the neoplastic process [165].
The acidic pHe has a number of important consequences that are directly related to cancer [157,166]. One of the direct consequences of the extracellular acidity is the degradation of the ECM by the activation of proteolytic enzymes, such as metalloproteases and cathepsins [167,168]. This mechanism is associated with migration, invasion, and metastasis [169,170]. In several tumors, including PDAC, the acidic microenvironment promotes the activity of metalloprotease 1, 2, and 9 [171] and drives EMT, invasion, and metastasis [172]. Indeed, the exposition of PDAC cells to low pHe promotes the invasive phenotype [172] by stimulating the expression of mesenchymal markers, such as N-cadherin, and reducing the expression of epithelial markers such as E-cadherin [172]. In this way, acidic pHe may also affect other processes such as angiogenesis, evasion from the immune system, and drug resistance [173,174].
3.2. Multidrug Resistance and the Acidic Tumor Microenvironment
One of the main therapeutic problems to solve in cancer is the frequent emergence of drug resistance, which can be enhanced by the acidic TME [175,176,177,178].
One of the mechanisms modulating the entry of an ionic drug into the cell is the existence of a reversed extracellular to intracellular pH gradient compared to normal cells. The pHe in normal cells is generally around 7.32 with a slightly more acidic pHi, about 7.10–7.20 [179]. This pHe to pHi gradient usually allows weakly basic drugs to enter passively into the cells. This inverted pH gradient has a crucial role in the “ion trapping” hypothesis, which predicts that weakly basic chemotherapeutic drugs such as anthracyclines, anthraquinones and vinca alkaloids will concentrate in more acidic cell compartments, such as the extracellular fluid [180,181]. Therefore, the acid pHe of tumors will effectively hinder weak basic drugs from reaching their intracellular target, thereby reducing cytotoxicity. The ion trapping model also predicts that the acid pHe of tumors will improve the uptake of weak acids such as chlorambucil [182]. The weak basic chemotherapeutic drugs will be seriously impaired by the inverted pH gradient. Acidic and neutral drugs will not be influenced negatively. Multidrug resistant cancer cells display an even higher inverted pH gradient than non-resistant cells [183]. Acidic pHe induces the activity of MDR and a series of related-effects including reduction in cell cycling fraction, selection for an apoptosis-resistant phenotype and increased ion trapping [184,185]. Extracellular acidity was previously considered to be directly related to an excessive lactic acid production, while nowadays it is clear that it is strongly dependent on CO2 production and the hyperactivation of proton extruders such as NHE1 and NHE3, vacuolar ATPase proton pump, and membrane enzymes such as CAIX and CAXII and the multidrug transporter P-glycoprotein [186,187,188] (Figure 3 and Figure 4). Therefore, the inhibition of acidity through, for example, sodium bicarbonate or through proton pump inhibitors has been shown to confer greater susceptibility to chemotherapy in tumor cells [189].
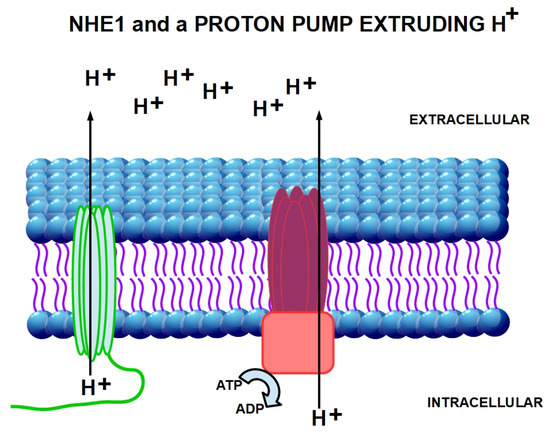
Figure 3.
Activity of the proton extruders NHE1 ATPase proton pump. On the left side in green, it is represented the active secretion of cellular protons into the extracellular space by NHE1. On the other side in red, the active extrusion of cellular protons into the extracellular space by V-ATPase proton exporter is shown. Interestingly, both extrude protons against the gradient; however, while proton pumps need energy (ATP) for their activity, NHE1 does not need ATP to achieve the same purpose.
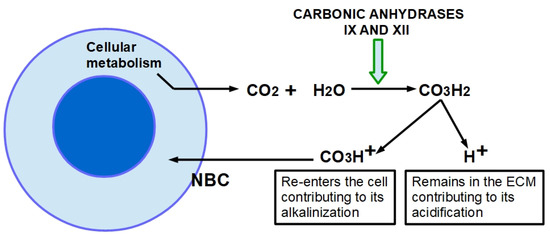
Figure 4.
Contribution of carbonic anhydrases to tumor acidification. The cellular metabolism produces an excess of CO2 that diffuses from the cell into the extracellular space. Membrane carbonic anhydrases IX and XII convert it in carbonic acid (CO3H2) through hydration. CO3H2 spontaneously ionizes into a molecule of ionized hydrogen (proton) that remains in the matrix, contributing to its acidification. The bicarbonate ion is reintroduced into the cell through the activity of the sodium bicarbonate cotransporter (NBC) contributing to cytoplasmic alkalinity.
3.3. Hypoxia: A Promoting Factor in Cancer Survival and Proliferation
The term hypoxia means a significant reduction in oxygen tissue levels, which characterizes 50–60% of locally advanced solid tumors, such as PDAC. PDAC tissue has a partial oxygen pressure, with the median pO2 of 0–5.3 mmHg (0–0.7%), while the median pO2 in the normal pancreas is 24.3–92.7 mmHg (3.2–12.3%) [190,191]. Since the seminal works by Semenza and Wang [192,193], we know that the main molecular mechanism by which oxygen homeostasis is maintained in tissues is regulated by hypoxia-inducible factors (HIFs) [194]. There are three isoforms HIF-1α, HIF-2α, and HIF-3α, and each of them can heterodimerize with HIF-1β and form HIF-1, HIF-2, and HIF-3. HIF-1, the most important, is a heterodimer consisting of a constitutively expressed HIF-1β subunit and an oxygen-regulated HIF-α [195]. During normoxia, HIF-1α protein levels are negatively regulated by the ubiquitin ligase Von Hippel Lindau protein (VHL) which polyubiquitinates HIF-1α for its rapid proteasomal degradation. Hydroxylation of HIF-α by prolyl hydroxylases (PHD1-3) in the presence of oxygen is required to promote its polyubiquitination by pVHL. In contrast, under hypoxic conditions PHDs do not hydroxylate HIF-α which is therefore not ubiquitinated by pVHL. Non-hydroxylated HIF-α accumulates and translocates into the nucleus to dimerize with HIF-β [196]. The heterodimer HIF1α:HIF1β induces a large number of downstream transactivating genes that will allow the cellular adaptation to low oxygen levels.
Among these genes are those that code for erythropoietin (EPO), enzymes of the glycolytic pathway such as GLUT1, the pro-angiogenic vascular endothelial growth factor (VEGF), the platelet-derived growth factor (PDGF) and NHE1 [197,198,199] (Figure 5).
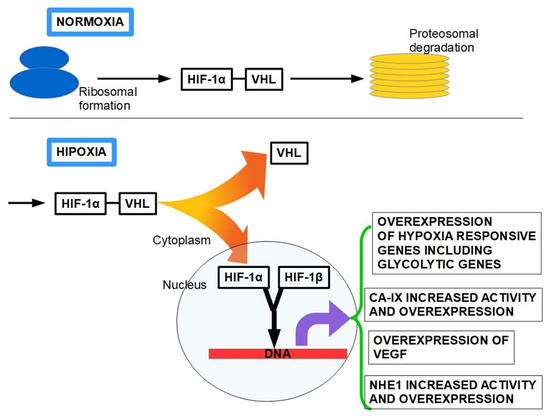
Figure 5.
The “fate” of the HIF-1α protein with the oxygen tissue level. Upper panel: HIF-1α is unstable in normoxia because due its binding to the VHL protein, it is carried to proteasomal degradation. Lower panel: The situation changes under hypoxic conditions. When HIF-1α is released from VHL (stabilization) and translocates to the nucleus, it dimerizes with the constitutional HIF-1β. This dimer acts as a transcription factor for a set of genes that contain a Hypoxia Responsive Element (HRE) sequence in their promoter region. On the right side there are some of the genes that are promoted by the dimer.
The increased expression of VEGF and, therefore, angiogenesis due to HIF-1α could promote the progression of pancreatic tumors [200]. PDAC has been shown to have reduced vascularization compared to the normal pancreatic tissue [201]. Hypoxia in tumors increases with the distance from the nearest capillary blood network, while the proliferative index of tumors decreases [202]. Indeed, tumor growth and size are strongly influenced by vascularization and angiogenesis. The hypoxic environment occurs when there is a high oxygen demand of the cancer cells, and poor lymphatic drainage; consequently, the growth rate of the tumor is greater than the rate of new vessels [203]. Recognized as a hallmark of most solid tumors, hypoxia profoundly influences multiple facets of cancer biology, through the HIF-1 mediated induction of metabolic reprogramming, neovascularization, EMT and metastasis [204,205]. Furthermore, it has been shown that HIF-1α expression increases pancreatic cancer cell motility and metastasis and it is associated with a negative prognosis [206,207]. Importantly, in PDAC cells, hypoxia increases the formation of invasive protrusions, known as invadopodia, and their mediated-focal ECM proteolysis, thereby increasing tumor aggressiveness [208,209,210].
EMT is a morphologic cellular program simply defined as the phenotypic transition from a stable epithelial state to a mesenchymal state, with “pro-metastatic” characteristics [211]. Numerous studies have demonstrated that the invasive capacity of PDAC correlates with EMT [212,213] together with their increased ability to migrate while remodeling the ECM barriers (e.g., invasion). These ECM barriers include the epithelial and endothelial basement membranes and the interstitial collagen rich stroma [214]. The hypoxic environment promotes EMT and cells undergoing EMT acquire stem-like features as well as drug resistance in different types of cancer, including PDAC [215]. PDAC stroma is frequently hypoxic, and the PSCs respond to hypoxia by increasing HIF-1α expression [216,217], motility and alpha-smooth muscle actin expression. Furthermore, increased HIF-1α expression stimulates the synthesis of collagen I, fibronectin, and periostin indicating a correlation between hypoxia and the densely fibrotic desmoplastic reaction [201,218] (Figure 6). In addition, the desmoplastic reaction can be considered as an effector of hypoxia, which, in turn, activates invasion and metastasis. Among the various effects of hypoxia on cancer malignancy, there is also the reduction in the effectiveness of chemo and radiotherapy. Since the existence of this malignant interplay between desmoplasia and hypoxia, further studies are required to establish a possible synergetic therapy by using modulators of both pro-carcinogenic events.
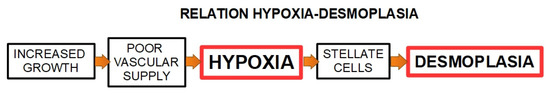
Figure 6.
Schematic representation of the relationship between hypoxia and desmoplasia.
3.4. Hypoxia: Induction of Chemoresistance in Cancer Cells
Hypoxia is an important hindrance in the development of successful cancer chemotherapies [219,220]. It is believed that hypoxia and HIFs can mediate chemotherapy resistance through mechanisms such as the extrinsic resistance, the regulation of drug efflux, metabolic reprogramming, alterations in apoptosis and cell survival, and induction of stemness. Recent reports have demonstrated that hypoxia selects for cells with increased apoptotic resistance to chemotherapeutic drugs by the overexpression of Bcl-2 and/or the downregulation of proapoptotic proteins (BNIP3, NOXA, and NIX), together with a diminished drug sensitivity through upregulation of the MDR protein expression [221,222,223,224]. Indeed, in PDAC cells it was observed that hypoxia enhanced apoptosis resistance induced by gemcitabine via PI3K/Akt/NF-kappa B pathways and partially through the MAPK(Erk) signaling pathway [225]. Other studies have shown an association between HIF expression and the regulation of drug efflux. HIF-1α stimulates the expression of the MDR1 gene coding for P-glycoprotein 1, a predominant membrane transporter associated with chemotherapy resistance [226] which is also involved in the reduction of intracellular level of drugs, such as paclitaxel and anthracyclines [227].
Another mechanism by which hypoxia induces chemoresistance is through metabolic reprogramming and the modulation of reactive oxygen species (ROS) production [228]. For example, Mayer Y. Abdalla et al. suggested that in pancreatic cancer cells hypoxia could upregulate Heme oxygenase 1 expression [229]. Inhibiting Heme oxygenase 1 with zinc protoporfiphyrin and tin protoporphyrin IX increased both ROS production and apoptosis, thus sensitizing pancreatic cancer cells to gemcitabine [229]. Moreover, the glycolytic enzyme enolase 1 alters ROS production and promotes chemoresistance [230].
3.5. Therapeutic Strategies Targeting the Acidic Extracellular pH and Hypoxia
As the contribution of the TME to the lethal outcomes of PDAC is substantial, the complex relationship between acidic pHe, hypoxia, ROS, and treatment resistance requires further research. Consequently, hypoxia and HIF-1α are key factors to have in mind while studying new therapeutic strategies. One of the possible strategies might be improving tumor perfusion through anti-fibrotic therapies, since PDAC is one of the most desmoplastic epithelial tumors [231]. Indeed, the addition of PEGPH20 to nabpaclitaxel/gemcitabine doubled the progression-free survival in a group of patients who had tumors with high hyaluronic-acid content [232]. However, it is necessary to better understand the functional characteristics of the stroma in order to be able to apply therapy to different cell types such as PSCs, that are the dominant producers of VEGF and the main contributors to the fibrotic/hypoxic milieu through abnormal ECM deposition [201]. Another strategy to target hypoxia-induced pathways in PDAC is by inhibiting HIF-1α signaling. A plant-derived agent, triptolide, has been shown to decrease both HIF-1α levels as well as the CSCs subset of PDAC [233,234]. One of its derivatives is now being tested in advanced gastrointestinal cancers including PDAC [235]. While the well-known cardiac glycoside, digoxin, which effectively inhibits HIF-1α synthesis at a relatively low concentrations, could be used as a sensitizer to reverse chemoresistance in PDAC, there are no registered clinical trials testing this hypothesis [236]. In a recent study, Lang and colleagues showed that a compound extracted from melphalan (PX-478) combined with arsenic trioxide could be a promising strategy to promote ROS-induced apoptosis in the treatment of PDAC [237]. Another therapeutic mechanism could be to block the signal transduction that upregulate HIF-1α expression [238]. In this regard, both Mek/Erk pathway and mTOR play a role in the regulation of HIF-1 expression. While the agent, everolimus, alone had minimal clinical activity in gemcitabine-refractory PDAC [239], when in combination with capecitabine, displayed a moderate clinical response. This indicates that the combination of mTOR inhibition with another targeted therapy or cytotoxic agent may show clinical benefits [240]. Moreover, the high production of ROS stabilizes HIF-1α and, consequently, the attenuation of this production with antioxidants or superoxide dismutase can inhibit tumor growth and metastasis [241,242]. However, decreasing ROS levels also reduces the effects of chemotherapeutic drugs [243]. In order to improve or develop new therapies in PDAC, it could be important to identify a subset of patients and cancer cell subpopulations expressing and hypoxia markers for HIF-1α -interfering substances. A better understanding of the mechanisms related to acid microenvironment and hypoxia is the basis for clinical and therapeutic improvements of PDAC in the future.
4. The Tumor Metabolic Microenvironment Promotes Resistance against Chemotherapy
4.1. Metabolic Rewiring and Nutrient Scavenging in Cancer Cells
Lack of vascularization in the tumor core not only causes hypoxia, but it also triggers metabolic stress due to nutrient deprivation. In the last two decades, metabolic reprogramming has been recognized as one of the hallmarks of cancer cells [244]. It has been proposed that cancer cells display a higher metabolic rate than their normal counterparts. Moreover, tumor cells are able to use glucose, glutamine, fatty acids, and even other amino acids as substrates to support their energy needs [245,246]. Cancer cells have the ability to reprogram their metabolism through several different ways with the purpose of supporting their unrestricted proliferation. As a result of their metabolic reprogramming, cancer cells quickly adapt to the characteristic physical changes occurring in the microenvironment, which reciprocally contributes to the heterogeneous cellular metabolic landscape of the tumor niche [247]. The most-well known metabolic alteration in cancer cells is the Warburg effect which postulates that cancer cells have enhanced aerobic glycolysis and less glucose oxidation compared to their normal counterparts [248]. Therefore, tumor cells have a higher flux through the pentose phosphate pathway, the anabolic side branches of glycolysis [249]. Indeed, the most important metabolic change observed in cancer cells is the shift of fuel use through anabolic pathways, with the objective to provide the cells with enough substrates to increase biomass [250].
As mentioned before, it is well known that PDAC is a hypovascular tumor, which results in inadequate tumor perfusion, meaning less availability of glucose, amino acids, and lipids [251]. To overcome this obstacle, cancer cells frequently exploit various scavenging strategies to harvest macromolecules from the microenvironment and break them down in the lysosome, creating substrates for ATP generation and anabolism [252]. Autophagy is the major mechanism used for nutrient scavenging by cancer cells. Moreover, it is a cellular mechanism which culminates in the lysosomal degradation of intracellular material and provides metabolic and cellular homeostasis through the recycling of cytoplasmic elements to cellular building blocks [253]. The subsequent autophagosome after merging with the lysosome delivers the recycled material back to the cytosol, via the degradation of its cargo [254,255]. Indeed, autophagy is a key cellular process since its dysfunction is associated with several disorders, including neurodegenerative diseases, inflammation, and cancer [256]. In PDAC, it has been demonstrated that autophagy is increased compared with normal cells/tissues [257]. Metabolism has been proposed as the possible mechanism with which autophagy may interact and the result of this interplay contributes to PDAC progression. This interplay relies on the ability of the autophagy to feed the metabolism by supplying recycled intracellular components, and this promotes a pro-tumorigenic effect [257]. Since autophagy is able to degrade a substantial range of substrates, it is evident that this cellular mechanism has the potential to fuel almost all pathways in central carbon metabolism. Therefore, the resultant metabolic plasticity confers to these tumors a survival advantage in the harsh TME which is characteristic of PDAC [258]. In PDAC cells, autophagy inhibition results in an impaired mitochondrial function, causing a reduced oxidative phosphorylation and a consequent fall in ATP levels [258].
Importantly, autophagy is not able to form a new biomass, since these cells are also degrading themselves [253]. To overcome this disadvantage, PDAC cells also rely on other lysosomal-dependent pathways to fuel their high metabolic requirements. Macropinocytosis consists in the engulfment and uptake of large amounts of extracellular fluid, containing protein, lipid, virus and bacteria [259]. This mechanism culminates with the release of the digested cargo into the cytosol; however, contrary to autophagy, the breakdown of this macromolecular cargo into their monomeric constituents will create a new intracellular source of diverse nutrients, leading to an increased biomass [260]. Interestingly, nutrients obtained by macropinocytosis have been demonstrated to display a crucial role in PDAC metabolism. Some reports demonstrated evidence of macropinocytosis occurring in human PDAC tumors [261]. It was shown that PDAC cells can take up and degrade collagen from the ECM through macropinocytosis [262] and, importantly, the collagen-derived proline contributed to central carbon metabolism and promoted PDAC cell survival even under nutrient poor conditions [262].
Overall, these studies demonstrate the critical role of recycling and scavenging mechanisms in modulating PDAC metabolism and its subsequent uncontrolled growth in the harsh tumor microenvironment.
4.2. Fuel Source Plasticity towards Resistance to Therapy
4.2.1. Glucose
Glucose is the primary metabolic energy source for sustaining several biochemical processes, including cancer cell proliferation by both supplying carbon for anabolic reactions and by generating ATP [263]. Cancer cells essentially rely on the “Warburg effect”, in which cells depend on mitochondrial oxidative phosphorylation to produce energy for cellular processes, rather than on mitochondrial aerobic glycolysis [264]. Although this change in glucose metabolism is less efficient in producing cellular energy, it confers a survival advantage to cancer cells via a rapid increase in ATP production [265]. Recently, a new two-compartment model has emerged, the “reverse Warburg effect”, in which cancer cells stimulate aerobic glycolysis in the stromal cells, whose glycolysis end-products are then used by cancer cells to feed mitochondrial oxidative phosphorylation [266]. The heterogeneity of PDAC TME might also be partially explained by this model, where the glycolytic differentiated cancer cells could provide substrates to oxidative CSCs thus creating a symbiotic relationship with them [267].
The shift towards enhanced glycolysis as a supplier of ATP, reduces neovascularization creating an adverse milieu, where both oxygen and nutrients are restricted [190,268]. These extreme microenvironmental conditions exert a drastic selective pressure on cancer cell growth and survival leading to the expansion of the most aggressive cellular clones. However, to overcome these stressful conditions, PDAC cells are forced to reprogram their metabolism with the objective to handle their bioenergetic demands for their proliferation and spread towards less harsh environments [217]. These metabolic changes are directly linked to the aberrant activity of specific oncogenes which drive the switching of nutrient preference [269]. Mutations in KRAS and other oncogenes (such as MYC) and tumor suppressors (TP53, RB and PTEN) have been identified as the principal drivers of PDAC reprogramming cellular metabolism towards enhanced cancer growth [270].
Indeed, oncogenic KRAS supports substantial alterations in the glycolytic pathway, including the upregulation of the glucose transporter (GLUT1) as well as the enzymes hexokinase (HK1 and HK2), phosphofructokinase-1 (PFK1) and lactate dehydrogenase (LDHA), aiming to satisfy the increased necessity for glucose required in PDAC [271,272]. Moreover, another metabolic advantage promoted by KRAS is the synthesis of monomeric constituents essential for cancer cell proliferation, namely amino acids, and nucleic acids, by deviating glucose toward anabolic pathways, including the pentose phosphate pathway [271]. As expected, a significant reduction in glucose uptake and consequently in the glycolytic flux was observed after silencing the oncogenic KRAS gene in tumors [271,273].
Another metabolism-related gene and one of the major genetic alterations in pancreatic cancer is TP53, which is mutated in >70% of all PDAC cases. TP53 contributes to the glycolytic shift via upregulation of GLUTs, particularly GLUT1 [274]. At the end of the glycolytic pathway, the generated pyruvate is mainly converted to lactate instead of undergoing oxidative phosphorylation, a change driven together with KRAS-mediated upregulation of LDHA [271]. Moreover, LDHA plays a key role in the renewal of the glycolytic cofactor NAD+, supporting the increased NAD+/NADH ratio and allowing an intensified glycolysis in cancer cells [275]. These common observed metabolic changes in cancer cells provide the necessary substrates to maintain the enhanced glycolytic flux and lactate production. Interestingly, lactate was recently discovered to be more than a waste product of glycolytic metabolism as it can be used as an energy source [276]. According to this mechanism known as “lactate shuttle”, glycolytic cancer cells generate lactate, which is extruded to the extracellular environment by the lactate transporter MCT4. Lactate is then taken up by oxidative cancer cells expressing MCT1, thus conserving the available glucose for glycolytic cancer cells [277,278]. Last but not least, microenvironmental acidosis due to protons extruded by the cell via these lactate/H+ cotransporters, contributes to the suppression of immune cells by supporting chronic inflammation, while suppressing the T-cell mediated adaptive immune response [174]. It is now becoming clear that the co-presence of lactate and acidic pH, being closely linked with chemoresistance, is associated with poor prognosis, metastasis and more aggressive tumor phenotypes [279].
4.2.2. Glutamine and Other Amino Acids
Another challenge faced by PDAC cells is the lack of amino acids in their characteristic harsh nutrient-poor microenvironment. To counteract this amino acid depletion and support their metabolic demands, cancer cells utilize different processes. The most abundant free amino acid in humans is glutamine and recent studies have demonstrated that glutamine can be used by cancer cells to support anabolic processes to fuel proliferation [280]. Indeed, the TCA cycle is continuously supported by glutamine-derived carbon in cancer cells [281]. Recently, it has been demonstrated that PDAC cells are able to use a non-canonical pathway of glutamine to satisfy their needs for tumor growth [282]. Indeed, most KRAS-mutated PDAC cells utilize glutamate dehydrogenase to convert glutamine-derived glutamate into α-ketoglutarate in the mitochondria to fuel the TCA cycle. On the other hand, in these cells mitochondrial glutamine-derived aspartate is transported via the mitochondrial uncoupling protein 2 (UCP2) from the matrix to the cytosol, where it is converted into oxaloacetate by aspartase transaminase [283]. One of the resultant products of this pathway is NADH, which helps in the maintenance of the cellular redox state, as well as in the production of metabolites needed for a de novo synthesis of macromolecule and lipids [284]. Interestingly, knocking down UCP2 in KRAS-mutated PDAC cells, results in strong suppression of tumor growth both in vitro and in vivo [273]. Furthermore, a strong relationship between glucose and glutamine metabolism has been reported, in which cell survival in the acidic TME triggered by lactate production from heightened glycolysis relies on amplified expression of aspartate transaminase and on the above mentioned non-canonical glutamine pathway [285]. Indeed, disruption at multiple steps of this pathway results in redox imbalance and diminished cellular proliferation [284]. Besides glutamine, several reports have shown a strong correlation between elevated plasma amino acids (leucine, isoleucine and valine) and pancreatic cancer risk [286]. Importantly, an intensified consumption of amino acids may arise about 10 days before PDAC diagnosis, suggesting that this increased plasma amino acids concentration should be considered as pre-diagnostic and diagnostic tool [287]. All these findings suggest that the level of some amino acids could be used as a tool to stratify PDAC patients after diagnosis, also considering the significant differences in their expression observed between normal versus malignant tissue and different disease stages [287].
The bidirectional interaction cancer cells–stromal cells in PDAC stroma represents another source of amino acid uptake for PDAC cells. Indeed, a large portion of the alanine, used by cancer cells to fuel their glutamine and glucose metabolism is secreted by PSCs [288,289]. Moreover, CAFs display an up-regulated amino acid catabolism and are able to fuel PDAC cells with branched-chain α-ketoacid [290]. This cellular communication between PDAC cells and the surroundings is extremely dependent on the expression of the L-type Amino Acid Transporter or the Cystine/Glutamate Exchanger, linking the expression of amino acid transporters to poor prognosis and drug resistance [291,292].
Recently ferroptosis, a regulated cell death mediated by iron accumulation and lipid peroxidation, has been recognized to have an important role in PDAC progression and treatment response; however, the underlying mechanisms are still not completely understood [293,294,295,296,297,298]. Interestingly, cystine handling/metabolism and particularly the cystine transporter (xCT) has recently been recognized to play a role in PDAC ferroptosis [299,300]. xCT knockdown [292] or inhibitors [301,302] have been studied as therapeutic compounds although there is some evidence that it could be a double-edged sword [303,304]. This is an interesting aspect of overcoming therapeutic resistance that deserves further study.
4.2.3. Lipids and Fatty Acids
Lipids and fatty acid metabolism are becoming a promising object of research in cancer metabolism. Indeed, lipid synthesis is involved in sustaining the bilayer lipid membrane formation, especially controlling the fluidity and shape of the membrane, producing a denser membrane which might diminish the uptake of anticancer drugs and promote chemoresistance [305]. Moreover, lipids are involved in signal transduction by building lipid rafts regulating protein recruitments and interactions, as well as by the production of lipidic signaling molecules [305]. Lastly, ATP production through beta-oxidation is necessary to support cancer cell division and proliferation and it is highly dependent on lipidic metabolism [305,306,307,308]. Fatty acids have a vital role in supporting PDAC development and cancer cell growth [309]. Compared to the normal pancreas, the pancreatic TME is thought to be deficient in lipid levels, which can be explained either by the scarcity of lipids or excess utilization [310]. Curiously, it has been observed that PDAC cells have an augmented tendency to take up exogenous cholesterol through elevated levels of LDLR [311]. Additionally, there is also evidence in both in vitro and in mouse models of PDAC that a high fat diet is able to increase PDAC cell growth [312,313]. Overall, these reports support the theory that exogenous lipids are beneficial for PDAC tumors.
In PDAC, normal human pancreatic ductal epithelial cells with the mutated KRAS oncogene enhance their lipid uptake from the environment [314]. Furthermore, fatty acid synthase (FASN), a pivotal enzyme in fatty acid synthesis, is upregulated by EGFR/ERK signaling and its inhibition is detrimental to PDAC cells [315]. It seems that the enhanced ability of PDAC cells to either take up or synthetize lipids and fatty acids plays a key role in cancer progression and development. In fact, high transcriptional levels of FASN or the low-density lipoprotein receptor (LDLR), responsible for the uptake of cholesterol, are correlated with poor overall survival or reoccurrence in PDAC, respectively [316].
It has been shown that lipid synthesis accounts for 75–90% of the cellular palmitate pools in PDAC [314]. Most cancer types need high levels of acetyl-CoA to sustain their high rates of fatty acid synthesis and to maintain this high demand there are different sources of acetyl-CoA production [317]. The first process of acetyl-CoA synthesis is due to the citrate metabolism. Citrate is exported from mitochondria via the tricarboxylate transporter SLC25A1 and in the cytosol is metabolized by ATP citrate lyase to produce acetyl-CoA and oxaloacetate [318]. Another pathway for acetyl-CoA production is related to acetate metabolism obtained from the diet or other external sources. Alternatively, MCT1/4, which are mainly recognized as transporters of lactate and pyruvate, were discovered to act also as transporters of acetate into the cell [319,320]. Indeed, these transporters are upregulated in PDAC, and it is believed that, besides being associated with lactate shuttle these transporters might also be associated with de novo lipid synthesis [321].
Additionally, it has been demonstrated that PDAC cells consume acetate from the environment and the ability to take it up is determined by the expression of acyl-CoA synthetase short-chain 2 [322]. The following steps in fatty acid synthesis consist of the action of the enzyme acetyl-CoA carboxylase (ACC), converting acetyl-CoA into malonyl-CoA, producing C16 palmitate through reactions catalyzed by FASN. The resultant palmitate has multiple intracellular functions, involving the regulation of signaling networks in PDAC. Therefore, interfering with these networks might help to mitigate PDAC growth [323].
Besides the above-mentioned direct effects, there is an increased complexity of lipid metabolism in PDAC, since some lipids and fatty acids have a role in supporting PDAC tumor progression while others seem to have anti-tumor effects. Indeed, while obesity and dietary factors are established risk factors for PDAC, there are some fatty acids that displayed anti-tumor effects by decreasing tumor cell proliferation [324]. Furthermore, polyunsaturated fatty acids of the n-6 variety are considered to promote the proliferation of PDAC cells, whereas n-3 polyunsaturated fatty acids have the opposite effect [325]. Overall, it is mandatory to further understand the mechanisms involved in the regulation and response to different fatty acids with the objective to disclose and identify novel therapeutic and treatment approaches for PDAC treatment.
Figure 7 summarizes all the metabolic pathways discussed above.
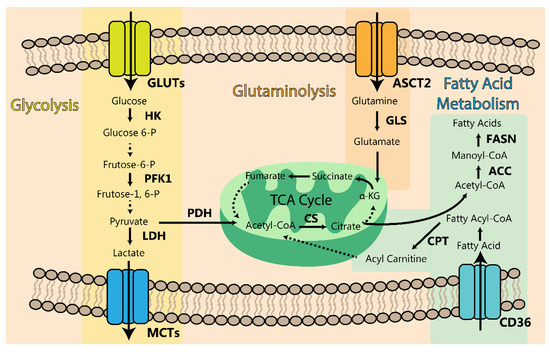
Figure 7.
Schematic representation of the discussed metabolic pathways in PDAC. The glycolytic pathway (yellow shade), glutaminolysis (orange shade), and the fatty acid metabolism (blue shade) are represented. The enzymes and transporters (bold) are the key intermediated targets which can be envisaged for new promising therapeutic strategies. Dashed arrows indicate more reactions not explored in the review. Legend: glucose transporters (GLUT1 and GLUT3), hexokinase (HK), phosphofructokinase 1 (PFK1), pyruvate dehydrogenase (PHD), lactate dehydrogenase (LDH), monocarboxylate transporters (MCT4 and MCT1), fatty acid transporter CD36, fatty-acid synthase (FASN), acetyl-CoA carboxylase (ACC), carnitine palmitoyl transferase (CPT), citrate synthetase (CS), ASCT2 (glutamine transporter) and glutaminase (GLS).
4.3. Targeting Metabolism to Overwhelm Chemoresistance
As described above, multiple metabolic changes, driven by genetic and epigenetic factors, have been associated to drug effects and clinical outcome, strengthening the hypothesis that cancer metabolism is deeply related with chemoresistance [326]. Moreover, metabolic reprogramming can promote several key tumor features, thus shaping cancer cell differentiation, proliferation and/or apoptosis, as well as therapeutic response [327]. Various enzymes and transporters that participate in the multiple metabolic pathways have been implicated in stimulating the drug resistant phenotype. The next sub-sections will discuss, in detail, the most important alterations leading to chemoresistance and the possible benefits of manipulating the metabolism as a complementary therapy in PDAC.
4.3.1. Glucose Transporters (GLUT Family)
GLUT1 is an ATP-independent transmembrane protein, and it is responsible for the facilitated diffusion of glucose across the plasma membranes of mammalian due to a glucose gradient from the extracellular compartment to the cytoplasmic compartment [328]. Its expression is higher in PDAC compared to normal pancreas and it is strongly correlated with PDAC stages, since the levels of GLUT1 increase as the disease progresses and becomes more aggressive. Indeed, high GLUT1 expression in resected tumors correlates highly with tumor size, nodal involvement, and shorter patient survival, suggesting that GLUT1 may serve as a prognostic marker [329,330,331,332]. Some reports have demonstrated the ability of apigenin, a dietary flavonoid, to inhibit the GLUT1 action as a contributor to cancer progression [333]. In PDAC, several in vitro studies support the beneficial effect of using the inhibitor of GLUT1 activity, apigenin, to reduce proliferation and angiogenesis by interfering with the PI3K/Akt and VEGF-HIF-1α pathways, respectively [334,335,336]. More studies are needed specially using animal models to confirm the tumor suppressor activity of apigenin and its possible potential as a future complementary chemopreventive agent for pancreatic cancer.
4.3.2. Hexokinase (HK)
HK is the enzyme responsible for the first glycolytic step and has two isoforms, with HK1 predominantly found in cytoplasm, whereas HK2 is mainly present in mitochondria [337]. HK2 is upregulated in multiple cancer types and has the ability to prevent mitochondrial apoptosis through its direct insertion in the mitochondrial outer membrane [245]. Moreover, the expression of HK2 is elevated in PDAC metastatic cells, demonstrating its relationship with the aggressiveness and progression of the disease [338]. Cancer associated-survival pathways such as PI3K/Akt/mTOR pathway are able to stimulate HK2 in cancer cells, inducing drug resistance [339]. HK2 is considered to be a crucial target for anticancer drug therapy due to its role in modulating apoptosis and cellular bioenergetics. The non-specific HK2 inhibitor, 3-bromopyruvate, can reduce ATP reserves and, therefore, reverse chemoresistance in pancreatic cancer cells [340]. It should be noted that elevated ATP levels resulting from increased glycolysis are also linked to activation of HIF-1α and chemoresistance [341]. Regarding the CSCs, which have the most aggressive and drug resistant phenotype, the utilization of 3-bromopyruvate to inhibit the glucose turnover sensitized this aggressive population to gemcitabine [342]. Moreover, this inhibitor has also been tested in vivo in an orthotopic mouse model of human pancreatic cancer, where it was found that ultrasound-guided delivery of 3 bromo-pyruvate blocked tumor progression through a decrease in proliferative potential and apoptosis induction [343].
4.3.3. Fructose Biphosphate Aldolase
Another enzyme overexpressed in PDAC is the fructose biphosphate aldolase (FBA), which encompasses the step of converting fructose 1,6-biphosphate into glyceraldehyde-3-phosphate (G3P) and dihydroxyacetone phosphate [344]. Both the overexpression of FBA and high levels of G3P are known to downregulate apoptosis by suppressing caspase-3 activity [345]. Despite the strong evidence that both proteins may have a role in PDAC progression and chemoresistance, there are no studies exploring this area. However, in other tumor types it was found that FBA correlates with the aggressiveness of the tumor [346]. Therefore, the use of either FBA or G3P as targets for new chemicals should be encouraged, with the objective to reduce the cancer-associated drug resistance.
4.3.4. Lactate Dehydrogenase (LDH)
Another important enzyme is LDH since it is responsible for the conversion of pyruvate into lactate, the endpoint of fermentative glycolysis. LDH is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells [161]. Moreover, the expression of this enzyme is closely associated with aggressiveness and poor prognosis, suggesting that it might be a good prognostic marker [347]. Lactate dehydrogenase 5 (LDH-5) catalyzes the reduction of pyruvate by NADH to form lactate, hence maintaining a constant availability of NAD+ to support glycolysis [348]. Tumor-dependent upregulation of the LDH-5 level was observed in pancreatic cancers and was found to correlate with metastases, tumor stage, recurrence of the tumor, and patient survival [349]. On the other hand, a recent report on new LDH inhibitors in PDAC demonstrated a synergistic effect of LDH inhibition with gemcitabine, probably by increasing the expression of deoxycytidine kinase (dCK), which is an important mediator of apoptosis-induced gemcitabine [350]. Moreover, the inhibition of LDH both in vitro and in vivo, with the small-molecule inhibitor, FX11, induced oxidative stress, cell death and inhibited tumor progression [351,352]. Considering these promising results, a deeper analysis is needed to disclose the clinical potential of the inhibition of LDH as a new encouraging target for PDAC treatment.
4.3.5. Pyruvate Kinase (PK) and Monocarboxylate Transporters (MCTs)
PK is a tetrameric enzyme divided into four subtypes (M1, M2, L and R), which are diversely expressed in various cell types [353]. PKM2, the isoform that converts phosphoenolpyruvate (PEP) and ADP into pyruvate and ATP, is the rate-limiting enzyme of glycolysis [354]. This enzyme is overexpressed in several types of cancer, possibly to drive enhanced glycolytic fluxes due to its high affinity for PEP. In pancreatic cancer cells, PKM2 promotes cell survival and invasion, especially under metabolic stress by enhancing Warburg effect and modulation of ROS production [355,356], thus becoming directly associated with pancreatic chemoresistance [357]. By increasing aerobic glycolysis, PKM2 maintains increased levels of lactate, which is known to have an important role in driving cancer metastasis [358]. Indeed, the knockdown of PKM2 significantly sensitizes pancreatic cancer cells to gemcitabine, by activating several caspase family members leading to apoptosis [359]. Lactate plays also a role as a signaling intermediate in hypoxic conditions leading to the induction of survival pathways [360]. Furthermore, lactate is also able to attenuate the immune response and, in particular, tumor-derived lactate can prevent the response of human T-cells, which are the predominant infiltrated immune cell in the human PDAC TME [361]. In addition, the presence of MCT4 in high levels defines a glycolytic subtype of pancreatic cancer which is correlated with a poor prognosis and outcome [362]. Therefore, the inhibition of the lactate transporters of the MCT family is being considered as a promising potential therapeutic option for cancer treatment, including PDAC [363,364].
4.3.6. Fatty Acid Transporter CD36
The intracellular uptake of fatty acids is highly dependent on the presence and activity of the transporter CD36 [365]. In PDAC, CD36 is able to influence gemcitabine resistance through the regulation of anti-apoptotic proteins [366]. Indeed, it has been suggested that PDAC patients with elevated CD36 expression have lower overall survival and recurrence-free survival rates compared to patients with reduced expression [366]. Overall, CD36 levels can be suggested as a prognostic factor for more aggressive stages of PDAC and the use of anti-CD36 strategies together with chemotherapy seems to be an encouraging therapeutic strategy and studies on this topic should be strongly encouraged.
4.3.7. Fatty Acid Synthase (FASN) and Carnitine Palmitoyl Transferase 1A (CPT1A)
PDAC cells are characterized by an increased expression of some lipid metabolism enzymes and transporters, such as FASN and CPT1A, respectively. Firstly, FASN was discovered to be upregulated in high-grade pancreatic tumors, suggesting that this enzyme might be helpful as a prognostic factor and a promising therapeutic target [367,368]. Indeed, the levels of FASN are correlated with both chemotherapy resistance, especially to gemcitabine, and to radiation resistance in PDAC [357,369]. In line with this, the use of FASN-siRNA or a FASN inhibitor, such as orlistat, decreased gemcitabine-associated resistance in PDAC cells [370]. Although these findings demonstrated a relationship between FASN and chemotherapy resistance, the underlying mechanisms are not yet elucidated, and further studies are needed. Lastly, the inhibition of the enzyme responsible for the entrance of fatty acids via mitochondria (CPT1A), by using etomoxir has been shown to restore the sensitivity of pancreatic CSCs to gemcitabine [371]. Altogether these data indicate that inhibition of FASN and CPT1A might represent important therapeutic targets. Further studies are essential to better understand the role of both enzymes on cancer metabolism reprogramming and whether this plasticity is important for the cancer cell to supply ATP under energy stress.
4.3.8. Clinical Trials: First Evidence in PDAC Patients
Some metabolic inhibitors of glycolysis have reached clinical trials for PDAC treatment. 2-deoxyglucose (2-DG), 3-bromopyruvate and lonidamine are HK inhibitors that were tested in early phase clinical trials, including some trials in PDAC patients [370]. Moreover, an LDH inhibitor, FLX11, was shown to reduce growth of both lymphoma and PDAC xenografts [372]. Despite the fact that PDAC has multiple metabolic modifications to meet the needs of unrestrained proliferation, there are still several aspects to be explored, such as how the complexity of the TME shapes metabolism and might contribute to carcinogenesis and progression of the disease. PDAC is characterized as a highly hypoxic tumor with significantly impaired drug perfusion and low nutrient availability [373,374]. Aberrant metabolism allows cancer cells to acquire resistance to standard treatments by modulating apoptosis, angiogenesis, invasion, and by affecting drug transport and targets. Several studies have demonstrated encouraging results by combining chemotherapy with metabolic inhibitors, where a synergistic effect for PDAC treatment was found [142,143,309,311,375,376]. However, most inhibitors are still in the preclinical phase and the glycolytic pathways are the only ones that have been exhaustively studied and are, at the moment, the most promising for targeting as alternative PDAC treatments. Further studies are necessary especially disclosing the possible role of other metabolic pathways such as glutaminolysis and other amino acids and fatty acids and lipids pathways, on the development and progression of PDAC with the objective to discover novel targets for PDAC treatment.
5. Conclusions
Despite the great effort made to date in the scientific research, PDAC still poses a major therapeutic challenge. So far, only small improvements have been achieved in the diagnosis and management of patients with this disease. PDAC TME stroma is characterized by an intense desmoplastic reaction, hypoxia, acidosis, high interstitial pressure and heterogeneity. The desmoplastic reaction is strongly related with cancer invasion, progression, and metastasis through a complex crosstalk between malignant and stellate cells. Moreover, desmoplasia combined with high interstitial pressure obliterates the poorly vascularized tumor increasing further hypoxia, which is usually found in all tumors. Altogether, these events create a mechanical barrier for the access of chemotherapeutic drugs.
In the last decade, much progress has been made and it has been found that extracellular acidosis supplies a beneficial environment for cancer cells, by creating a chemical barrier to immune surveillance and chemotherapy. This acidic environment results mainly from the dysregulated metabolism of cancers. The secreted metabolic end-products and cytokines stimulates the interaction between cancer cells and stromal cells, which contribute to the tumor progression and aggressiveness. Indeed, the acidic microenvironment functions as a trigger to the metastatic cascade, stimulating EMT, ECM degradation, and tumor cell migration.
Due to the complex TME which comprises the mechanisms discussed in this review, namely, desmoplastic reaction, hypoxia, lack of vascular access, low extracellular pH, and the production of drug extruder proteins, working together or separately, PDAC still displays low chemosensitivity to most of the chemotherapeutic drugs. The complexity of the components of TME makes evident their importance in immune system suppression and tumor progression. Indeed, since PDAC drug resistance relies on this specific hostile microenvironment and metabolic reprogramming, this offers potentially innovative strategies for treating patients with PDAC in the future.
Therefore, targeting desmoplasia and/or cancer metabolism in combination with other targeted agents or cytotoxic compounds represent promising therapeutic strategies that could be beneficial for PDAC.
Author Contributions
Conceptualization, T.M.A.C.; T.K.; D.D.M. and S.J.R.; Resources, T.K.; K.O.A.; M.R.G.; Writing—Original Draft Preparation, T.K.; T.M.A.C.; D.D.M.; Writing—Review & Editing, R.A.C.; T.M.A.C.; D.D.M.; S.J.R.; T.K.; K.O.A.; Visualization, M.R.G.; Supervision, S.J.R.; R.A.C.; T.K.; Project Administration, S.J.R.; R.A.C.; T.K.; Funding Acquisition, S.J.R. and R.A.C.; All authors have read and approved of the version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the European Marie Skłodowska-Curie Innovative Training Network (ITN) pH and Ion Transport in Pancreatic Cancer–pHioniC (Grant Agreement number: 813834; H2020-MSCA-ITN-2018), by PRIN n. 20174TB8KW002 “Lioness” from Italian Ministry for Education, University and Research (MIUR) and by the Programma Operativo Complementare Ricerca e Innovazione, Asse I “Investimenti in Capitale Umano”—Azione I.1 “Dottorati innovativi con caratterizzazione industriale” del PON R&I 2014–2020.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Binkley, C.E.; Zhang, L.; Greenson, J.K.; Giordano, T.J.; Kuick, R.; Misek, D.; Hanash, S.; Logsdon, C.D.; Simeone, D.M. The molecular basis of pancreatic fibrosis: Common stromal gene expression in chronic pancreatitis and pancreatic adenocarcinoma. Pancreas 2004, 29, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, C.; Xie, K.P. Therapeutic resistance of pancreatic cancer: Roadmap to its reversal. Biochim. Biophys. Acta. Rev. Cancer 2021, 1875, 188461. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Löhr, J.M. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014, 6, 2137–2154. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T. The Solid Mechanics of Cancer and Strategies for Improved Therapy. J. Biomech. Eng. 2017, 139, 021004. [Google Scholar] [CrossRef] [PubMed]
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed]
- Stanisavljevic, J.; Loubat-Casanovas, J.; Herrera, M.; Luque, T.; Peña, R.; Lluch, A.; Albanell, J.; Bonilla, F.; Rovira, A.; Peña, C.; et al. Snail1-expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res. 2015, 75, 284–295. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Ioannides, C.G.; Whiteside, T.L. T cell recognition of human tumors: Implications for molecular immunotherapy of cancer. Clin. Immunol. Immunopathol. 1993, 66, 91–106. [Google Scholar] [CrossRef]
- Apte, M.V.; Xu, Z.; Pothula, S.; Goldstein, D.; Pirola, R.C.; Wilson, J.S. Pancreatic cancer: The microenvironment needs attention too! Pancreatology 2015, 15 (Suppl. S4), S32–S38. [Google Scholar] [CrossRef]
- Chang, J.H.; Jiang, Y.; Pillarisetty, V.G. Role of immune cells in pancreatic cancer from bench to clinical application: An updated review. Medicine 2016, 95, e5541. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.-M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 1–13. [Google Scholar] [CrossRef]
- Yeldag, G.; Rice, A.; del Río Hernández, A. Chemoresistance and the self-maintaining tumor microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; di Magliano, M.P.; Simeone, D.M. Deciphering the role of stroma in pancreatic cancer. Curr. Opin. Gastroenterol. 2013, 29, 537. [Google Scholar] [CrossRef]
- Hall, B.R.; Cannon, A.; Atri, P.; Wichman, C.S.; Smith, L.M.; Ganti, A.K.; Are, C.; Sasson, A.R.; Kumar, S.; Batra, S.K. Advanced pancreatic cancer: A meta-analysis of clinical trials over thirty years. Oncotarget 2018, 9, 19396. [Google Scholar] [CrossRef]
- Apte, M.; Park, S.; Phillips, P.; Santucci, N.; Goldstein, D.; Kumar, R.; Ramm, G.; Buchler, M.; Friess, H.; McCarroll, J. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Lu, J.; Zhou, S.; Siech, M.; Habisch, H.; Seufferlein, T.; Bachem, M. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen I-mediated signalling pathway. Br. J. Cancer 2014, 110, 409–420. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Dunér, S.; Lindman, J.L.; Ansari, D.; Gundewar, C.; Andersson, R. Pancreatic cancer: The role of pancreatic stellate cells in tumor progression. Pancreatology 2010, 10, 673–681. [Google Scholar] [CrossRef]
- Sherman, M.H.; Ruth, T.Y.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Carapuça, E.F.; Gemenetzidis, E.; Feig, C.; Bapiro, T.E.; Williams, M.D.; Wilson, A.S.; Delvecchio, F.R.; Arumugam, P.; Grose, R.P.; Lemoine, N.R. Anti-stromal treatment together with chemotherapy targets multiple signalling pathways in pancreatic adenocarcinoma. J. Pathol. 2016, 239, 286–296. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Masamune, A.; Hamada, S.; Kikuta, K.; Takikawa, T.; Miura, S.; Nakano, E.; Shimosegawa, T. The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand. J. Gastroenterol. 2013, 48, 602–609. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Posner, R.G.; Von Hoff, D.D.; Han, H. Desmoplasia and chemoresistance in pancreatic cancer. In Pancreatic Cancer and Tumor Microenvironment; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- Yen, T.W.; Aardal, N.P.; Bronner, M.P.; Thorning, D.R.; Savard, C.E.; Lee, S.P.; Bell, R.H., Jr. Myofibroblasts are responsible for the desmoplastic reaction surrounding human pancreatic carcinomas. Surgery 2002, 131, 129–134. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schneider, E.; Groß, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Apte, M.; Haber, P.; Darby, S.; Rodgers, S.; McCaughan, G.; Korsten, M.; Pirola, R.; Wilson, J. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, Y.; Oda, T.; Kinoshita, T.; Nakahashi, C.; Hasebe, T.; Ohkohchi, N.; Ochiai, A. Overexpression of TGF-β by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br. J. Cancer 2004, 91, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Löhr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Müller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-β1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar]
- Awaji, M.; Futakuchi, M.; Heavican, T.; Iqbal, J.; Singh, R.K. Cancer-associated fibroblasts enhance survival and progression of the aggressive pancreatic tumor Via FGF-2 and CXCL8. Cancer Microenviron. 2019, 12, 37–46. [Google Scholar] [CrossRef]
- Hartel, M.; di Mola, F.F.; Gardini, A.; Zimmermann, A.; Di Sebastiano, P.; Guweidhi, A.; Innocenti, P.; Giese, T.; Giese, N.; Büchler, M.W. Desmoplastic reaction influences pancreatic cancer growth behavior. World J. Surg. 2004, 28, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL-1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef]
- Stern, R. Hyaluronidases in cancer biology. Hyaluronan Cancer Biol. 2008, 207–220. [Google Scholar] [CrossRef]
- Marastoni, S.; Ligresti, G.; Lorenzon, E.; Colombatti, A.; Mongiat, M. Extracellular matrix: A matter of life and death. Connect. Tissue Res. 2008, 49, 203–206. [Google Scholar] [CrossRef]
- Kaspar, M.; Zardi, L.; Neri, D. Fibronectin as target for tumor therapy. Int. J. Cancer 2006, 118, 1331–1339. [Google Scholar] [CrossRef]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The extracellular matrix of animals. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Imamura, T.; Iguchi, H.; Manabe, T.; Ohshio, G.; Yoshimura, T.; Wang, Z.; Suwa, H.; Ishigami, S.; Imamura, M. Quantitative analysis of collagen and collagen subtypes I, III, and V in human pancreatic cancer, tumor-associated chronic pancreatitis, and alcoholic chronic pancreatitis. Pancreas 1995, 11, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, J.; Roether, I.; Kern, H. Distribution of extracellular matrix proteins in pancreatic ductal adenocarcinoma and its influence on tumor cell proliferation in vitro. Pancreas 1987, 2, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Castaños-Velez, E.; Biberfeld, P. Immunohistochemical expression of extracellular matrix proteins and adhesion molecules in pancreatic carcinoma. Hepatogastroenterology 2001, 48, 1321–1327. [Google Scholar]
- Verrecchia, F.; Mauviel, A. TGF-β and TNF-α: Antagonistic cytokines controlling type I collagen gene expression. Cell. Signal. 2004, 16, 873–880. [Google Scholar] [CrossRef]
- Nieskoski, M.D.; Marra, K.; Gunn, J.R.; Hoopes, P.J.; Doyley, M.M.; Hasan, T.; Trembly, B.S.; Pogue, B.W. Collagen complexity spatially defines microregions of total tissue pressure in pancreatic cancer. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Laurent, T.C.; Fraser, J. Hyaluronan. FASEB J. 1992, 6, 2397–2404. [Google Scholar] [CrossRef]
- Yu, M.; Tannock, I.F. Targeting tumor architecture to favor drug penetration: A new weapon to combat chemoresistance in pancreatic cancer? Cancer Cell 2012, 21, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.; Hingorani, S. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 2013, 108, 1–8. [Google Scholar] [CrossRef]
- Johnsson, C.; Hällgren, R.; Tufveson, G. Role of hyaluronan in acute pancreatitis. Surgery 2000, 127, 650–658. [Google Scholar] [CrossRef]
- Voutouri, C.; Polydorou, C.; Papageorgis, P.; Gkretsi, V.; Stylianopoulos, T. Hyaluronan-derived swelling of solid tumors, the contribution of collagen and cancer cells, and implications for cancer therapy. Neoplasia 2016, 18, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Kultti, A.; Zhao, C.; Singha, N.C.; Zimmerman, S.; Osgood, R.J.; Symons, R.; Jiang, P.; Li, X.; Thompson, C.B.; Infante, J.R. Accumulation of extracellular hyaluronan by hyaluronan synthase 3 promotes tumor growth and modulates the pancreatic cancer microenvironment. BioMed Res. Int. 2014, 2014, 817613. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Akiyama, S.K.; Olden, K.; Yamada, K.M. Fibronectin and integrins in invasion and metastasis. Cancer Metastasis Rev. 1995, 14, 173–189. [Google Scholar] [CrossRef]
- Lenselink, E.A. Role of fibronectin in normal wound healing. Int. Wound J. 2015, 12, 313–316. [Google Scholar] [CrossRef] [PubMed]
- George, E.L.; Georges-Labouesse, E.N.; Patel-King, R.S.; Rayburn, H.; Hynes, R.O. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 1993, 119, 1079–1091. [Google Scholar] [CrossRef]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenes. Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.S.; Williams, L.V.; Schwarzbauer, J.E. Tissue repair and the dynamics of the extracellular matrix. Int. J. Biochem. Cell Biol. 2004, 36, 1031–1037. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef]
- Forciniti, S.; Dalla Pozza, E.; Greco, M.R.; Amaral Carvalho, T.M.; Rolando, B.; Ambrosini, G.; Carmona-Carmona, C.A.; Pacchiana, R.; Di Molfetta, D.; Donadelli, M. Extracellular Matrix Composition Modulates the Responsiveness of Differentiated and Stem Pancreatic Cancer Cells to Lipophilic Derivate of Gemcitabine. Int. J. Mol. Sci. 2021, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Chand, S.N.; Zarei, M.; Schiewer, M.J.; Kamath, A.R.; Romeo, C.; Lal, S.; Cozzitorto, J.A.; Nevler, A.; Scolaro, L.; Londin, E. Posttranscriptional regulation of PARG mRNA by HuR facilitates DNA repair and resistance to PARP inhibitors. Cancer Res. 2017, 77, 5011–5025. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; de Almeida, F.K.; Simões, C.B.; Wiesmüller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef]
- Fujita, Y.; Kitagawa, M.; Nakamura, S.; Azuma, K.; Ishii, G.; Higashi, M.; Kishi, H.; Hiwasa, T.; Koda, K.; Nakajima, N. CD44 signaling through focal adhesion kinase and its anti-apoptotic effect. FEBS Lett. 2002, 528, 101–108. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.; Koutsilieris, M. The Akt pathway: Molecular targets for anti-cancer drug development. Curr. Cancer Drug Targets 2004, 4, 235–256. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [PubMed]
- Biondani, G.; Zeeberg, K.; Greco, M.R.; Cannone, S.; Dando, I.; Dalla Pozza, E.; Mastrodonato, M.; Forciniti, S.; Casavola, V.; Palmieri, M. Extracellular matrix composition modulates PDAC parenchymal and stem cell plasticity and behavior through the secretome. FEBS J. 2018, 285, 2104–2124. [Google Scholar] [CrossRef]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar]
- Lieleg, O.; Baumgärtel, R.M.; Bausch, A.R. Selective filtering of particles by the extracellular matrix: An electrostatic bandpass. Biophys. J. 2009, 97, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B.; Shepard, H.M.; O’Connor, P.M.; Kadhim, S.; Jiang, P.; Osgood, R.J.; Bookbinder, L.H.; Li, X.; Sugarman, B.J.; Connor, R.J. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol. Cancer Ther. 2010, 9, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.A.; Coker, R. Transforming growth factor-beta (TGF-beta). Int. J. Biochem. Cell Biol. 1998, 30, 293–298. [Google Scholar] [CrossRef]
- Hilbig, A.; Oettle, H. Transforming growth factor beta in pancreatic cancer. Curr. Pharm. Biotechnol. 2011, 12, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- García-Montero, A.C.; Vasseur, S.; Giono, L.E.; Canepa, E.; Moreno, S.; Dagorn, J.C.; Iovanna, J.L. Transforming growth factor β-1 enhances Smad transcriptional activity through activation of p8 gene expression. Biochem. J. 2001, 357, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Shields, M.A.; Dangi-Garimella, S.; Krantz, S.B.; Bentrem, D.J.; Munshi, H.G. Pancreatic cancer cells respond to type I collagen by inducing snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion. J. Biol. Chem. 2011, 286, 10495–10504. [Google Scholar] [CrossRef]
- Kim, Y.J.; Hwang, J.S.; Hong, Y.B.; Bae, I.; Seong, Y.-S. Transforming growth factor beta receptor I inhibitor sensitizes drug-resistant pancreatic cancer cells to gemcitabine. Anticancer Res. 2012, 32, 799–806. [Google Scholar] [PubMed]
- Toole, B.P.; Slomiany, M.G. Hyaluronan, CD44 and Emmprin: Partners in cancer cell chemoresistance. Drug Resist. Updates 2008, 11, 110–121. [Google Scholar] [CrossRef]
- Edward, M.; Quinn, J.; Pasonen-Seppänen, S.; McCann, B.; Tammi, R. 4-Methylumbelliferone inhibits tumour cell growth and the activation of stromal hyaluronan synthesis by melanoma cell-derived factors. Br. J. Dermatol. 2010, 162, 1224–1232. [Google Scholar] [CrossRef]
- Kultti, A.; Pasonen-Seppänen, S.; Jauhiainen, M.; Rilla, K.J.; Kärnä, R.; Pyöriä, E.; Tammi, R.H.; Tammi, M.I. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp. Cell Res. 2009, 315, 1914–1923. [Google Scholar] [CrossRef]
- Urakawa, H.; Nishida, Y.; Wasa, J.; Arai, E.; Zhuo, L.; Kimata, K.; Kozawa, E.; Futamura, N.; Ishiguro, N. Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. Int. J. Cancer 2012, 130, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Hajime, M.; Shuichi, Y.; Makoto, N.; Masanori, Y.; Ikuko, K.; Atsushi, K.; Mutsuo, S.; Keiichi, T. Inhibitory effect of 4-methylesculetin on hyaluronan synthesis slows the development of human pancreatic cancer in vitro and in nude mice. Int. J. Cancer 2007, 120, 2704–2709. [Google Scholar] [CrossRef]
- Morohashi, H.; Kon, A.; Nakai, M.; Yamaguchi, M.; Kakizaki, I.; Yoshihara, S.; Sasaki, M.; Takagaki, K. Study of hyaluronan synthase inhibitor, 4-methylumbelliferone derivatives on human pancreatic cancer cell (KP1-NL). Biochem. Biophys. Res. Commun. 2006, 345, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; Yoshihara, S.; Kudo, D.; Morohashi, H.; Kakizaki, I.; Kon, A.; Takagaki, K.; Sasaki, M. 4-methylumbelliferone, a hyaluronan synthase suppressor, enhances the anticancer activity of gemcitabine in human pancreatic cancer cells. Cancer Chemother. Pharmacol. 2006, 57, 165–170. [Google Scholar] [CrossRef]
- Michl, P.; Gress, T.M. Improving drug delivery to pancreatic cancer: Breaching the stromal fortress by targeting hyaluronic acid. Gut 2012, 61, 1377–1379. [Google Scholar] [CrossRef]
- Kohno, N.; Ohnuma, T.; Truog, P. Effects of hyaluronidase on doxorubicin penetration into squamous carcinoma multicellular tumor spheroids and its cell lethality. J. Cancer Res. Clin. Oncol. 1994, 120, 293–297. [Google Scholar] [CrossRef]
- Magzoub, M.; Jin, S.; Verkman, A. Enhanced macromolecule diffusion deep in tumors after enzymatic digestion of extracellular matrix collagen and its associated proteoglycan decorin. FASEB J. 2008, 22, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Koltai, T.; Reshkin, S.J.; Carvalho, T.; Cardone, R.A. Targeting the Stromal Pro-Tumoral Hyaluronan-CD44 Pathway in Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 3953. [Google Scholar] [CrossRef]
- Kelleher, F.C. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis 2011, 32, 445–451. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Bai, Y.; Dong, J.; Li, Q.; Jin, Y.; Chen, B.; Zhou, M. Hedgehog signaling in pancreatic fibrosis and cancer. Medicine 2016, 95, e2996. [Google Scholar] [CrossRef]
- Feldmann, G.; Fendrich, V.; McGovern, K.; Bedja, D.; Bisht, S.; Alvarez, H.; Koorstra, J.-B.M.; Habbe, N.; Karikari, C.; Mullendore, M. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 2008, 7, 2725–2735. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, F.C.; McDermott, R. Aberrations and therapeutics involving the developmental pathway Hedgehog in pancreatic cancer. Vitam. Horm. 2012, 88, 355–378. [Google Scholar] [CrossRef]
- Bisht, S.; Brossart, P.; Maitra, A.; Feldmann, G. Agents targeting the Hedgehog pathway for pancreatic cancer treatment. Curr. Opin. Investig. Drugs 2010, 11, 1387–1398. [Google Scholar]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Stephenson, J.; Wolpin, B.M.; Becerra, C.; Hamm, J.T.; Messersmith, W.A.; Devens, S.; Cushing, J.; Schmalbach, T.; Fuchs, C.S. A phase Ib trial of IPI-926, a hedgehog pathway inhibitor, plus gemcitabine in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2012, 30 (Suppl. S4), 213. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Phillips, P.A.; Santucci, N.; Pirola, R.C.; Wilson, J.; Apte, M. Vitamin A inhibits pancreatic stellate cell activation: Implications for treatment of pancreatic fibrosis. Gut 2006, 55, 79–89. [Google Scholar] [CrossRef]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid–induced pancreatic stellate cell quiescence reduces paracrine Wnt–β-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Carter, N.J. Pirfenidone. Drugs 2011, 71, 1721–1732. [Google Scholar] [CrossRef]
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2010, 35, 821–829. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Garcıa, L.; Hernández, I.; Sandoval, A.; Salazar, A.; Garcia, J.; Vera, J.; Grijalva, G.; Muriel, P.; Margolin, S.; Armendariz-Borunda, J. Pirfenidone effectively reverses experimental liver fibrosis. J. Hepatol. 2002, 37, 797–805. [Google Scholar] [CrossRef]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506. [Google Scholar] [CrossRef]
- Knüppel, L.; Ishikawa, Y.; Aichler, M.; Heinzelmann, K.; Hatz, R.; Behr, J.; Walch, A.; Bächinger, H.P.; Eickelberg, O.; Staab-Weijnitz, C.A. A novel antifibrotic mechanism of nintedanib and pirfenidone. Inhibition of collagen fibril assembly. Am. J. Respir. Cell Mol. Biol. 2017, 57, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Inoue, H.; Nakazawa, R.; Azuma, N.; Suzuki, M.; Yamauchi, S.; Margolin, S.; Tsubota, K.; Saito, I. Pirfenidone induces intercellular adhesion molecule-1 (ICAM-1) down-regulation on cultured human synovial fibroblasts. Clin. Exp. Immunol. 1998, 113, 72–76. [Google Scholar] [CrossRef]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Pirfenidone treatment decreases transforming growth factor-β1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am. J. Transplant. 2002, 2, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, H.; Oku, H.; Yamane, S.; Tsuruta, Y.; Suzuki, R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-α at the translational level. Eur. J. Pharmacol. 2002, 446, 177–185. [Google Scholar] [CrossRef]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef]
- Oku, H.; Nakazato, H.; Horikawa, T.; Tsuruta, Y.; Suzuki, R. Pirfenidone suppresses tumor necrosis factor-α, enhances interleukin-10 and protects mice from endotoxic shock. Eur. J. Pharmacol. 2002, 446, 167–176. [Google Scholar] [CrossRef]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef]
- Usugi, E.; Ishii, K.; Hirokawa, Y.; Kanayama, K.; Matsuda, C.; Uchida, K.; Shiraishi, T.; Watanabe, M. Antifibrotic agent pirfenidone suppresses proliferation of human pancreatic cancer cells by inducing G0/G1 cell cycle arrest. Pharmacology 2019, 103, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Kuno, A.; Masuda, K.; Ogawa, K.; Sogawa, M.; Nakamura, S.; Ando, T.; Sano, H.; Nakazawa, T.; Ohara, H. Candesartan, an angiotensin II receptor antagonist, suppresses pancreatic inflammation and fibrosis in rats. J. Pharmacol. Exp. Ther. 2003, 307, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Richards, D.; Wolpin, B.; Becerra, C.; Hamm, J.; Messersmith, W.; Devens, S.; Cushing, J.; Goddard, J.; Schmalbach, T. The safety of IPI-926, a novel hedgehog pathway inhibitor, in combination with gemcitabine in patients (pts) with metastatic pancreatic cancer. J. Clin. Oncol. 2011, 29 (Suppl. S15), 4114. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kelley, R.K.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V. A phase I study of FOLFIRINOX plus IPI-926, a hedgehog pathway inhibitor, for advanced pancreatic adenocarcinoma. Pancreas 2016, 45, 370. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284. [Google Scholar] [CrossRef]
- Yoshida, E.; Kudo, D.; Nagase, H.; Shimoda, H.; Suto, S.; Negishi, M.; Kakizaki, I.; Endo, M.; Hakamada, K. Antitumor effects of the hyaluronan inhibitor 4-methylumbelliferone on pancreatic cancer. Oncol. Lett. 2016, 12, 2337–2344. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zheng, L.-C.; Zhang, H.-J.; Tsang, S.W.; Bian, Z.-X. Anti-fibrotic effects of phenolic compounds on pancreatic stellate cells. BMC Complement. Altern. Med. 2015, 15, 259. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Yoshimura, K.; Asada, M.; Imaizumi, A.; Suzuki, C.; Matsumoto, S.; Nishimura, T.; Mori, Y.; Masui, T.; Kawaguchi, Y. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother. Pharmacol. 2011, 68, 157–164. [Google Scholar] [CrossRef]
- Shehzad, A.; Qureshi, M.; Anwar, M.N.; Lee, Y.S. Multifunctional curcumin mediate multitherapeutic effects. J. Food Sci. 2017, 82, 2006–2015. [Google Scholar] [CrossRef]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharmacol. 2008, 75, 787–809. [Google Scholar] [CrossRef] [PubMed]
- Gundewar, C.; Ansari, D.; Tang, L.; Wang, Y.; Liang, G.; Rosendahl, A.H.; Saleem, M.A.; Andersson, R. Antiproliferative effects of curcumin analog L49H37 in pancreatic stellate cells: A comparative study. Ann. Gastroenterol. Q. Publ. Hell. Soc. Gastroenterol. 2015, 28, 391. [Google Scholar]
- Tsang, S.W.; Zhang, H.; Lin, C.; Xiao, H.; Wong, M.; Shang, H.; Yang, Z.-J.; Lu, A.; Yung, K.K.-L.; Bian, Z. Rhein, a natural anthraquinone derivative, attenuates the activation of pancreatic stellate cells and ameliorates pancreatic fibrosis in mice with experimental chronic pancreatitis. PLoS ONE 2013, 8, e82201. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.W.; Bian, Z.X. Anti-fibrotic and anti-tumorigenic effects of rhein, a natural anthraquinone derivative, in mammalian stellate and carcinoma cells. Phytother. Res. 2015, 29, 407–414. [Google Scholar] [CrossRef]
- Yan, B.; Cheng, L.; Jiang, Z.; Chen, K.; Zhou, C.; Sun, L.; Cao, J.; Qian, W.; Li, J.; Shan, T. Resveratrol inhibits ROS-promoted activation and glycolysis of pancreatic stellate cells via suppression of miR-21. Oxid. Med. Cell. Longev. 2018. [Google Scholar] [CrossRef]
- Wei, W.-T.; Lin, S.-Z.; Liu, D.-L.; Wang, Z.-H. The distinct mechanisms of the antitumor activity of emodin in different types of cancer. Oncol. Rep. 2013, 30, 2555–2562. [Google Scholar] [CrossRef]
- Cha, T.L.; Chuang, M.J.; Tang, S.H.; Wu, S.T.; Sun, K.H.; Chen, T.T.; Sun, G.H.; Chang, S.Y.; Yu, C.P.; Ho, J.Y. Emodin modulates epigenetic modifications and suppresses bladder carcinoma cell growth. Mol. Carcinog. 2015, 54, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, X.-B.; Yu, Q.-M.; Luo, Q.-Y.; Zhang, Z.-Z. Synergistic cancer growth-inhibitory effect of emodin and low-dose cisplatin on gastric cancer cells in vitro. Trop. J. Pharm. Res. 2015, 14, 1427–1434. [Google Scholar] [CrossRef]
- Jia, X.; Yu, F.; Wang, J.; Iwanowycz, S.; Saaoud, F.; Wang, Y.; Hu, J.; Wang, Q.; Fan, D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res. Treat. 2014, 148, 291–302. [Google Scholar] [CrossRef]
- Masamune, A.; Satoh, M.; Kikuta, K.; Suzuki, N.; Satoh, K.; Shimosegawa, T. Ellagic acid blocks activation of pancreatic stellate cells. Biochem. Pharmacol. 2005, 70, 869–878. [Google Scholar] [CrossRef]
- Aono, Y.; Nishioka, Y.; Inayama, M.; Ugai, M.; Kishi, J.; Uehara, H.; Izumi, K.; Sone, S. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care Med. 2005, 171, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Gioni, V.; Karampinas, T.; Voutsinas, G.; Roussidis, A.E.; Papadopoulos, S.; Karamanos, N.K.; Kletsas, D. Imatinib mesylate inhibits proliferation and exerts an antifibrotic effect in human breast stroma fibroblasts. Mol. Cancer Res. 2008, 6, 706–714. [Google Scholar] [CrossRef]
- Shiha, G.E.; Abu-Elsaad, N.M.; Zalata, K.R.; Ibrahim, T.M. Tracking anti-fibrotic pathways of nilotinib and imatinib in experimentally induced liver fibrosis: A n insight. Clin. Exp. Pharmacol. Physiol. 2014, 41, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Noguchi, R.; Kuriyama, S.; Ikenaka, Y.; Yoshii, J.; Yanase, K.; Namisaki, T.; Kitade, M.; Masaki, T.; Fukui, H. Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. Am. J. Physiol.-Gastrointest. Liver Physiol. 2005, 288, G907–G913. [Google Scholar] [CrossRef]
- Li, M.; Abdollahi, A.; Gröne, H.-J.; Lipson, K.E.; Belka, C.; Huber, P.E. Late treatment with imatinib mesylate ameliorates radiation-induced lung fibrosis in a mouse model. Radiat. Oncol. 2009, 4, 1–9. [Google Scholar] [CrossRef]
- Vittal, R.; Zhang, H.; Han, M.K.; Moore, B.B.; Horowitz, J.C.; Thannickal, V.J. Effects of the protein kinase inhibitor, imatinib mesylate, on epithelial/mesenchymal phenotypes: Implications for treatment of fibrotic diseases. J. Pharmacol. Exp. Ther. 2007, 321, 35–44. [Google Scholar] [CrossRef]
- Moss, R.A.; Moore, D.; Mulcahy, M.F.; Nahum, K.; Saraiya, B.; Eddy, S.; Kleber, M.; Poplin, E.A. A multi-institutional phase 2 study of imatinib mesylate and gemcitabine for first-line treatment of advanced pancreatic cancer. Gastrointest. Cancer Res. GCR 2012, 5, 77. [Google Scholar] [PubMed]
- Gharibo, M.; Patrick-Miller, L.; Zheng, L.; Guensch, L.; Juvidian, P.; Poplin, E. A phase II trial of imatinib mesylate in patients with metastatic pancreatic cancer. Pancreas 2008, 36, 341–345. [Google Scholar] [CrossRef]
- Duan, W.; Chen, K.; Jiang, Z.; Chen, X.; Sun, L.; Li, J.; Lei, J.; Xu, Q.; Ma, J.; Li, X. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett. 2017, 385, 225–233. [Google Scholar] [CrossRef]
- Qian, W.; Li, J.; Chen, K.; Jiang, Z.; Cheng, L.; Zhou, C.; Yan, B.; Cao, J.; Ma, Q.; Duan, W. Metformin suppresses tumor angiogenesis and enhances the chemosensitivity of gemcitabine in a genetically engineered mouse model of pancreatic cancer. Life Sci. 2018, 208, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Elahi-Gedwillo, K.Y.; Carlson, M.; Zettervall, J.; Provenzano, P.P. Antifibrotic therapy disrupts stromal barriers and modulates the immune landscape in pancreatic ductal adenocarcinoma. Cancer Res. 2019, 79, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.M.; Horton, K.J.; Coveler, A.L.; Hingorani, S.R.; Harris, W.P. Targeting the tumor stroma: The biology and clinical development of pegylated recombinant human hyaluronidase (PEGPH20). Curr. Oncol. Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; del Monte-Nieto, G.; Conway, J.R. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Ng, S.; Barrett, M.T.; Hostetter, G.; Von Hoff, D.D.; Han, H. Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS ONE 2017, 12, e0183871. [Google Scholar] [CrossRef]
- Vennin, C.; Rath, N.; Pajic, M.; Olson, M.F.; Timpson, P. Targeting ROCK activity to disrupt and prime pancreatic cancer for chemotherapy. Small GTPases 2020, 11, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Okada, M.; Suzuki, S.; Kuramoto, K.; Sakaki, H.; Watarai, H.; Sanomachi, T.; Seino, S.; Yoshioka, T.; Kitanaka, C. Rho-associated protein kinase (ROCK) inhibitors inhibit survivin expression and sensitize pancreatic cancer stem cells to gemcitabine. Anticancer Res. 2016, 36, 6311–6318. [Google Scholar] [CrossRef]
- Rath, N.; Munro, J.; Cutiongco, M.F.; Jagiełło, A.; Gadegaard, N.; McGarry, L.; Unbekandt, M.; Michalopoulou, E.; Kamphorst, J.J.; Sumpton, D. Rho kinase inhibition by AT13148 blocks pancreatic ductal adenocarcinoma invasion and tumor growth. Cancer Res. 2018, 78, 3321–3336. [Google Scholar] [CrossRef]
- Wen, W.X.; Lee, S.Y.; Siang, R.; Koh, R.Y. Repurposing pentoxifylline for the treatment of fibrosis: An overview. Adv. Ther. 2017, 34, 1245–1269. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, J.; Doche, C.; Gutowski, M.-C.; Lenoble, M.; Lepape, A.; Perdrix, J.-P. Production of proinflammatory cytokines and cytokines involved in the TH1/TH2 balance is modulated by pentoxifylline. J. Cardiovasc. Pharmacol. 1995, 25, S80–S84. [Google Scholar] [CrossRef]
- Abdel-Salam, O.M.; Baiuomy, A.R.; El-Shenawy, S.M.; Arbid, M.S. The anti-inflammatory effects of the phosphodiesterase inhibitor pentoxifylline in the rat. Pharmacol. Res. 2003, 47, 331–340. [Google Scholar] [CrossRef]
- Zeng, X.-P.; Wang, L.-J.; Guo, H.-L.; He, L.; Bi, Y.-W.; Xu, Z.-L.; Li, Z.-S.; Hu, L.-H. Dasatinib ameliorates chronic pancreatitis induced by caerulein via anti-fibrotic and anti-inflammatory mechanism. Pharmacol. Res. 2019, 147, 104357. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH sensing and regulation in cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- De Milito, A.; Fais, S. Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol. 2005, 1, 779–786. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Strapcova, S.; Takacova, M.; Csaderova, L.; Martinelli, P.; Lukacikova, L.; Gal, V.; Kopacek, J.; Svastova, E. Clinical and pre-clinical evidence of carbonic anhydrase ix in pancreatic cancer and its high expression in pre-cancerous lesions. Cancers 2020, 12, 2005. [Google Scholar] [CrossRef]
- Rong, Y.; Wu, W.; Ni, X.; Kuang, T.; Jin, D.; Wang, D.; Lou, W. Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumor Biol. 2013, 34, 1523–1530. [Google Scholar] [CrossRef]
- Kong, S.C.; Gianuzzo, A.; Novak, I.; Pedersen, S.F. Acid-base transport in pancreatic cancer: Molecular mechanisms and clinical potential. Biochem. Cell Biol. 2014, 92, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Pedersen, S.F. Roles of pH and the Na+/H+ exchanger NHE1 in cancer: From cell biology and animal models to an emerging translational perspective. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2017; pp. 5–16. [Google Scholar]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Reshkin, S.J.; Bellizzi, A.; Caldeira, S.; Albarani, V.; Malanchi, I.; Poignee, M.; Alunni-Fabbroni, M.; Casavola, V.; Tommasino, M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000, 14, 2185–2197. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The acidic tumor microenvironment as a driver of cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]
- Duffy, M. The role of proteolytic enzymes in cancer invasion and metastasis. Clin. Exp. Metastasis 1992, 10, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Palermo, C.; Joyce, J.A. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol. Sci. 2008, 29, 22–28. [Google Scholar] [CrossRef]
- Harguindey, S.; Arranz, J.L.; Wahl, M.L.; Orive, G.; Reshkin, S.J. Proton transport inhibitors as potentially selective anticancer drugs. Anticancer Res. 2009, 29, 2127–2136. [Google Scholar]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar] [CrossRef]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The extracellular matrix and pancreatic cancer: A complex relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef]
- Shin, S.C.; Thomas, D.; Radhakrishnan, P.; Hollingsworth, M.A. Invasive phenotype induced by low extracellular pH requires mitochondria dependent metabolic flexibility. Biochem. Biophys. Res. Commun. 2020, 525, 162–168. [Google Scholar] [CrossRef]
- Dhup, S.; Kumar Dadhich, R.; Ettore Porporato, P.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.C.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: A regulatory, immunosuppressive metabolite? J. Pathol. 2013, 230, 350–355. [Google Scholar] [CrossRef]
- Gonen, N.; Assaraf, Y.G. Antifolates in cancer therapy: Structure, activity and mechanisms of drug resistance. Drug Resist. Updates 2012, 15, 183–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-S. Mechanisms of chemotherapeutic drug resistance in cancer therapy—A quick review. Taiwan. J. Obstet. Gynecol. 2009, 48, 239–244. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.M.; Wojtkowiak, J.W.; Hashim, A.I.; Gillies, R.J. Targeting the metabolic microenvironment of tumors. Adv. Pharmacol. 2012, 65, 63–107. [Google Scholar] [CrossRef] [PubMed]
- De Milito, A.; Fais, S. Proton pump inhibitors may reduce tumour resistance. Expert Opin. Pharmacother. 2005, 6, 1049–1054. [Google Scholar] [CrossRef]
- Roos, A. Weak acids, weak bases, and intracellular pH. Respir. Physiol. 1978, 33, 27–30. [Google Scholar] [CrossRef]
- Gillies, R.; Deamer, D. Intracellular pH changes during the cell cycle in Tetrahymena. J. Cell. Physiol. 1979, 100, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, B.P.; Raghunand, N.; Baggett, B.; Gillies, R.J. Tumor acidity, ion trapping and chemotherapeutics: I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem. Pharmacol. 2003, 66, 1207–1218. [Google Scholar] [CrossRef]
- Belhoussine, R.; Morjani, H.; Sharonov, S.; Ploton, D.; Manfait, M. Characterization of intracellular pH gradients in human multidrug-resistant tumor cells by means of scanning microspectrofluorometry and dual-emission-ratio probes. Int. J. Cancer 1999, 81, 81–89. [Google Scholar] [CrossRef]
- Newell, K.; Wood, P.; Stratford, I.; Tannock, I. Effects of agents which inhibit the regulation of intracellular pH on murine solid tumours. Br. J. Cancer 1992, 66, 311–317. [Google Scholar] [CrossRef][Green Version]
- Ohtsubo, T.; Igawa, H.; Saito, T.; Matsumoto, H.; Park, H.J.; Song, C.W.; Kano, E.; Saito, H. Acidic environment modifies heat-or radiation-induced apoptosis in human maxillary cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 1391–1398. [Google Scholar] [CrossRef]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist. Updates 2015, 23, 69–78. [Google Scholar] [CrossRef]
- Luciani, F.; Molinari, A.; Lozupone, F.; Calcabrini, A.; Lugini, L.; Stringaro, A.; Puddu, P.; Arancia, G.; Cianfriglia, M.; Fais, S. P-glycoprotein–actin association through ERM family proteins: A role in P-glycoprotein function in human cells of lymphoid origin. Blood J. Am. Soc. Hematol. 2002, 99, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, D.; Zamboni, S.; Federici, C.; Lugini, L.; Lozupone, F.; Milito, A.D.; Cecchetti, S.; Cianfriglia, M.; Fais, S. P-glycoprotein binds to ezrin at amino acid residues 149–242 in the FERM domain and plays a key role in the multidrug resistance of human osteosarcoma. Int. J. Cancer 2012, 130, 2824–2834. [Google Scholar] [CrossRef] [PubMed]
- Luciani, F.; Spada, M.; De Milito, A.; Molinari, A.; Rivoltini, L.; Montinaro, A.; Marra, M.; Lugini, L.; Logozzi, M.; Lozupone, F. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl. Cancer Inst. 2004, 96, 1702–1713. [Google Scholar] [CrossRef]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- Colbert, L.E.; Fisher, S.B.; Balci, S.; Saka, B.; Chen, Z.; Kim, S.; El-Rayes, B.F.; Adsay, N.V.; Maithel, S.K.; Landry, J.C. High nuclear hypoxia-inducible factor 1 alpha expression is a predictor of distant recurrence in patients with resected pancreatic adenocarcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and Characterization of Hypoxia-inducible Factor 1 (∗). J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 2009, 24, 97–106. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Tan, Z.; Xu, J.; Zhang, B.; Shi, S.; Yu, X.; Liang, C. Hypoxia: A barricade to conquer the pancreatic cancer. Cell. Mol. Life Sci. 2020, 77, 1–7. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its α subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Gassmann, M. Oxygen (es) and the hypoxia-inducible factor-1. Biol. Chem. 1997, 378, 609–616. [Google Scholar]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000. [Google Scholar] [CrossRef]
- Itakura, J.; Ishiwata, T.; Friess, H.; Fujii, H.; Matsumoto, Y.; Büchler, M.W.; Korc, M. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin. Cancer Res. 1997, 3, 1309–1316. [Google Scholar] [PubMed]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis inhibitors generated by tumors. Mol. Med. 1995, 1, 120–122. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Koukourakis, M.; Sivridis, E.; Turley, H.; Talks, K.; Pezzella, F.; Gatter, K.; Harris, A. Relation of hypoxia inducible factor 1 α and 2 α in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br. J. Cancer 2001, 85, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Qiu, G.Z.; Jin, M.Z.; Dai, J.X.; Sun, W.; Feng, J.H.; Jin, W.L. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 669–686. [Google Scholar] [CrossRef]
- Shibaji, T.; Nagao, M.; Ikeda, N.; Kanehiro, H.; Hisanaga, M.; Ko, S.; Fukumoto, A.; Nakajima, Y. Prognostic significance of HIF-1 alpha overexpression in human pancreatic cancer. Anticancer Res. 2003, 23, 4721–4727. [Google Scholar] [PubMed]
- Couvelard, A.; O’Toole, D.; Leek, R.; Turley, H.; Sauvanet, A.; Degott, C.; Ruszniewski, P.; Belghiti, J.; Harris, A.; Gatter, K. Expression of hypoxia-inducible factors is correlated with the presence of a fibrotic focus and angiogenesis in pancreatic ductal adenocarcinomas. Histopathology 2005, 46, 668–676. [Google Scholar] [CrossRef]
- Díaz, B.; Yuen, A.; Iizuka, S.; Higashiyama, S.; Courtneidge, S.A. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J. Cell Biol. 2013, 201, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Lucien, F.; Brochu-Gaudreau, K.; Arsenault, D.; Harper, K.; Dubois, C.M. Hypoxia-induced invadopodia formation involves activation of NHE-1 by the p90 ribosomal S6 kinase (p90RSK). PLoS ONE 2011, 6, e28851. [Google Scholar] [CrossRef]
- Hanna, S.C.; Krishnan, B.; Bailey, S.T.; Moschos, S.J.; Kuan, P.-F.; Shimamura, T.; Osborne, L.D.; Siegel, M.B.; Duncan, L.M.; O’Brien, E.T. HIF1α and HIF2α independently activate SRC to promote melanoma metastases. J. Clin. Investig. 2013, 123, 2078–2093. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, S.; Sun, Y.L. Epithelial-mesenchymal transition in pancreatic cancer: A review. BioMed Res. Int. 2017, 2017, 2646148. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Li, W.; Kang, Y. Probing the fifty shades of EMT in metastasis. Trends Cancer 2016, 2, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T. Membrane proteases: Roles in tissue remodeling and tumour invasion. Curr. Opin. Cell Biol. 1992, 4, 802–809. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Kitajima, Y.; Miyoshi, A.; Ohtsuka, T.; Mitsuno, M.; Ohtaka, K.; Miyazaki, K. The hypoxic environment in tumor-stromal cells accelerates pancreatic cancer progression via the activation of paracrine hepatocyte growth factor/c-Met signaling. Ann. Surg. Oncol. 2007, 14, 2600–2607. [Google Scholar] [CrossRef]
- Spivak-Kroizman, T.R.; Hostetter, G.; Posner, R.; Aziz, M.; Hu, C.; Demeure, M.J.; Von Hoff, D.; Hingorani, S.R.; Palculict, T.B.; Izzo, J. Hypoxia triggers hedgehog-mediated tumor–stromal interactions in pancreatic cancer. Cancer Res. 2013, 73, 3235–3247. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol.—Gastrointest. Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef]
- Greco, O.; Marples, B.; Joiner, M.C.; Scott, S.D. How to overcome (and exploit) tumor hypoxia for targeted gene therapy. J. Cell. Physiol. 2003, 197, 312–325. [Google Scholar] [CrossRef]
- Brown, J.M. Exploiting the hypoxic cancer cell: Mechanisms and therapeutic strategies. Mol. Med. Today 2000, 6, 157–162. [Google Scholar] [CrossRef]
- Papandreou, I.; Krishna, C.; Kaper, F.; Cai, D.; Giaccia, A.J.; Denko, N.C. Anoxia is necessary for tumor cell toxicity caused by a low-oxygen environment. Cancer Res. 2005, 65, 3171–3178. [Google Scholar] [CrossRef]
- Kinoshita, M.; Johnson, D.L.; Shatney, C.H.; Lee, Y.L.; Mochizuki, H. Cancer cells surviving hypoxia obtain hypoxia resistance and maintain anti-apoptotic potential under reoxygenation. Int. J. Cancer 2001, 91, 322–326. [Google Scholar] [CrossRef]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and-independent mechanisms and contributes to drug resistance. Mol. Cell. Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Bacon, A.; Harris, A. Hypoxia-inducible factors and hypoxic cell death in tumour physiology. Ann. Med. 2004, 36, 530–539. [Google Scholar] [CrossRef]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef]
- Fardel, O.; Lecureur, V.; Guillouzo, A. The P-glycoprotein multidrug transporter. Gen. Pharmacol. Vasc. Syst. 1996, 27, 1283–1291. [Google Scholar] [CrossRef]
- Comerford, K.M.; Colgan, S.P. Assessing oxygen sensitivity of the multidrug resistance (MDR) gene. Methods Enzymol. 2004, 381, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bi, R.; Yin, H.; Liu, H.; Li, L. ENO1 silencing impaires hypoxia-induced gemcitabine chemoresistance associated with redox modulation in pancreatic cancer cells. Am. J. Transl. Res. 2019, 11, 4470. [Google Scholar] [PubMed]
- Abdalla, M.Y.; Britigan, B.E.; Wen, F.; Icardi, M.; McCormick, M.L.; LaBrecque, D.R.; Voigt, M.; Brown, K.E.; Schmidt, W.N. Down-regulation of heme oxygenase-1 by hepatitis C virus infection in vivo and by the in vitro expression of hepatitis C core protein. J. Infect. Dis. 2004, 190, 1109–1118. [Google Scholar] [CrossRef]
- Wang, L.; Qu, M.; Huang, S.; Fu, Y.; Yang, L.; He, S.; Li, L.; Zhang, Z.; Lin, Q.; Zhang, L. A novel α-enolase-targeted drug delivery system for high efficacy prostate cancer therapy. Nanoscale 2018, 10, 13673–13683. [Google Scholar] [CrossRef]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.; Gladkov, O.; Holcombe, R.F.; Jiang, P. Final results of a phase Ib study of gemcitabine plus PEGPH20 in patients with stage IV previously untreated pancreatic cancer. J. Clin. Oncol. 2015, 33, 359. [Google Scholar] [CrossRef]
- Ding, X.; Zhou, X.; Jiang, B.; Zhao, Q.; Zhou, G. Triptolide suppresses proliferation, hypoxia-inducible factor-1α and c-Myc expression in pancreatic cancer cells. Mol. Med. Rep. 2015, 12, 4508–4513. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Salnikov, A.V.; Bauer, N.; Aleksandrowicz, E.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Schemmer, P.; Werner, J. Triptolide reverses hypoxia-induced epithelial–mesenchymal transition and stem-like features in pancreatic cancer by NF-κB downregulation. Int. J. Cancer 2014, 134, 2489–2503. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov Identifier: NCT01927965. Study of Minnelide™ in Patients With Advanced GI Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT01927965 (accessed on 23 September 2021).
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Lang, M.; Wang, X.; Wang, H.; Dong, J.; Lan, C.; Hao, J.; Huang, C.; Li, X.; Yu, M.; Yang, Y. Arsenic trioxide plus PX-478 achieves effective treatment in pancreatic ductal adenocarcinoma. Cancer Lett. 2016, 378, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R. mTOR as a positive regulator of tumor cell responses to hypoxia. In TOR; Springer: Berlin/Heidelberg, Germany, 2004; pp. 299–319. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Hezel, A.F.; Abrams, T.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Chan, J.A.; Enzinger, P.C.; Allen, B.; Clark, J.W.; Ryan, D.P. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J. Clin. Oncol. 2009, 27, 193. [Google Scholar] [CrossRef] [PubMed]
- Kordes, S.; Klümpen, H.-J.; Weterman, M.J.; Schellens, J.H.; Richel, D.J.; Wilmink, J.W. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2015, 75, 1135–1141. [Google Scholar] [CrossRef]
- Shimojo, Y.; Akimoto, M.; Hisanaga, T.; Tanaka, T.; Tajima, Y.; Honma, Y.; Takenaga, K. Attenuation of reactive oxygen species by antioxidants suppresses hypoxia-induced epithelial-mesenchymal transition and metastasis of pancreatic cancer cells. Clin. Exp. Metastasis 2013, 30, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Sibenaller, Z.A.; Welsh, J.L.; Du, C.; Witmer, J.R.; Schrock, H.E.; Du, J.; Buettner, G.R.; Goswami, P.C.; Cieslak III, J.A.; Cullen, J.J. Extracellular superoxide dismutase suppresses hypoxia-inducible factor-1α in pancreatic cancer. Free Radic. Biol. Med. 2014, 69, 357–366. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Anastasiou, D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br. J. Cancer 2017, 116, 277–286. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Cantley, L.C. Cancer’s fuel choice: New flavors for a picky eater. Mol. Cell 2015, 60, 514–523. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Finicle, B.T.; Jayashankar, V.; Edinger, A.L. Nutrient scavenging in cancer. Nat. Rev. Cancer 2018, 18, 619–633. [Google Scholar] [CrossRef]
- Biancur, D.E.; Kimmelman, A.C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2018, 1870, 67–75. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Rajeshkumar, N.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef]
- Kerr, M.C.; Teasdale, R.D. Defining macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Ferreira, L.M. Cancer metabolism: The Warburg effect today. Exp. Mol. Pathol. 2010, 89, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef]
- Derle, A.; De Santis, M.C.; Gozzelino, L.; Ratto, E.; Martini, M. The role of metabolic adaptation to nutrient stress in pancreatic cancer. Cell Stress 2018, 2, 332. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-W.; Purkayastha, A.; Jones, K.T.; Thaker, S.K.; Banerjee, U. In vivo genetic dissection of tumor growth and the Warburg effect. eLife 2016, 5, e18126. [Google Scholar] [CrossRef] [PubMed]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD+ metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Waste not, want not: Lactate oxidation fuels the TCA cycle. Cell Metab. 2017, 26, 803–804. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.-N.; Vidal, N.; Berthezène, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.B.; Elhassan, G.O.; Ibrahim, M.E.; Orozco, J.D.P.; Cardone, R.A.; Reshkin, S.J. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar] [CrossRef]
- Guo, Y.; Deng, Y.; Li, X.; Ning, Y.; Lin, X.; Guo, S.; Chen, M.; Han, M. Glutaminolysis was induced by TGF-β1 through PP2Ac regulated Raf-MEK-ERK signaling in endothelial cells. PLoS ONE 2016, 11, e0162658. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Abrego, J.; Gunda, V.; Vernucci, E.; Shukla, S.K.; King, R.J.; Dasgupta, A.; Goode, G.; Murthy, D.; Yu, F.; Singh, P.K. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017, 400, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Tumas, J.; Baskirova, I.; Petrenas, T.; Norkuniene, J.; Strupas, K.; Sileikis, A. Towards a personalized approach in pancreatic cancer diagnostics through plasma amino acid analysis. Anticancer Res. 2019, 39, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.; Biancur, D.E. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Achreja, A.; Meurs, N.; Animasahun, O.; Owen, S.; Mittal, A.; Parikh, P.; Lo, T.-W.; Franco-Barraza, J.; Shi, J. Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat. Metab. 2020, 2, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Altan, B.; Kaira, K.; Watanabe, A.; Kubo, N.; Bao, P.; Dolgormaa, G.; Bilguun, E.-O.; Araki, K.; Kanai, Y.; Yokobori, T. Relationship between LAT1 expression and resistance to chemotherapy in pancreatic ductal adenocarcinoma. Cancer Chemother. Pharmacol. 2018, 81, 141–153. [Google Scholar] [CrossRef]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouysségur, J.; Vučetić, M. Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef] [PubMed]
- Tao, P.; Wang, H.; Gao, G. CYP2J2-produced epoxyeicosatrienoic acids contribute to the ferroptosis resistance of pancreatic ductal adenocarcinoma in a PPAR γ-dependent manner. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2021, 46, 932–941. [Google Scholar] [CrossRef]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Zeh, H.J.; Kang, R.; Bai, L.; Tang, D. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Zhong, M.; Yang, M.; Sun, X.; Liu, J.; Kroemer, G.; Lotze, M.; Zeh III, H.J.; Kang, R. Inhibition of Aurora kinase A induces necroptosis in pancreatic carcinoma. Gastroenterology 2017, 153, 1429–1443.e5. [Google Scholar] [CrossRef] [PubMed]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, Y.; Liu, J.; Kang, R.; Klionsky, D.J.; Tang, D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 2021, 17, 948–960. [Google Scholar] [CrossRef]
- Tang, R.; Wu, Z.; Rong, Z.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Ferroptosis-related lncRNA pairs to predict the clinical outcome and molecular characteristics of pancreatic ductal adenocarcinoma. Brief. Bioinform. 2021. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Santofimia-Castaño, P.; Liu, X.; Xia, Y.; Peng, L.; Gotorbe, C.; Neira, J.L.; Tang, D.; Pouyssegur, J.; Iovanna, J. NUPR1 inhibitor ZZW-115 induces ferroptosis in a mitochondria-dependent manner. Cell Death Discov. 2021, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kuang, F.; Liu, J.; Xie, Y.; Tang, D.; Kang, R. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem. Biol. 2021, 28, 765–775.e5. [Google Scholar] [CrossRef]
- Liu, J.; Dai, E.; Kang, R.; Kroemer, G.; Tang, D. The dark side of ferroptosis in pancreatic cancer. Oncoimmunology 2021, 10, 1868691. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Targeting ferroptosis in pancreatic cancer: A double-edged sword. Trends Cancer 2021. [Google Scholar] [CrossRef]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Castellanos, G.; Masoud, R.; Carrier, A. Mitochondrial metabolism in PDAC: From better knowledge to new targeting strategies. Biomedicines 2020, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhao, X.; Ouyang, H.; Sun, F.; Zhang, H.; Zhou, C.; Shen, H. The metabolic features of normal pancreas and pancreatic adenocarcinoma: Preliminary result of in vivo proton magnetic resonance spectroscopy at 3.0 T. J. Comput. Assist. Tomogr. 2011, 35, 539–543. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Kumagai-Braesch, M.; Herrington, M.K.; Larsson, J.; Permert, J. Increased lipid metabolism and cell turnover of MiaPaCa2 cells induced by high-fat diet in an orthotopic system. Metabolism 2009, 58, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; De Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef]
- Bian, Y.; Yu, Y.; Wang, S.; Li, L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015, 463, 612–617. [Google Scholar] [CrossRef]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers 2018, 10, 3. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2012, 1825, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Voorde, J.V.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.Y.; Lee, M.; Whang, S.H.; Kim, J.-W.; Cho, A.; Yun, M. Regulation of acetate utilization by monocarboxylate transporter 1 (MCT1) in hepatocellular carcinoma (HCC). Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Chiche, J.; Pouysségur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611. [Google Scholar] [CrossRef]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Voorde, J.V.; Blyth, K. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef]
- Nishi, K.; Suzuki, K.; Sawamoto, J.; Tokizawa, Y.; Iwase, Y.; Yumita, N.; Ikeda, T. Inhibition of fatty acid synthesis induces apoptosis of human pancreatic cancer cells. Anticancer Res. 2016, 36, 4655–4660. [Google Scholar] [CrossRef] [PubMed]
- Bracci, P.M. Obesity and pancreatic cancer: Overview of epidemiologic evidence and biologic mechanisms. Mol. Carcinog. 2012, 51, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Brewer, M.; Duff, A.; Lightfoot, S.; Brush, R.S.; Anderson, R.E.; Rao, C.V. Endogenous n-3 polyunsaturated fatty acids delay progression of pancreatic ductal adenocarcinoma in Fat-1-p48Cre/+-LSL-KrasG12D/+ mice. Neoplasia 2012, 14, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Duckwall, C.S.; Murphy, T.A.; Young, J.D. Mapping cancer cell metabolism with13C flux analysis: Recent progress and future challenges. J. Carcinog. 2013, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Wang, J.; Ye, C.; Chen, C.; Xiong, H.; Xie, B.; Zhou, J.; Chen, Y.; Zheng, S.; Wang, L. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 16875. [Google Scholar] [CrossRef] [PubMed]
- Sharen, G.; Peng, Y.; Cheng, H.; Liu, Y.; Shi, Y.; Zhao, J. Prognostic value of GLUT-1 expression in pancreatic cancer: Results from 538 patients. Oncotarget 2017, 8, 19760. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Yang, J.; Li, D.C.; He, S.B.; Zhu, D.M.; Zhang, L.F.; Zhang, X.; Chen, X.C.; Zhang, B.; Zhou, J. Expression and clinical significance of glucose transporter-1 in pancreatic cancer. Oncol. Lett. 2016, 12, 243–249. [Google Scholar] [CrossRef]
- Davis-Yadley, A.H.; Abbott, A.M.; Pimiento, J.M.; Chen, D.-T.; Malafa, M.P. Increased Expression of the GLUT-1 Gene is Associated With Worse Overall Survival in Resected Pancreatic Adenocarcinoma. Pancreas 2016, 45, 974. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, P.; Zhou, S.; Zhang, L.; Zhao, J.-h.; Tang, J.-h. Predictive value of glucose transporter-1 and glucose transporter-3 for survival of cancer patients: A meta-analysis. Oncotarget 2017, 8, 13206. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Madunić, J.; Madunić, I.V.; Gajski, G.; Popić, J.; Garaj-Vrhovac, V. Apigenin: A dietary flavonoid with diverse anticancer properties. Cancer Lett. 2018, 413, 11–22. [Google Scholar] [CrossRef]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.-Z.; Strouch, M.J.; Grippo, P.J.; Mirzoeva, S.; Pelling, J.C.; Bentrem, D.J. Apigenin down-regulates the hypoxia response genes: HIF-1α, GLUT-1, and VEGF in human pancreatic cancer cells. J. Surg. Res. 2011, 167, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.-Z.; Milam, B.M.; Strouch, M.; Pelling, J.C.; Bentrem, D.J. Apigenin inhibits the GLUT-1 glucose transporter and the phosphoinositide 3-kinase/Akt pathway in human pancreatic cancer cells. Pancreas 2008, 37, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Bakhoda, M.R.; Bahmanpour, Z.; Ilkhani, K.; Zarrabi, A.; Makvandi, P.; Khan, H.; Mazaheri, S.; Darvish, M.; Mirzaei, H. Apigenin as Tumor Suppressor in Cancers: Biotherapeutic Activity, Nanodelivery, and Mechanisms with Emphasis on Pancreatic Cancer. Front. Chem. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ren, B.; Yang, G.; Wang, H.; Chen, G.; You, L.; Zhang, T.; Zhao, Y. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell. Mol. Life Sci. 2020, 77, 305–321. [Google Scholar] [CrossRef]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2017, 8, 56081. [Google Scholar] [CrossRef] [PubMed]
- Min, J.W.; Kim, K.I.; Kim, H.-A.; Kim, E.-K.; Noh, W.C.; Jeon, H.B.; Cho, D.-H.; Oh, J.S.; Park, I.-C.; Hwang, S.-G. INPP4B-mediated tumor resistance is associated with modulation of glucose metabolism via hexokinase 2 regulation in laryngeal cancer cells. Biochem. Biophys. Res. Commun. 2013, 440, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Glycolysis in cancer: A potential target for therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Morten, K.J.; Badder, L.; Knowles, H.J. Differential regulation of HIF-mediated pathways increases mitochondrial metabolism and ATP production in hypoxic osteoclasts. J. Pathol. 2013, 229, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Isayev, O.; Rausch, V.; Bauer, N.; Liu, L.; Fan, P.; Zhang, Y.; Gladkich, J.; Nwaeburu, C.C.; Mattern, J.; Mollenhauer, M. Inhibition of glucose turnover by 3-bromopyruvate counteracts pancreatic cancer stem cell features and sensitizes cells to gemcitabine. Oncotarget 2014, 5, 5177. [Google Scholar] [CrossRef] [PubMed]
- Ota, S.; Geschwind, J.-F.H.; Buijs, M.; Wijlemans, J.W.; Kwak, B.K.; Ganapathy-Kanniappan, S. Ultrasound-guided direct delivery of 3-bromopyruvate blocks tumor progression in an orthotopic mouse model of human pancreatic cancer. Target. Oncol. 2013, 8, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.K.; Bruce, J.I.; Siriwardena, A.K. Glucose metabolic phenotype of pancreatic cancer. World J. Gastroenterol. 2016, 22, 3471. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kang, H.J.; Lee, S.Y.; Chung, S.J.; Kang, S.; Chi, S.W.; Cho, S.; Lee, S.C.; Lee, C.-K.; Park, B.C. Glyceraldehyde-3-phosphate, a glycolytic intermediate, plays a key role in controlling cell fate via inhibition of caspase activity. Mol. Cells 2009, 28, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, F.; Ge, Z.; Chen, B.; Yu, J.; Xin, M.; Wang, J.; An, L.; Wei, J.; Wu, L. Fructose-bisphosphate aldolase a regulates hypoxic adaptation in hepatocellular carcinoma and involved with tumor malignancy. Dig. Dis. Sci. 2019, 64, 3215–3227. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-L.; Xu, L.-T.; Qi, Q.; Geng, Y.-W.; Chen, H.; Meng, Z.-Q.; Wang, P.; Chen, Z. Serum lactate dehydrogenase predicts prognosis and correlates with systemic inflammatory response in patients with advanced pancreatic cancer after gemcitabine-based chemotherapy. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R. Lactate dehydrogenase 5: An old friend and a new hope in the war on cancer. Cancer Lett. 2015, 358, 1–7. [Google Scholar] [CrossRef]
- Kolev, Y.; Uetake, H.; Takagi, Y.; Sugihara, K. Lactate dehydrogenase-5 (LDH-5) expression in human gastric cancer: Association with hypoxia-inducible factor (HIF-1α) pathway, angiogenic factors production and poor prognosis. Ann. Surg. Oncol. 2008, 15, 2336–2344. [Google Scholar] [CrossRef]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.; Boggi, U. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.H.; Vassileva, V.; Acedo, P.; Olde Damink, S.W.; Malago, M.; Dhar, D.K.; Pereira, S.P. Targeting pyruvate kinase M2 and lactate dehydrogenase a is an effective combination strategy for the treatment of pancreatic cancer. Cancers 2019, 11, 1372. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef]
- Gupta, V.; Bamezai, R.N. Human pyruvate kinase M2: A multifunctional protein. Protein Sci. 2010, 19, 2031–2044. [Google Scholar] [CrossRef]
- Li, C.; Zhao, Z.; Zhou, Z.; Liu, R. PKM2 promotes cell survival and invasion under metabolic stress by enhancing Warburg effect in pancreatic ductal adenocarcinoma. Dig. Dis. Sci. 2016, 61, 767–773. [Google Scholar] [CrossRef]
- Yokoyama, M.; Tanuma, N.; Shibuya, R.; Shiroki, T.; Abue, M.; Yamamoto, K.; Miura, K.; Yamaguchi, K.; Sato, I.; Tamai, K. Pyruvate kinase type M2 contributes to the development of pancreatic ductal adenocarcinoma by regulating the production of metabolites and reactive oxygen species. Int. J. Oncol. 2018, 52, 881–891. [Google Scholar] [CrossRef]
- Tian, S.; Li, P.; Sheng, S.; Jin, X. Upregulation of pyruvate kinase M2 expression by fatty acid synthase contributes to gemcitabine resistance in pancreatic cancer. Oncol. Lett. 2018, 15, 2211–2217. [Google Scholar] [CrossRef]
- Van Veelen, C.W.; Verbiest, H.; Vlug, A.M.; Rijksen, G.; Staal, G.E. Isozymes of pyruvate kinase from human brain, meningiomas, and malignant gliomas. Cancer Res. 1978, 38, 4681–4687. [Google Scholar]
- Kim, D.J.; Park, Y.S.; Kang, M.G.; You, Y.-M.; Jung, Y.; Koo, H.; Kim, J.-A.; Kim, M.-J.; Hong, S.-M.; Lee, K.B. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp. Cell Res. 2015, 336, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.-Y.; Oh, S.; Kang, Y.K.; Lee, K.-m.; Kang, M.; Jang, Y.J.; Yang, S.-J.; Hong, Y.K. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Baek, G.; Yan, F.T.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Dadhich, R.K.; Dhup, S.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef]
- Lai, I.-L.; Chou, C.-C.; Lai, P.-T.; Fang, C.-S.; Shirley, L.A.; Yan, R.; Mo, X.; Bloomston, M.; Kulp, S.K.; Bekaii-Saab, T. Targeting the Warburg effect with a novel glucose transporter inhibitor to overcome gemcitabine resistance in pancreatic cancer cells. Carcinogenesis 2014, 35, 2203. [Google Scholar] [CrossRef][Green Version]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic β-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Gotoh, K.; Eguchi, H.; Kobayashi, S.; Iwagami, Y.; Tomimaru, Y.; Akita, H.; Asaoka, T.; Noda, T.; Takeda, Y. Impact of CD36 on chemoresistance in pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2020, 27, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Alo, P.L.; Amini, M.; Piro, F.; Pizzuti, L.; Sebastiani, V.; Botti, C.; Murari, R.; Zotti, G.; Di Tondo, U. Immnunohistochemical expression and prognostic significance of fatty acid synthase in pancreatic carcinoma. Anticancer Res. 2007, 27, 2523–2527. [Google Scholar] [PubMed]
- Walter, K.; Hong, S.-M.; Nyhan, S.; Canto, M.; Fedarko, N.; Klein, A.; Griffith, M.; Omura, N.; Medghalchi, S.; Kuhajda, F. Serum fatty acid synthase as a marker of pancreatic neoplasia. Cancer Epidemiol. Prev. Biomark. 2009, 18, 2380–2385. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, H.; Li, Z.; Zhao, Z.; Yip-Schneider, M.; Fan, Q.; Schmidt, C.M.; Chiorean, E.G.; Xie, J.; Cheng, L. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int. J. Biochem. Mol. Biol. 2011, 2, 89. [Google Scholar]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Hong, Y.; Tao, X.; Wei, X.; Zhang, L.; Li, Q. An indispensable role of CPT-1a to survive cancer cells during energy stress through rewiring cancer metabolism. Tumor Biol. 2016, 37, 15795–15804. [Google Scholar] [CrossRef]
- Butler, E.B.; Zhao, Y.; Muñoz-Pinedo, C.; Lu, J.; Tan, M. Stalling the engine of resistance: Targeting cancer metabolism to overcome therapeutic resistance. Cancer Res. 2013, 73, 2709–2717. [Google Scholar] [CrossRef]
- Whatcott, C.; Han, H.; Posner, R.G.; Von Hoff, D.D. Tumor-stromal interactions in pancreatic cancer. Crit. Rev. Oncog. 2013, 18, 135–151. [Google Scholar] [CrossRef]
- Banerjee, S.; Modi, S.; McGinn, O.; Zhao, X.; Dudeja, V.; Ramakrishnan, S.; Saluja, A.K. Impaired synthesis of stromal components in response to minnelide improves vascular function, drug delivery, and survival in pancreatic cancer. Clin. Cancer Res. 2016, 22, 415–425. [Google Scholar] [CrossRef]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathôt, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R., Jr.; Topaloglu, U. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).