
Cell-Type-Specific Profibrotic Scores across Multi-Organ Systems Predict Cancer Prognosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Multi-Organ Tissue Atlas at Single-Cell Resolution
2.2. Preprocessing the Single-Cell Tissue Atlas Data
2.3. Identifying Profibrotic Cell Populations and Their Transcriptomic Signatures
2.4. Constructing Cell-Type-Specific Profibrotic Co-Active Pathway Network
2.5. Validating Cell-Type-Specific Gene Signatures in Both Human and Mouse scRNA-Seq Data
2.6. Calculating Cell-Type-Specific Profibrotic Phenotype Scores in Bulk TCGA Tumors
2.7. Survival Analysis
3. Results
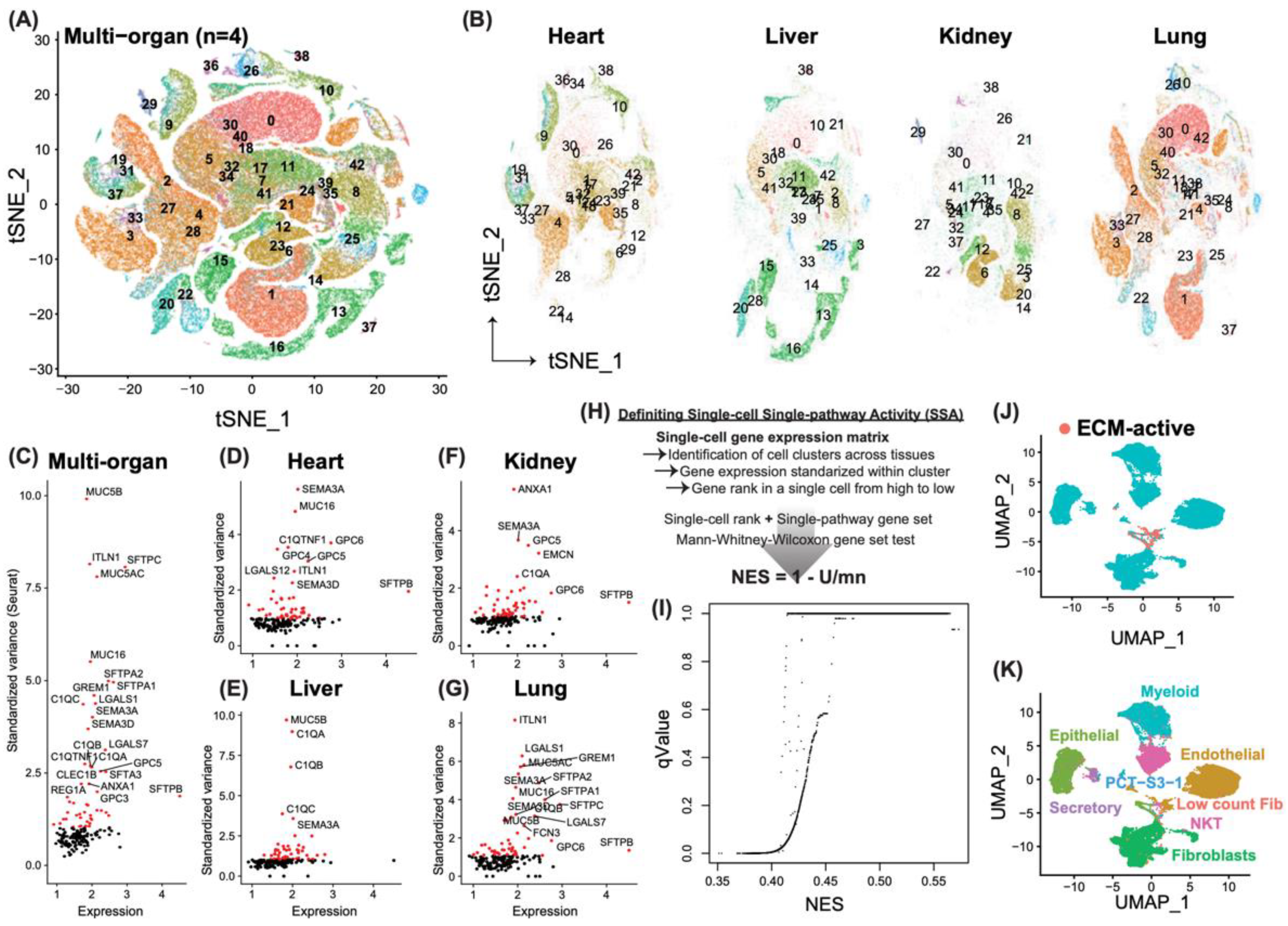
3.1. Identification of Novel Profibrotic Cell Populations Induced by Severe Acute Viral Infection
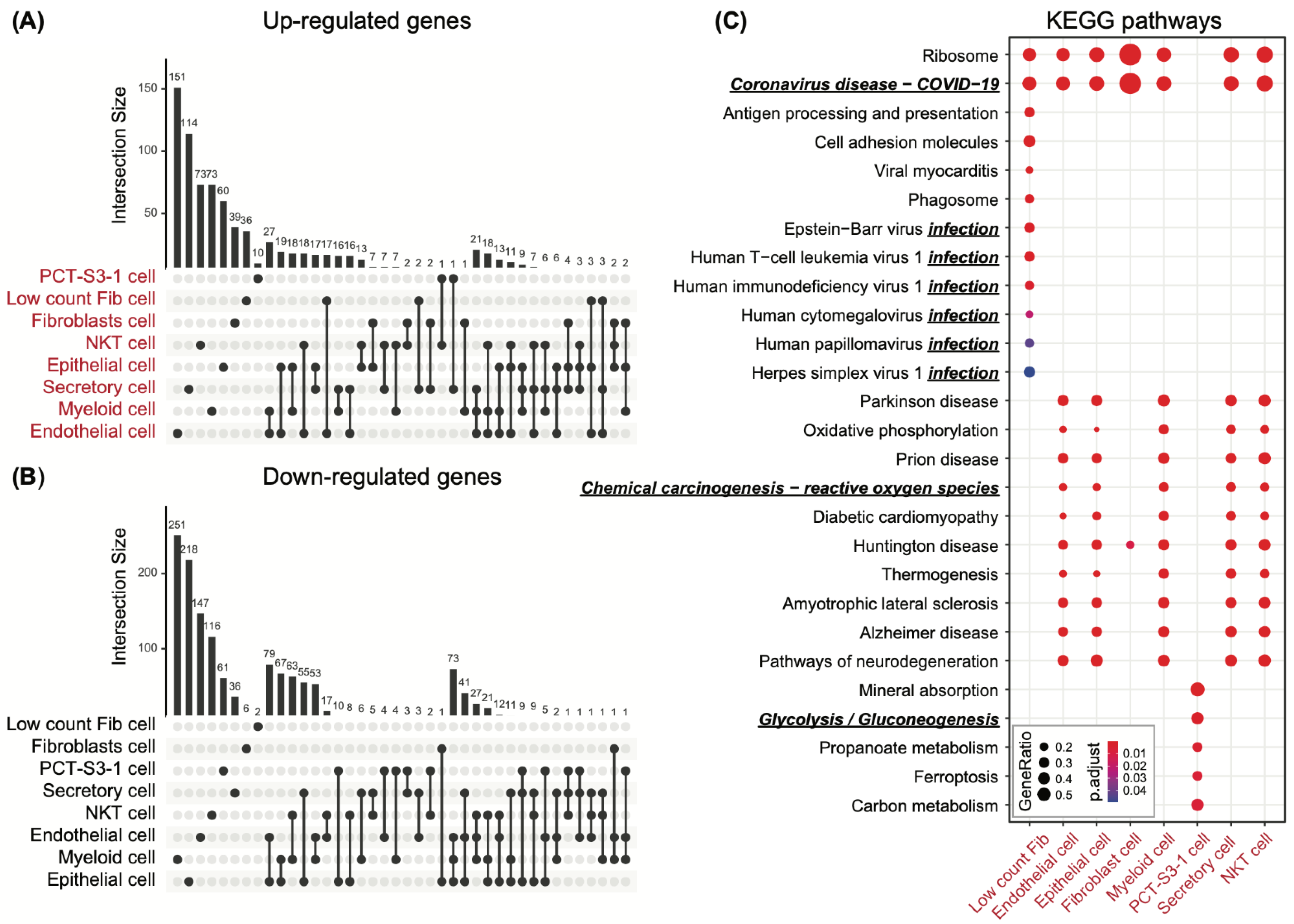
3.2. Cell-Type-Specific Differential Genes in Support of the Profibrotic Cellular Phenotype
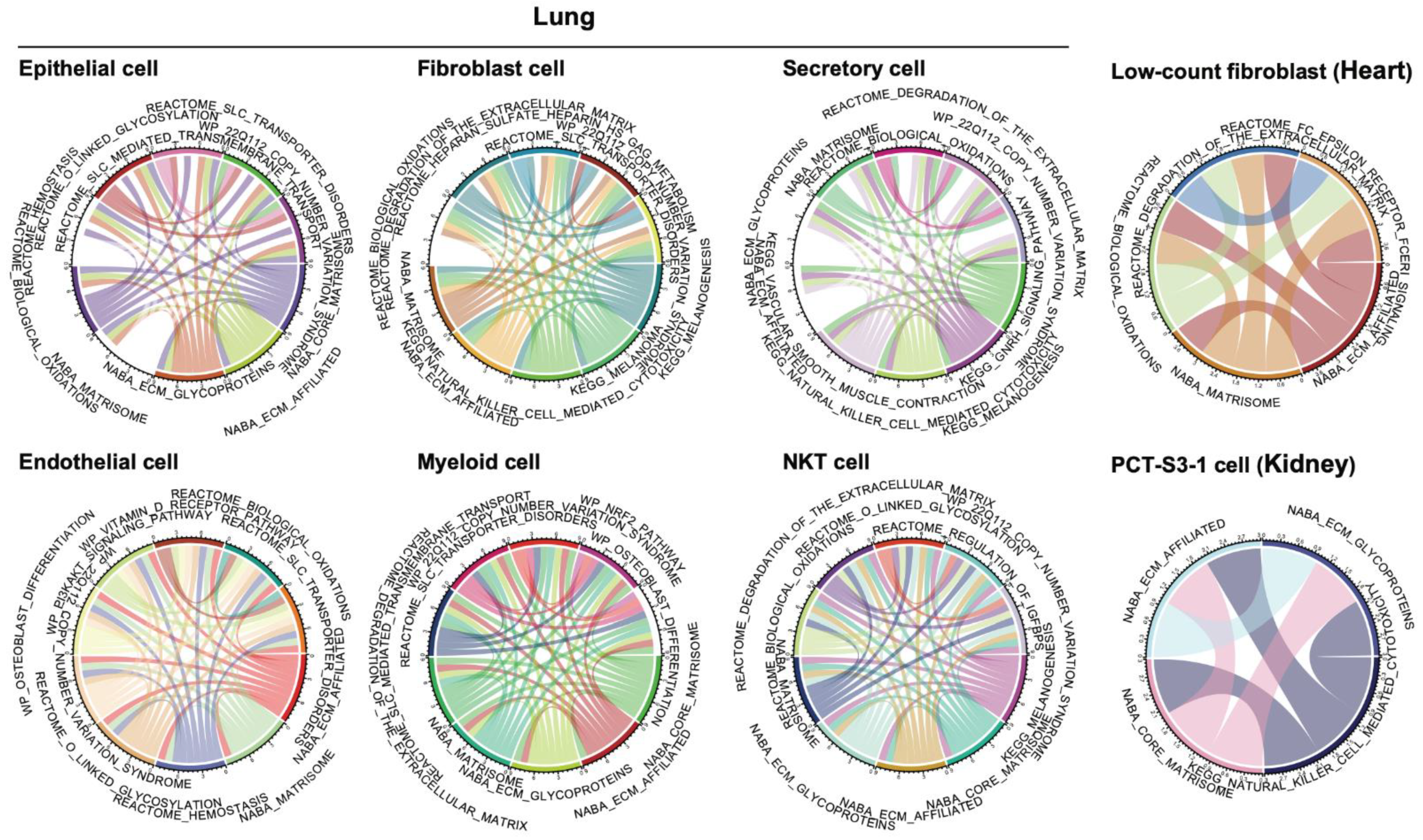
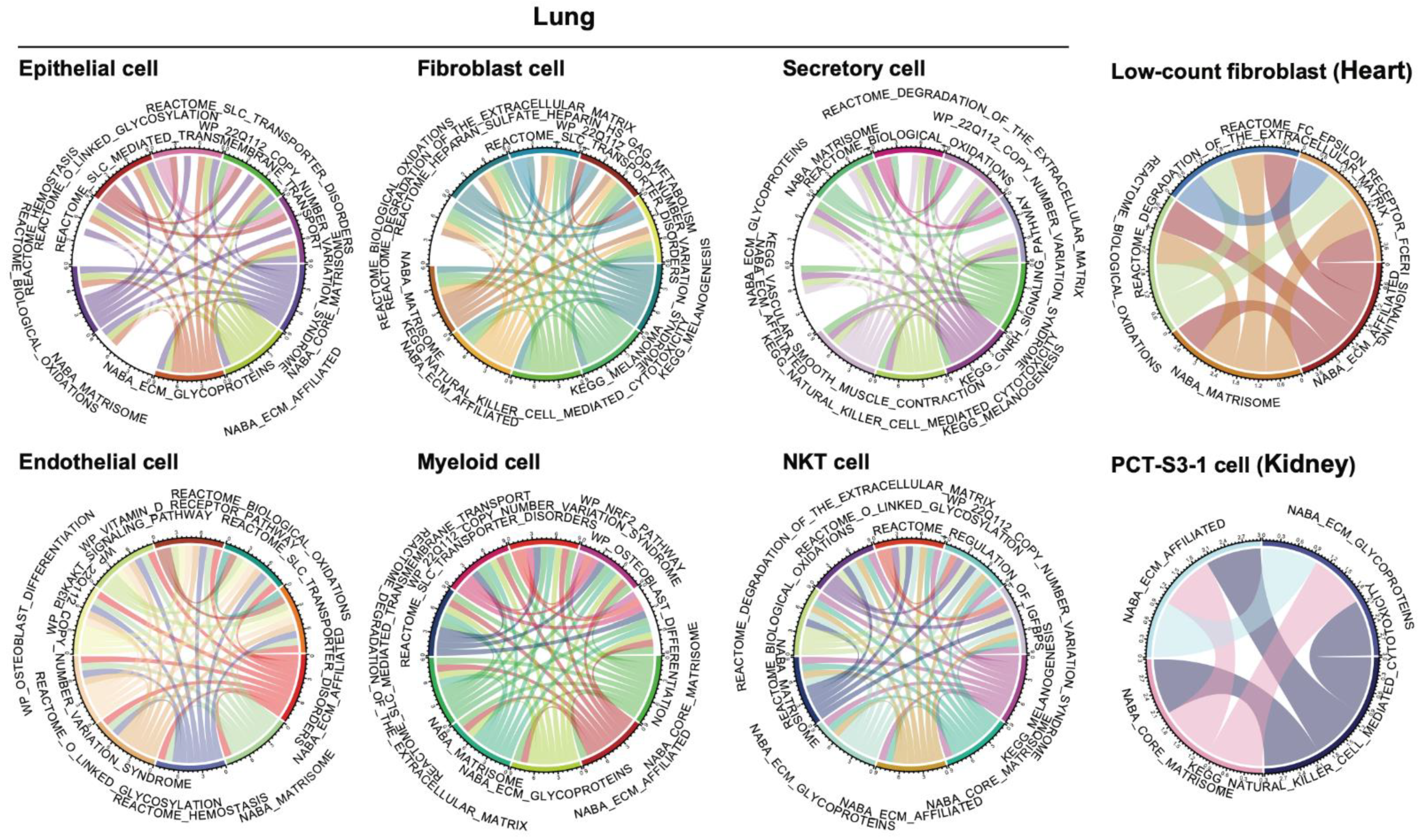
3.3. Co-Active Functional Pathways Driving the Cell-Type-Specific Profibrotic Phenotype
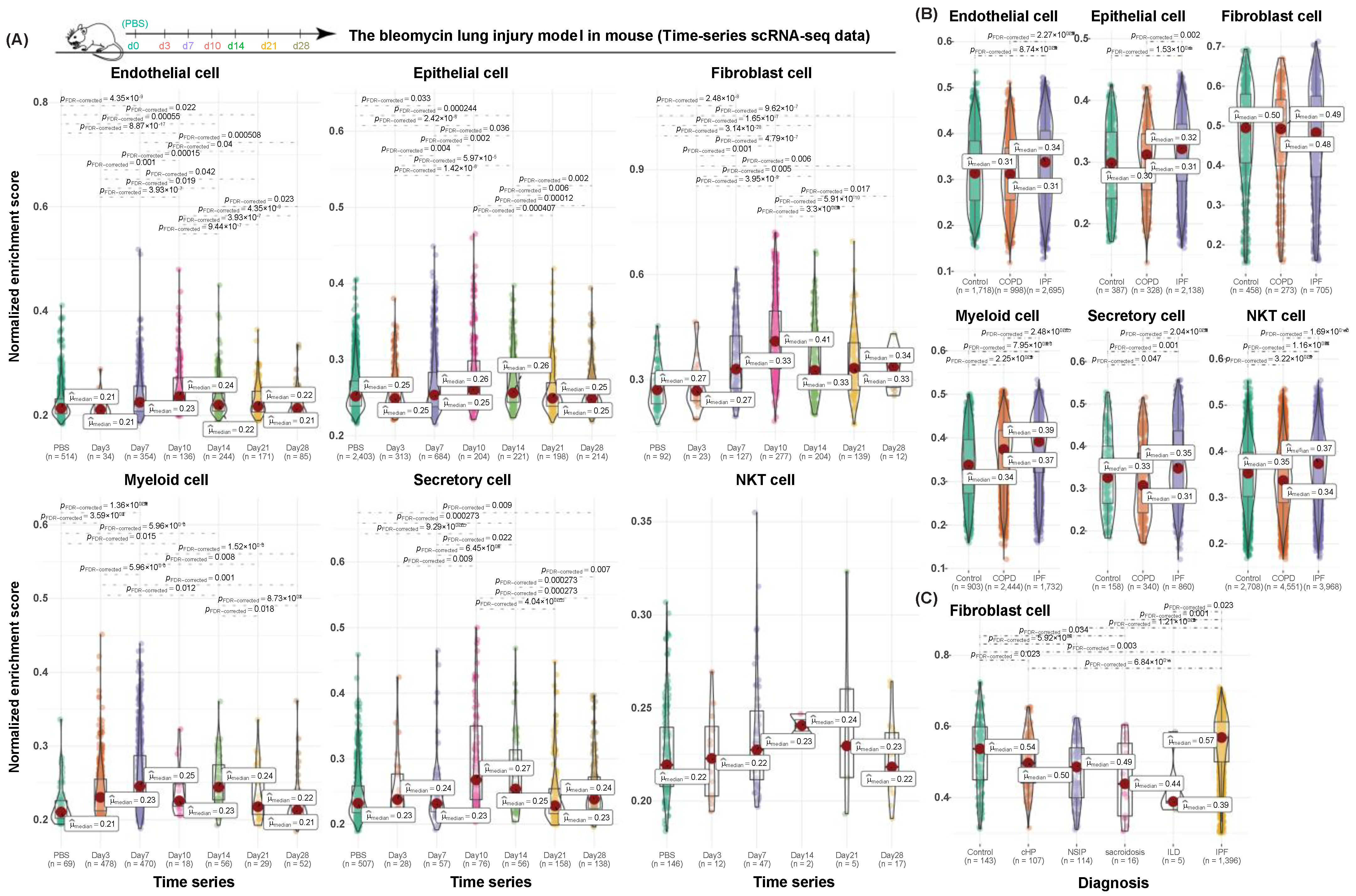
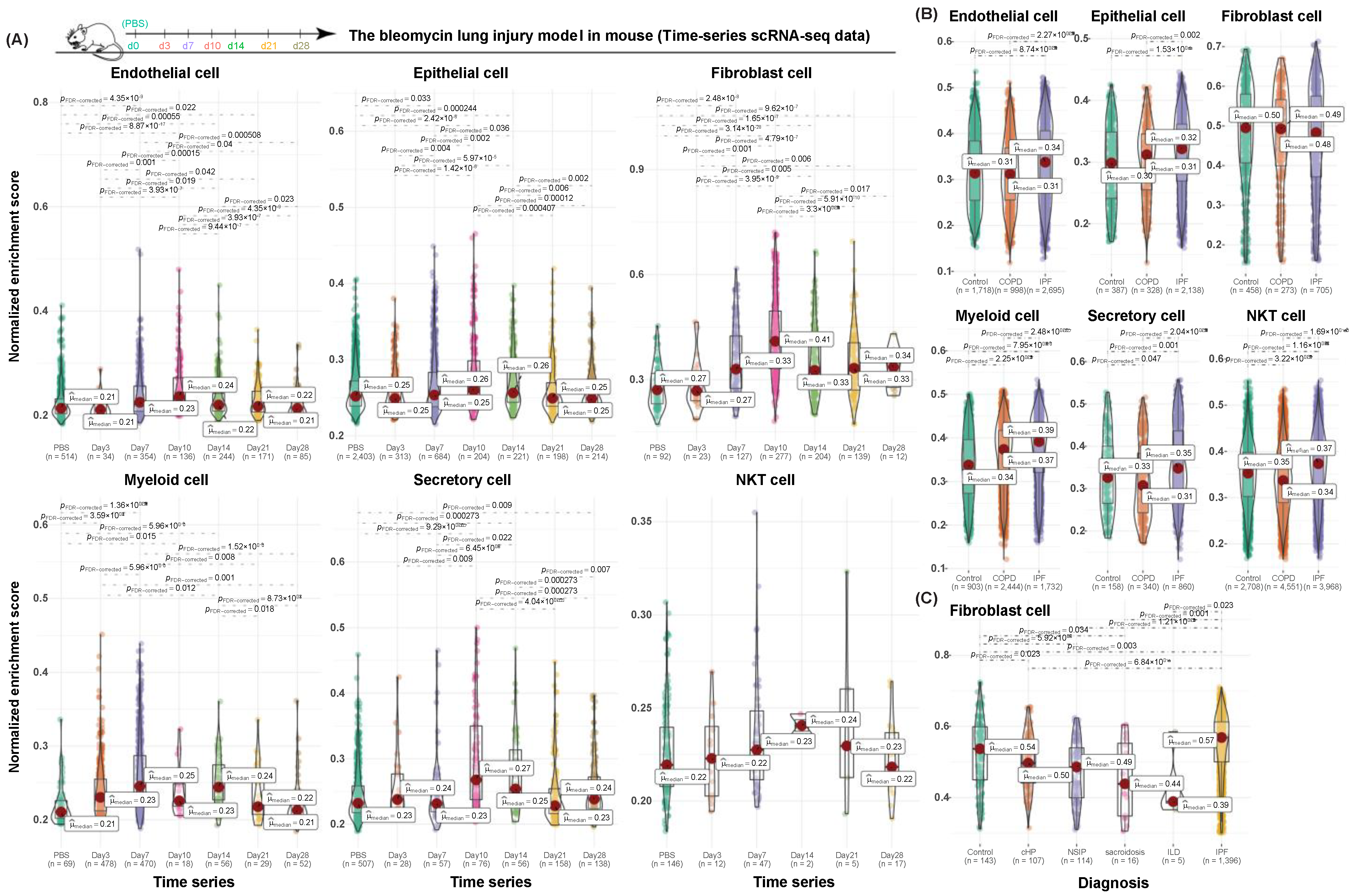
3.4. Validation of Cell-Type-Specific Profibrotic Gene Signatures in Both Human and Mouse Datasets
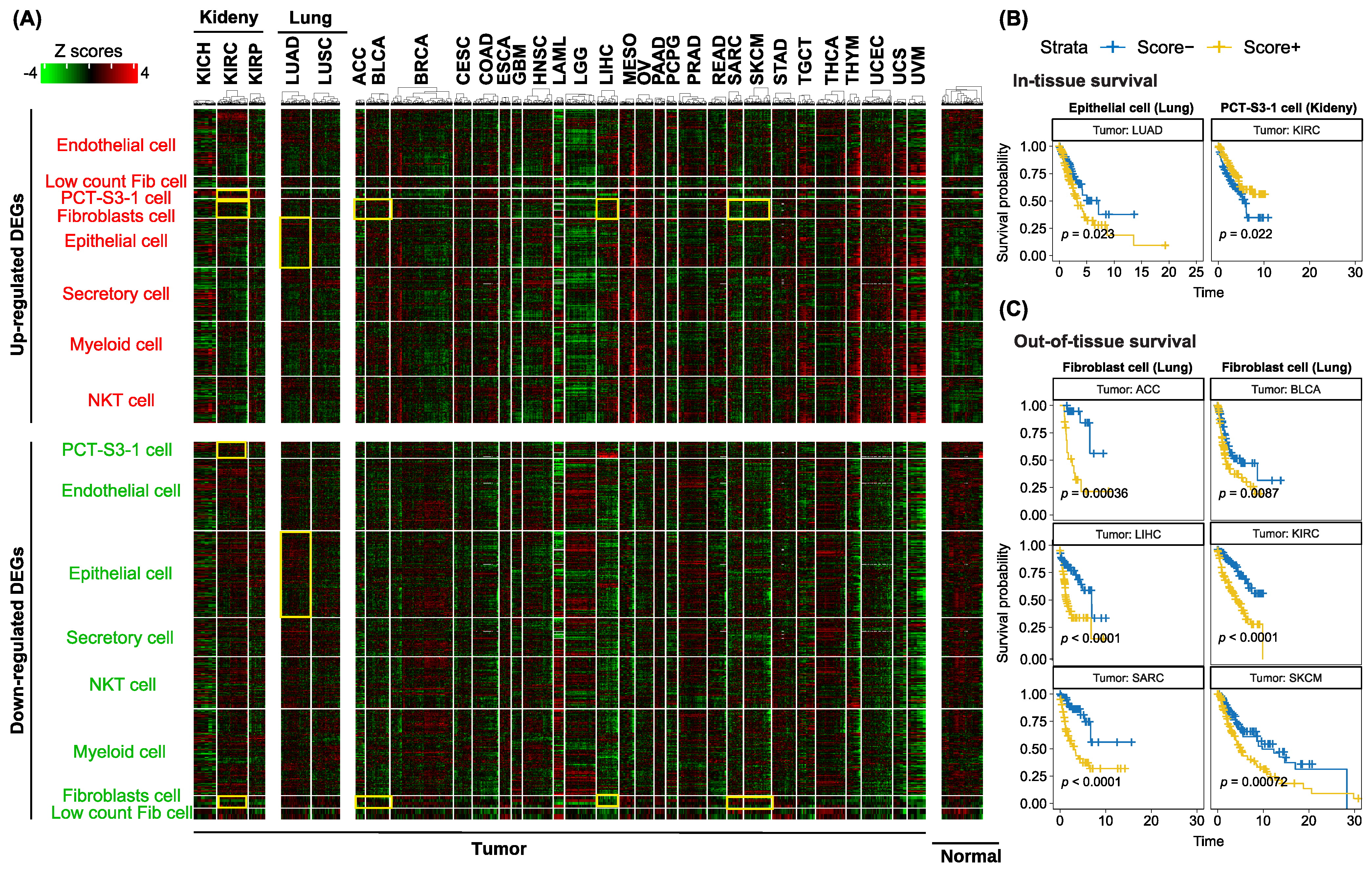
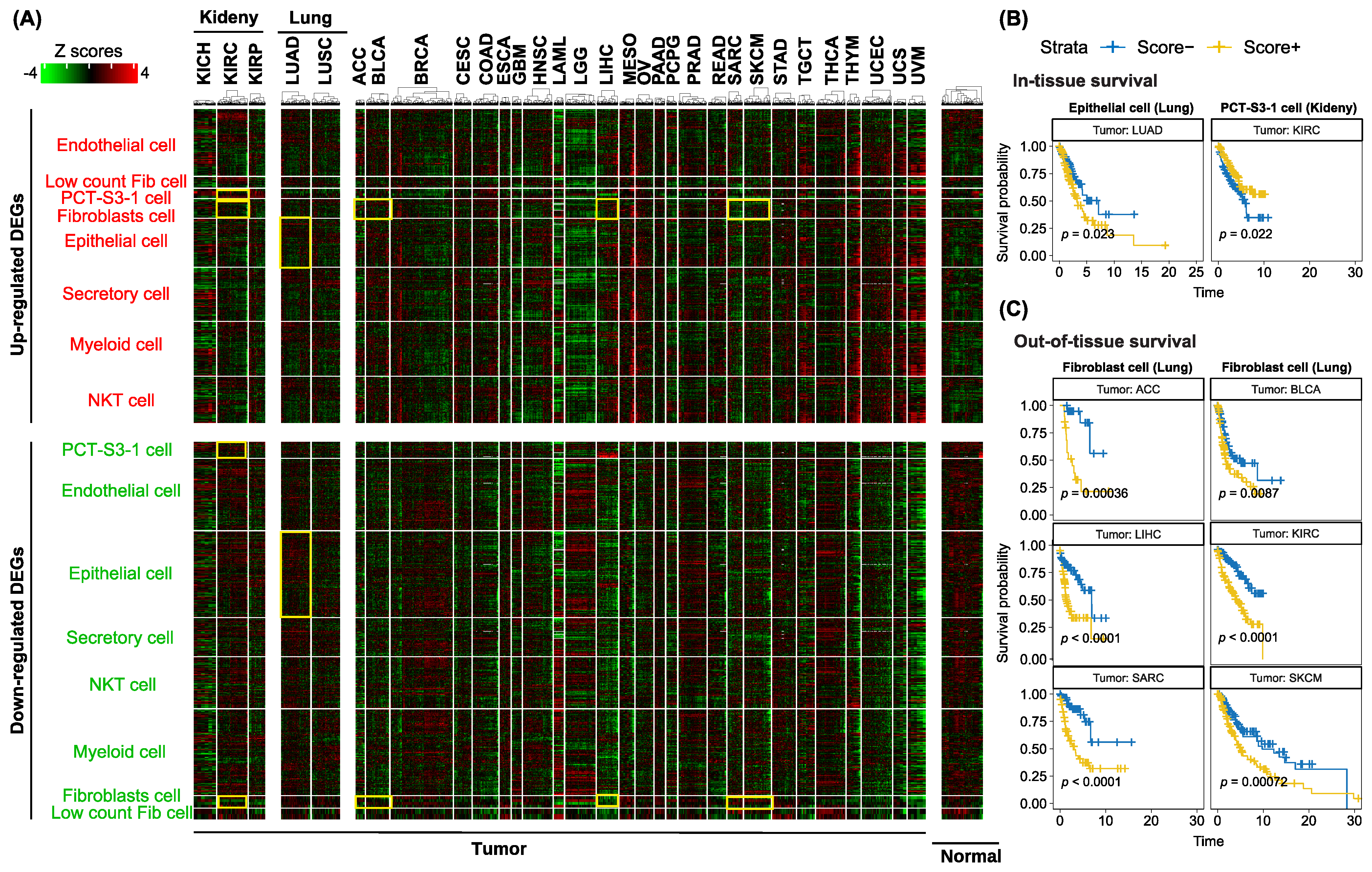
3.5. Utilization of Cell-Type-Specific Profibrotic Scores to Predict Survival in the TCGA Cohort
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, T.S.; Tager, A.M.; Borok, Z.; Moore, B.B.; Schwartz, D.A.; Anstrom, K.J.; Bar-Joseph, Z.; Bitterman, P.; Blackburn, M.R.; Bradford, W.; et al. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am. J. Respir. Crit. Care Med. 2014, 189, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Aspects Med. 2019, 65, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Bulow, R.D.; Boor, P. Extracellular matrix in kidney fibrosis: More than just a scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Brenner, D.A. Mechanisms of fibrogenesis. Exp. Biol. Med. 2008, 233, 109–122. [Google Scholar] [CrossRef]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, M.M.; Akcali, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell 2017, 20, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Pei, G.; Yao, J.; Ding, Q.; Jia, P.; Zhao, Z. Cell-type deconvolution analysis identifies cancer-associated myofibroblast component as a poor prognostic factor in multiple cancer types. Oncogene 2021, 40, 4686–4694. [Google Scholar] [CrossRef]
- Wohlfahrt, T.; Rauber, S.; Uebe, S.; Luber, M.; Soare, A.; Ekici, A.; Weber, S.; Matei, A.E.; Chen, C.W.; Maier, C.; et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 2019, 566, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Kral, J.B.; Kuttke, M.; Schrottmaier, W.C.; Birnecker, B.; Warszawska, J.; Wernig, C.; Paar, H.; Salzmann, M.; Sahin, E.; Brunner, J.S.; et al. Sustained PI3K Activation exacerbates BLM-induced Lung Fibrosis via activation of pro-inflammatory and pro-fibrotic pathways. Sci. Rep. 2016, 6, 23034. [Google Scholar] [CrossRef]
- Mancini, O.K.; Acevedo, M.; Fazez, N.; Cuillerier, A.; Ruiz, A.F.; Huynh, D.N.; Burelle, Y.; Ferbeyre, G.; Baron, M.; Servant, M.J. Oxidative stress-induced senescence mediates inflammatory and fibrotic phenotypes in fibroblasts from systemic sclerosis patients. Rheumatology 2021, keab477. [Google Scholar] [CrossRef]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.S.; Wilcox, C.S. NADPH oxidases in the kidney. Antioxid. Redox Signal. 2006, 8, 1597–1607. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Fleming, S.J.; Marioni, J.C.; Babadi, M. CellBender remove-background: A deep generative model for unsupervised removal of background noise from scRNA-seq datasets. BioRxiv 2019, 791699. [Google Scholar]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, A.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Wang, Y.Y.; Simon, L.M.; Dai, Y.; Zhao, Z.; Jia, P. Gene expression imputation and cell-type deconvolution in human brain with spatiotemporal precision and its implications for brain-related disorders. Genome Res. 2021, 31, 146–158. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; The R Foundation: Vienna, Austria, 2013. [Google Scholar]
- Lekkerkerker, A.N.; Aarbiou, J.; van Es, T.; Janssen, R.A. Cellular players in lung fibrosis. Curr. Pharm. Des. 2012, 18, 4093–4102. [Google Scholar] [CrossRef]
- Peyser, R.; MacDonnell, S.; Gao, Y.; Cheng, L.; Kim, Y.; Kaplan, T.; Ruan, Q.; Wei, Y.; Ni, M.; Adler, C.; et al. Defining the activated fibroblast population in lung fibrosis using single-cell sequencing. Am. J. Respir. Cell. Mol. Biol. 2019, 61, 74–85. [Google Scholar] [CrossRef]
- Kato, S.; Inui, N.; Hakamata, A.; Suzuki, Y.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; Watanabe, H.; Suda, T. Changes in pulmonary endothelial cell properties during bleomycin-induced pulmonary fibrosis. Respir. Res. 2018, 19, 127. [Google Scholar] [CrossRef]
- Kania, G.; Rudnik, M.; Distler, O. Involvement of the myeloid cell compartment in fibrogenesis and systemic sclerosis. Nat. Rev. Rheumatol. 2019, 15, 288–302. [Google Scholar] [CrossRef]
- Han, Q.J.; Mu, Y.L.; Zhao, H.J.; Zhao, R.R.; Guo, Q.J.; Su, Y.H.; Zhang, J. Fasudil prevents liver fibrosis via activating natural killer cells and suppressing hepatic stellate cells. World J. Gastroenterol. 2021, 27, 3581–3594. [Google Scholar] [CrossRef]
- Shuch, B.; Linehan, W.M.; Srinivasan, R. Aerobic glycolysis: A novel target in kidney cancer. Expert Rev. Anticancer Ther. 2013, 13, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezkalla, S.H.; Kloner, R.A. Viral myocarditis: 1917–2020: From the Influenza A to the COVID-19 pandemics. Trends Cardiovasc. Med. 2021, 31, 163–169. [Google Scholar] [CrossRef]
- Huang, S.Y.; Fang, C.Y.; Wu, C.C.; Tsai, C.H.; Lin, S.F.; Chen, J.Y. Reactive oxygen species mediate Epstein-Barr virus reactivation by N-methyl-N’-nitro-N-nitrosoguanidine. PLoS ONE 2013, 8, e84919. [Google Scholar] [CrossRef] [Green Version]
- Miskowiak, K.W.; Johnsen, S.; Sattler, S.M.; Nielsen, S.; Kunalan, K.; Rungby, J.; Lapperre, T.; Porsberg, C.M. Cognitive impairments four months after COVID-19 hospital discharge: Pattern, severity and association with illness variables. Eur. Neuropsychopharmacol. 2021, 46, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, J.; Jeong, H.H.; Chen, W.; Jia, P.; Zhao, Z. Association of CXCR6 with COVID-19 severity: Delineating the host genetic factors in transcriptomic regulation. Hum. Genet. 2021, 140, 1313–1328. [Google Scholar] [CrossRef]
- Jeong, H.H.; Jia, J.; Dai, Y.; Simon, L.M.; Zhao, Z. Investigating cellular trajectories in the severity of COVID-19 and their transcriptional programs using machine learning approaches. Genes 2021, 12, 635. [Google Scholar] [CrossRef]
- Hiroi, N.; Takahashi, T.; Hishimoto, A.; Izumi, T.; Boku, S.; Hiramoto, T. Copy number variation at 22q11.2: From rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol. Psychiatry 2013, 18, 1153–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative stress in cancer and fibrosis: Opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Aljabali, A.A.; Tambuwala, M.M.; Tiwari, S.; Serrano-Aroca, A.; Alzahrani, K.J.; Silva Andrade, B.; Azevedo, V.; Ganguly, N.K.; Lundstrom, K. Predicting COVID-19-comorbidity pathway crosstalk-based targets and drugs: Towards personalized COVID-19 management. Biomedicines 2021, 9, 556. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-beta1 signaling and tissue fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.L. Role of extracellular vesicles in renal inflammation and fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 455–466. [Google Scholar] [CrossRef]
- Carraro, G.; Mulay, A.; Yao, C.; Mizuno, T.; Konda, B.; Petrov, M.; Lafkas, D.; Arron, J.R.; Hogaboam, C.M.; Chen, P.; et al. Single-cell reconstruction of human basal cell diversity in normal and idiopathic pulmonary fibrosis lungs. Am. J. Respir. Crit. Care Med. 2020, 202, 1540–1550. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in fibrosis and fibrotic diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Jeong, W.I.; Park, O.; Suh, Y.G.; Byun, J.S.; Park, S.Y.; Choi, E.; Kim, J.K.; Ko, H.; Wang, H.; Miller, A.M.; et al. Suppression of innate immunity (natural killer cell/interferon-gamma) in the advanced stages of liver fibrosis in mice. Hepatology 2011, 53, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Lahnemann, D.; Koster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, H.; Jia, P.; Zhao, Z. Cell-Type-Specific Profibrotic Scores across Multi-Organ Systems Predict Cancer Prognosis. Cancers 2021, 13, 6024. https://doi.org/10.3390/cancers13236024
Fan H, Jia P, Zhao Z. Cell-Type-Specific Profibrotic Scores across Multi-Organ Systems Predict Cancer Prognosis. Cancers. 2021; 13(23):6024. https://doi.org/10.3390/cancers13236024
Chicago/Turabian StyleFan, Huihui, Peilin Jia, and Zhongming Zhao. 2021. "Cell-Type-Specific Profibrotic Scores across Multi-Organ Systems Predict Cancer Prognosis" Cancers 13, no. 23: 6024. https://doi.org/10.3390/cancers13236024
APA StyleFan, H., Jia, P., & Zhao, Z. (2021). Cell-Type-Specific Profibrotic Scores across Multi-Organ Systems Predict Cancer Prognosis. Cancers, 13(23), 6024. https://doi.org/10.3390/cancers13236024