
NTRK1/TrkA Activation Overrides the G2/M-Checkpoint upon Irradiation

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
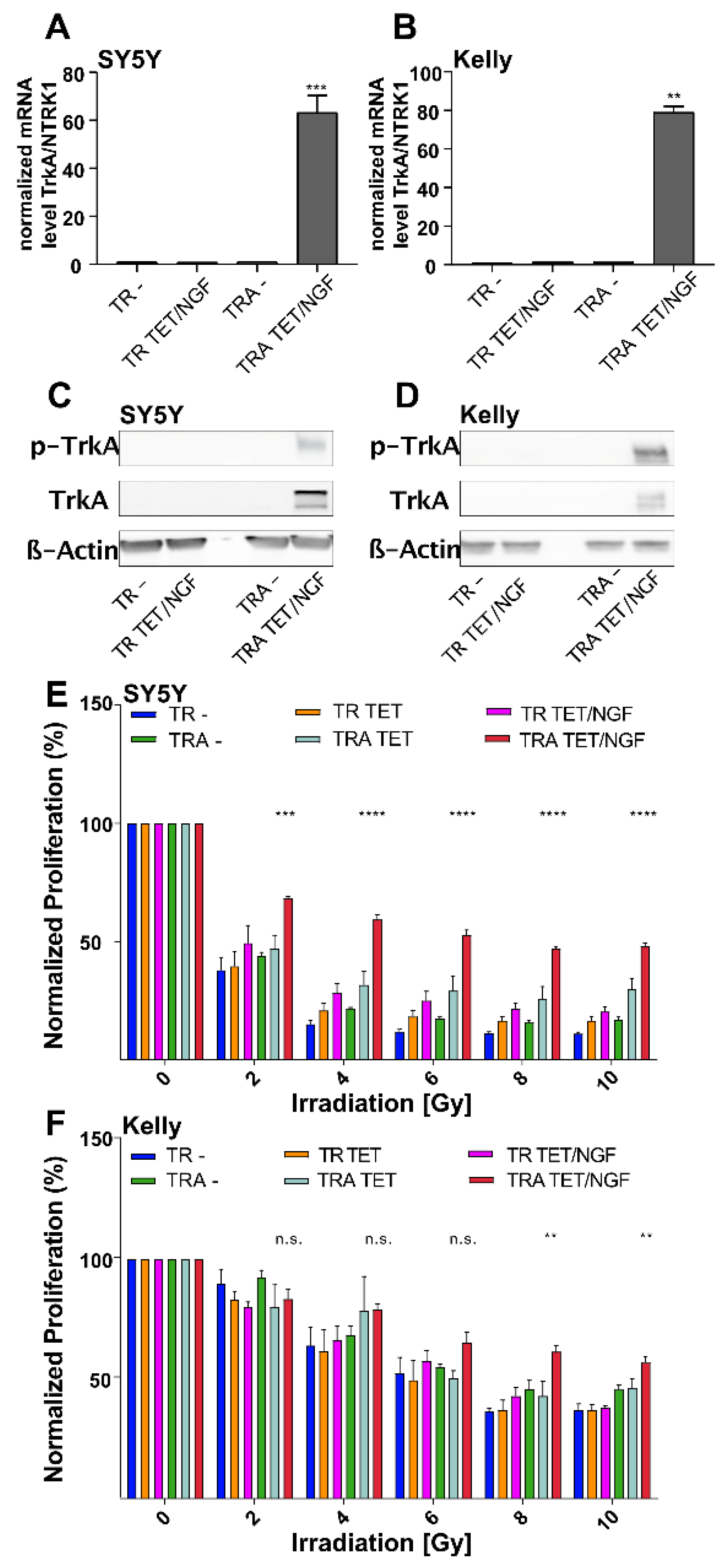
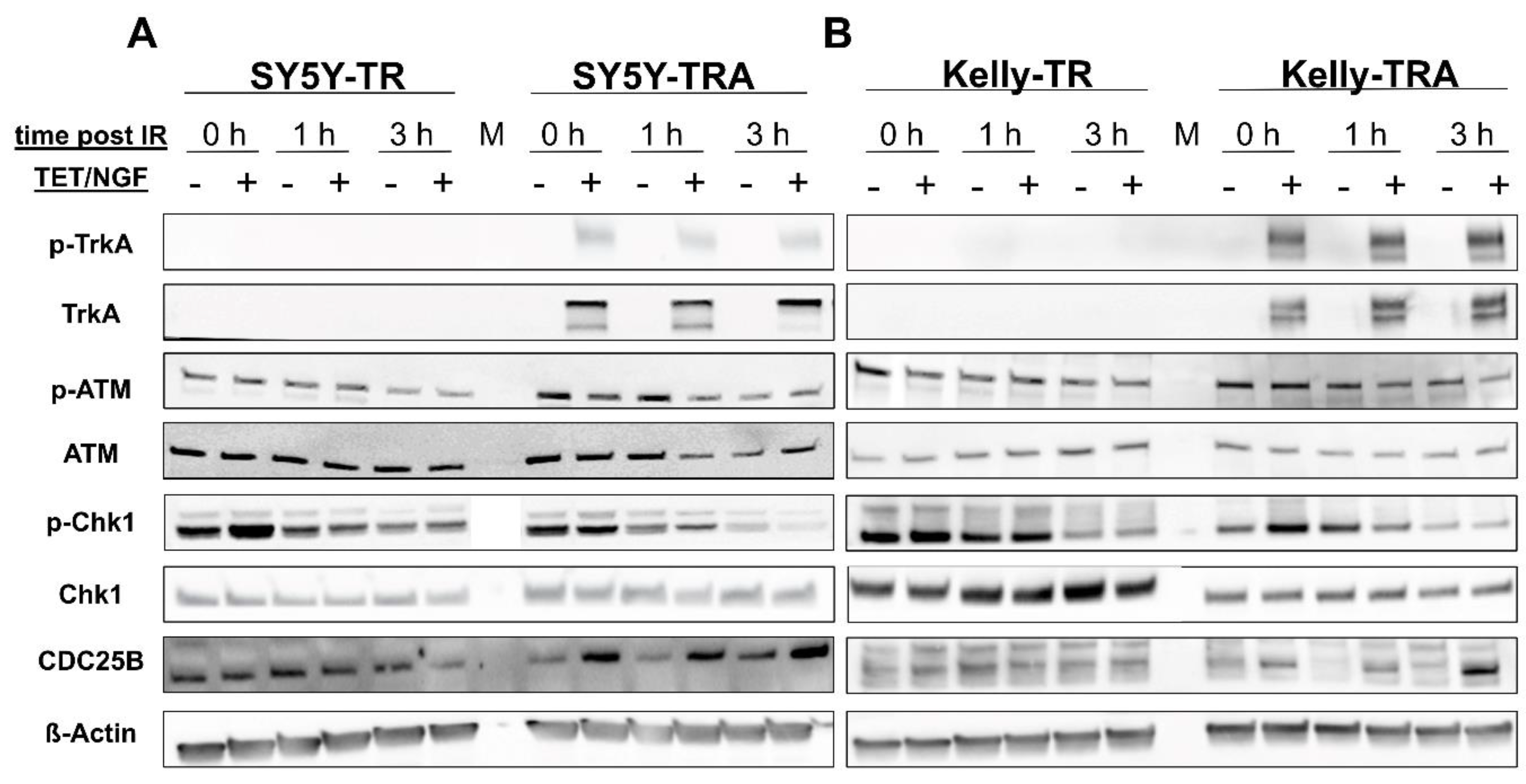
2.1. Inducible Expression of TrkA/NTRK1 in Human Neuroblastoma Cell Lines Increases Short-Term Survival upon Ionizing Radiation (IR)
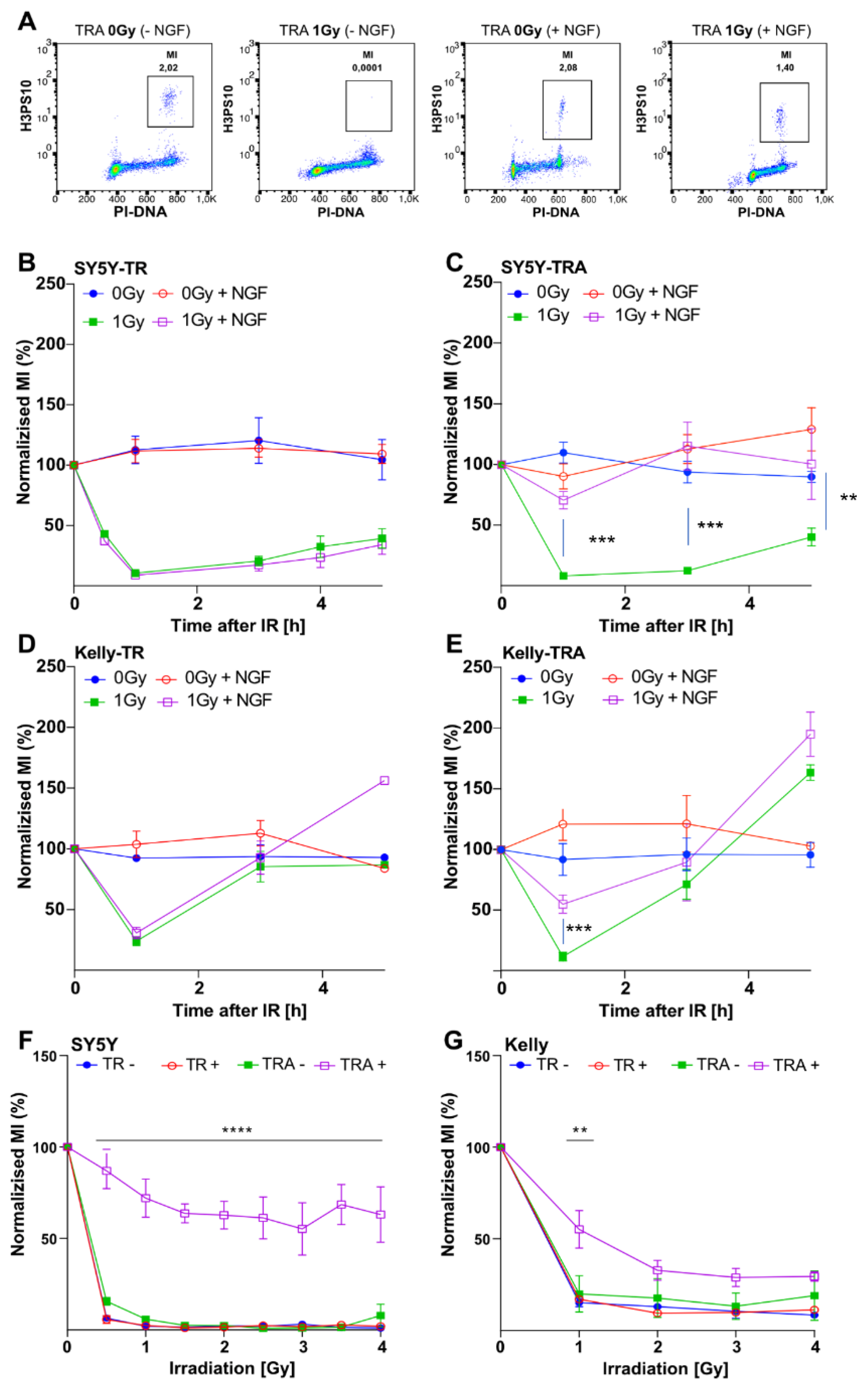
2.2. TrkA/NTRK1-Activation Impacts on Cell Cycle Distribution and Induces Impaired G2-Arrest in Irradiated Neuroblastoma Cells
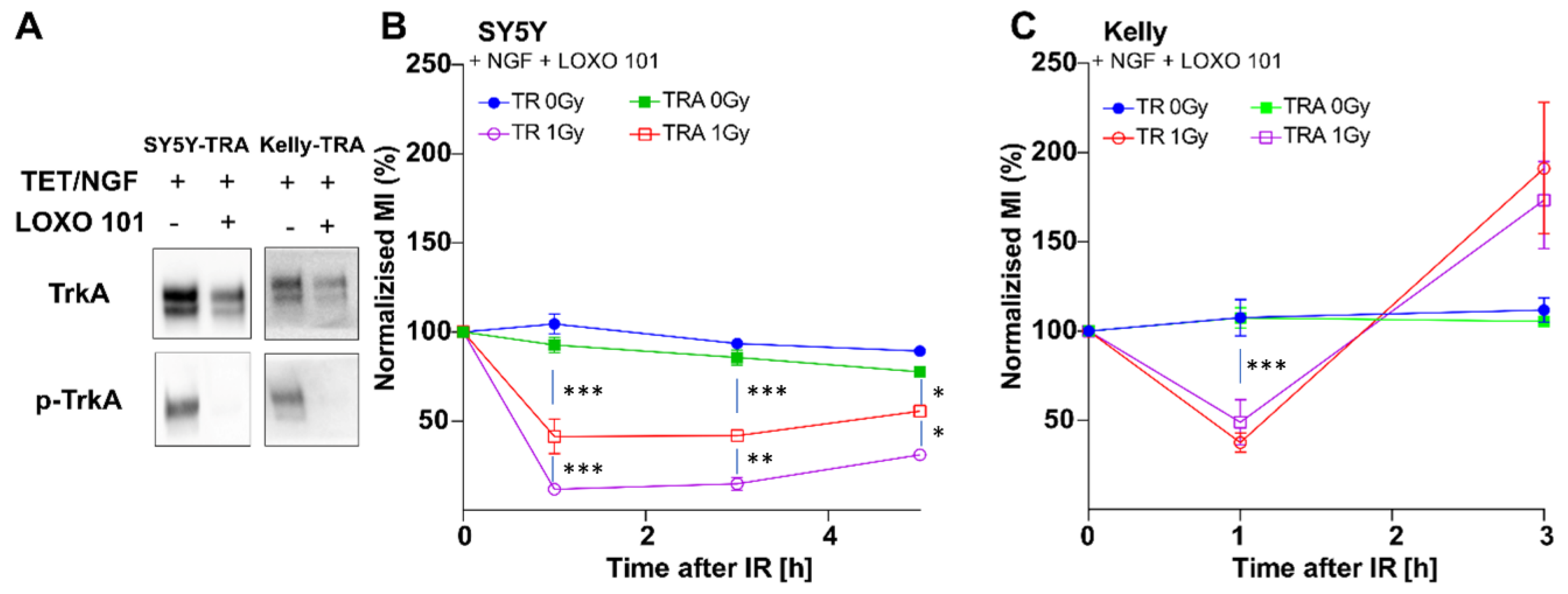
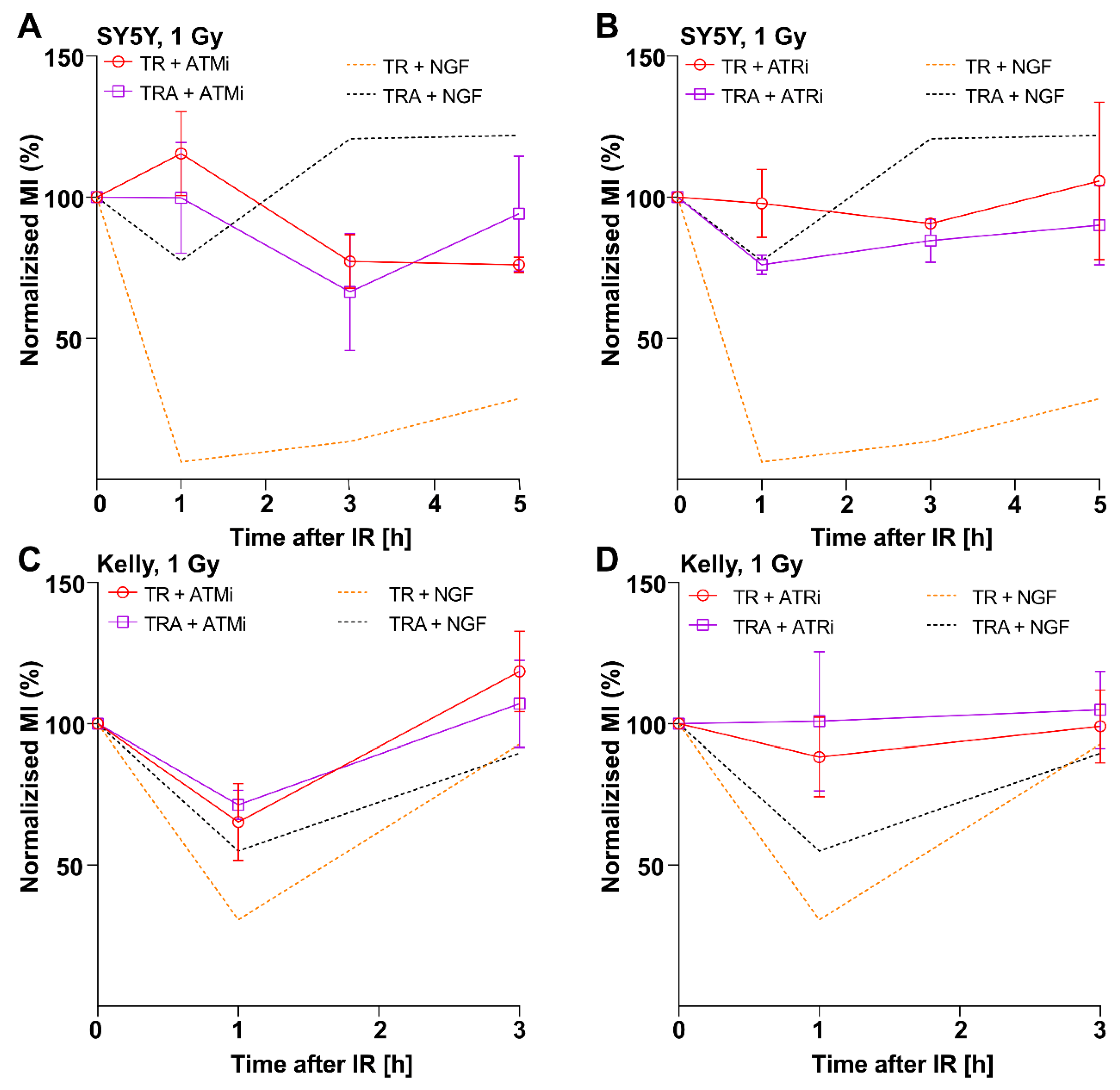
2.3. Inhibition of Cell Cycle Checkpoint Proteins Recapitulate the Effect of TrkA Activation on the G2 Checkpoint Response
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Inhibitors and Antibodies
4.3. Irradiation
4.4. Viability Assay
4.5. Calculation of the Mitotic Index Using Histone H3-pS10 (H3pS10) and PI Staining
4.6. Western Blot Analyses
4.7. EdU Incorporation and Detection
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Gatta, G.; Botta, L.; Rossi, S.; Aareleid, T.; Bielska-Lasota, M.; Clavel, J.; Dimitrova, N.; Jakab, Z.; Kaatsch, P.; Lacour, B.; et al. Childhood cancer survival in Europe 1999–2007: Results of EUROCARE-5—A population-based study. Lancet Oncol. 2014, 15, 35–47. [Google Scholar] [CrossRef]
- Diede, S.J. Spontaneous regression of metastatic cancer: Learning from neuroblastoma. Nat. Rev. Cancer 2014, 14, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K. Stage 4S neuroblastoma: What makes it special? J. Clin. Oncol. 1998, 16, 2003–2006. [Google Scholar] [CrossRef]
- Nakagawara, A. Molecular basis of spontaneous regression of neuroblastoma: Role of neurotrophic signals and genetic abnormalities. Hum. Cell 1998, 11, 115–124. [Google Scholar]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- George, R.E.; Sanda, T.; Hanna, M.; Fröhling, S.; Luther, W.; Zhang, J., II; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Khazi, D.; Carlisle, A.J.; Winter, C.L.; Rappaport, E.; Maris, J.M. Germline PHOX2B mutation in hereditary neuroblastoma. Am. J. Hum. Genet. 2004, 75, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Caron, H.; van Sluis, P.; de Kraker, J.; Bökkerink, J.; Egeler, M.; Laureys, G.; Slater, R.; Westerveld, A.; Voûte, P.A.; Versteeg, R. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N. Engl. J. Med. 1996, 334, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Srivatsan, E.S.; Ying, K.L.; Seeger, R.C. Deletion of chromosome 11 and of 14q sequences in neuroblastoma. Genes Chromosomes Cancer 1993, 7, 32–37. [Google Scholar] [CrossRef]
- Bown, N.; Cotterill, S.; Lastowska, M.; O’Neill, S.; Pearson, A.D.; Plantaz, D.; Meddeb, M.; Danglot, G.; Brinkschmidt, C.; Christiansen, H.; et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 1999, 340, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A.; Azar, C.G.; Scavarda, N.J.; Brodeur, G.M. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol. Cell. Biol. 1994, 14, 759–767. [Google Scholar]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Kanda, N.; Maseki, N.; Sakurai, M.; Tsuchida, Y.; Takeda, T.; Okabe, I.; Sakurai, M. Different karyotypic patterns in early and advanced stage neuroblastomas. Cancer Res. 1987, 47, 311–318. [Google Scholar]
- Nakagawara, A.; Arima-Nakagawara, M.; Scavarda, N.J.; Azar, C.G.; Cantor, A.B.; Brodeur, G.M. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N. Engl. J. Med. 1993, 328, 847–854. [Google Scholar] [CrossRef]
- Look, A.T.; Hayes, F.A.; Nitschke, R.; McWilliams, N.B.; Green, A.A. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N. Engl. J. Med. 1984, 311, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Schulte, J.H.; Schramm, A.; Klein-Hitpass, L.; Klenk, M.; Wessels, H.; Hauffa, B.P.; Eils, J.; Eils, R.; Brodeur, G.M.; Schweigerer, L.; et al. Microarray analysis reveals differential gene expression patterns and regulation of single target genes contributing to the opposing phenotype of TrkA- and TrkB-expressing neuroblastomas. Oncogene 2005, 24, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emdal, K.B.; Pedersen, A.K.; Bekker-Jensen, D.B.; Tsafou, K.P.; Horn, H.; Lindner, S.; Schulte, J.H.; Eggert, A.; Jensen, L.J.; Francavilla, C.; et al. Temporal proteomics of NGF-TrkA signaling identifies an inhibitory role for the E3 ligase Cbl-b in neuroblastoma cell differentiation. Sci. Signal. 2015, 8, ra40. [Google Scholar] [CrossRef]
- Schramm, A.; Schowe, B.; Fielitz, K.; Heilmann, M.; Martin, M.; Marschall, T.; Köster, J.; Vandesompele, J.; Vermeulen, J.; de Preter, K.; et al. Exon-level expression analyses identify MYCN and NTRK1 as major determinants of alternative exon usage and robustly predict primary neuroblastoma outcome. Br. J. Cancer 2012, 107, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Pajtler, K.W.; Mahlow, E.; Odersky, A.; Lindner, S.; Stephan, H.; Bendix, I.; Eggert, A.; Schramm, A.; Schulte, J.H. Neuroblastoma in dialog with its stroma: NTRK1 is a regulator of cellular cross-talk with Schwann cells. Oncotarget 2014, 5, 11180–11192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliakis, G.; Wang, Y.; Guan, J.; Wang, H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 2003, 22, 5834–5847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, M.; Martinez-Pastor, B.; Murga, M.; Toledo, L.I.; Gutierrez-Martinez, P.; Lopez, E.; Fernandez-Capetillo, O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med. 2006, 203, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E., III; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G(2)-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Mladenov, E.; Iliakis, G. Proficiency in homologous recombination repair is prerequisite for activation of G(2)-checkpoint at low radiation doses. DNA Repair. 2021, 101, 103076. [Google Scholar] [CrossRef]
- Moding, E.J.; Kastan, M.B.; Kirsch, D.G. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat. Rev. Drug Discov. 2013, 12, 526–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Kim, S.T.; Lim, D.-S.; Kastan, M.B. Two Molecularly Distinct G2/M Checkpoints Are Induced by Ionizing Irradiation. MCB 2002, 22, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, C.A.; Huggins, J.; Laquaglia, M.; Hulderman, C.E.; Russell, M.R.; Bosse, K.; Diskin, S.J.; Attiyeh, E.F.; Sennett, R.; Norris, G.; et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3336–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandel, E.S.; Skeen, J.; Majewski, N.; Di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell. Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtivelman, E.; Sussman, J.; Stokoe, D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr. Biol. 2002, 12, 919–924. [Google Scholar] [CrossRef] [Green Version]
- Nishijima, H.; Nishitani, H.; Seki, T.; Nishimoto, T. A dual-specificity phosphatase Cdc25B is an unstable protein and triggers p34(cdc2)/cyclin B activation in hamster BHK21 cells arrested with hydroxyurea. J. Cell Biol. 1997, 138, 1105–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieber, M.; Smith, B.; Szakal, A.; Nelson-Rees, W.; Todaro, G. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer 1976, 17, 62–70. [Google Scholar] [CrossRef]
- Yu, Z.; Boggon, T.J.; Kobayashi, S.; Jin, C.; Ma, P.C.; Dowlati, A.; Kern, J.A.; Tenen, D.G.; Halmos, B. Resistance to an irreversible epidermal growth factor receptor (EGFR) inhibitor in EGFR-mutant lung cancer reveals novel treatment strategies. Cancer Res. 2007, 67, 10417–10427. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassiepen, C.; Soni, A.; Rudolf, I.; Boron, V.; Oeck, S.; Iliakis, G.; Schramm, A. NTRK1/TrkA Activation Overrides the G2/M-Checkpoint upon Irradiation. Cancers 2021, 13, 6023. https://doi.org/10.3390/cancers13236023
Hassiepen C, Soni A, Rudolf I, Boron V, Oeck S, Iliakis G, Schramm A. NTRK1/TrkA Activation Overrides the G2/M-Checkpoint upon Irradiation. Cancers. 2021; 13(23):6023. https://doi.org/10.3390/cancers13236023
Chicago/Turabian StyleHassiepen, Christina, Aashish Soni, Ines Rudolf, Vivian Boron, Sebastian Oeck, George Iliakis, and Alexander Schramm. 2021. "NTRK1/TrkA Activation Overrides the G2/M-Checkpoint upon Irradiation" Cancers 13, no. 23: 6023. https://doi.org/10.3390/cancers13236023
APA StyleHassiepen, C., Soni, A., Rudolf, I., Boron, V., Oeck, S., Iliakis, G., & Schramm, A. (2021). NTRK1/TrkA Activation Overrides the G2/M-Checkpoint upon Irradiation. Cancers, 13(23), 6023. https://doi.org/10.3390/cancers13236023