
Changes in Methylation across Structural and MicroRNA Genes Relevant for Progression and Metastasis in Colorectal Cancer

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Tissue Material and Ethical Consent
2.2. Genomic DNA Isolation
2.3. Methylation Profiling
2.4. Bioinformatics Analysis
2.5. Data Availability
2.6. KEGG Analysis
2.7. Ingenuity Pathway Analysis (IPA)
3. Results
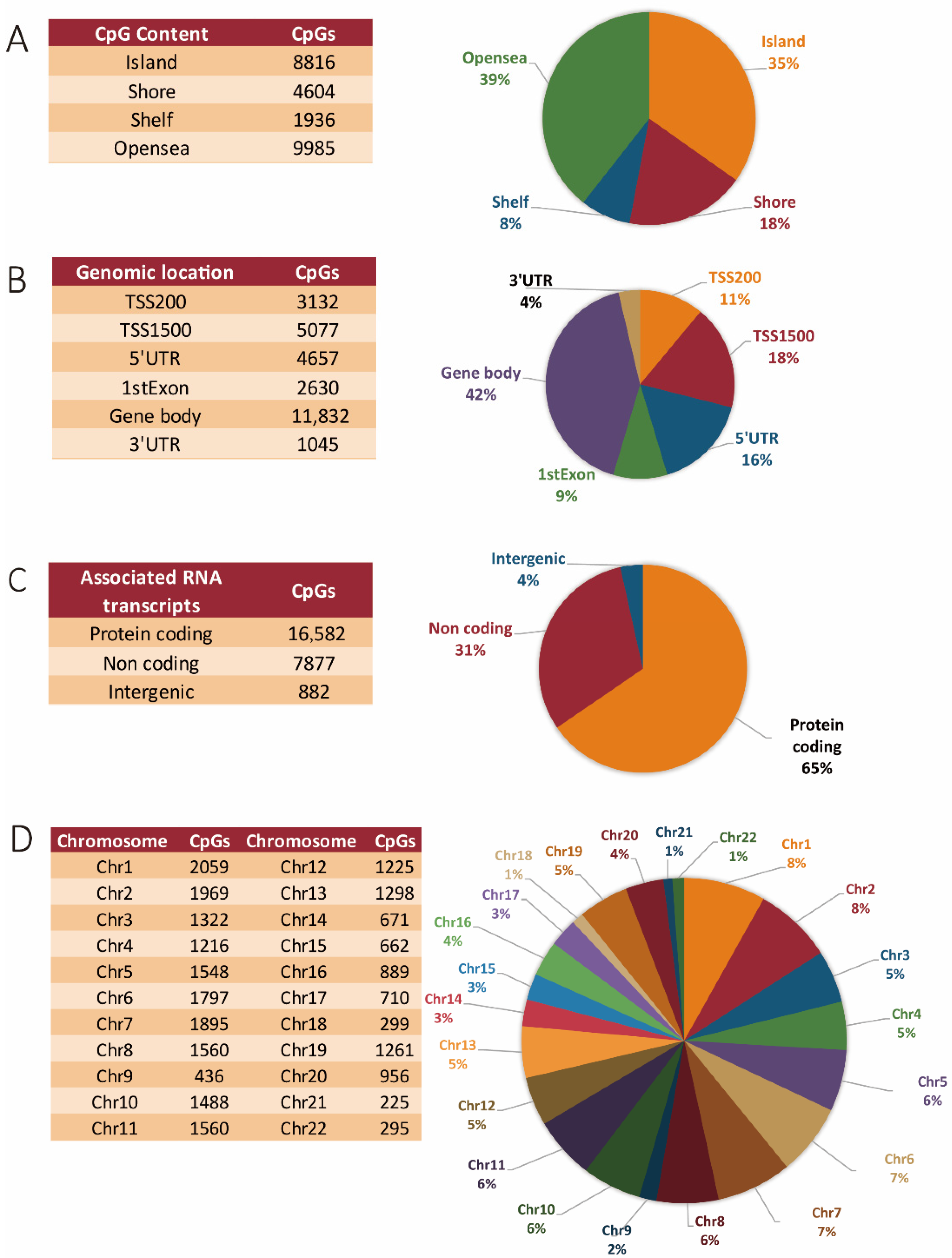
3.1. Methylation Array and Associated Bioinformatics: General Distribution of Differentially Methylated Sites between Coding, Non-Coding, and Intergenic Regions
3.2. Hypermethylated and Hypomethylated CpG Areas across Genomic Features
3.3. Methylation-Specific Patterns across miR Genes
3.4. Identification of Significant Differentially Methylated miRs
3.5. Pathway Enrichment Analysis of Differentially Methylated miR Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782–4795. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, Q.; Pan, X. Micrornas and their regulatory roles in animals and plants. J. Cell. Physiol. 2007, 210, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and micrornas. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef]
- Lang, F.; Contreras-Gerenas, M.F.; Gelléri, M.; Neumann, J.; Kröger, O.; Sadlo, F.; Berniak, K.; Marx, A.; Cremer, C.; Wagenknecht, H.-A.; et al. Tackling tumour cell heterogeneity at the super-resolution level in human colorectal cancer tissue. Cancers 2021, 13, 3692–3713. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 rna regulates developmental timing in caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Silva, B.; Gao, T.; Xu, Z.; Cui, J. Dynamic and modularized microrna regulation and its implication in human cancers. Sci. Rep. 2017, 7, 13356–13372. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Allgayer, H.; Leupold, J.H.; Patil, N. Defining the “metastasome”: Perspectives from the genome and molecular landscape in colorectal cancer for metastasis evolution and clinical consequences. Semin. Cancer Biol. 2020, 60, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mudduluru, G.; Abba, M.; Batliner, J.; Patil, N.; Scharp, M.; Lunavat, T.R.; Leupold, J.H.; Oleksiuk, O.; Juraeva, D.; Thiele, W.; et al. A systematic approach to defining the microrna landscape in metastasis. Cancer Res. 2015, 75, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. Microrna-21 (mir-21) post-transcriptionally downregulates tumor suppressor pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef]
- Abba, M.; Benner, A.; Patil, N.; Heil, O.; Allgayer, H. Differentially expressed micrornas in colorectal cancer metastasis. Genom. Data 2015, 6, 33–35. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of micrornas in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004–15012. [Google Scholar] [CrossRef]
- Kunej, T.; Godnic, I.; Horvat, S.; Zorc, M.; Calin, G.A. Cross talk between microrna and coding cancer genes. Cancer J. 2012, 18, 223–231. [Google Scholar] [CrossRef]
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: An intimate affair. Am. J. Cancer Res. 2020, 10, 1954–1978. [Google Scholar]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef]
- Goh, L.; Murphy, S.K.; Muhkerjee, S.; Furey, T.S. Genomic sweeping for hypermethylated genes. Bioinformatics 2007, 23, 281–288. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fleming, M.; Ravula, S.; Tatishchev, S.F.; Wang, H.L. Colorectal carcinoma: Pathologic aspects. J. Gastrointest. Oncol. 2012, 3, 153–173. [Google Scholar] [PubMed]
- Amin, M.B.; Edge, S.B.; American Joint Committee of Cancer. AJCC Cancer Staging Manual; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in h3f3a and idh1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Westfall, P.H.; Stanley, Y.S. Resampling-Based Multiple Testing: Examples and Methods for p-Value Adjustment, Wiley Series in Probability and Statistics ed.; John Wiley & Sons Inc.: New York, NY, USA, 1993. [Google Scholar]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. David: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single cpg site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef]
- Wilting, S.M.; van Boerdonk, R.A.A.; Henken, F.E.; Meijer, C.J.L.M.; Diosdado, B.; Meijer, G.A.; le Sage, C.; Agami, R.; Snijders, P.J.F.; Steenbergen, R.D.M. Methylation-mediated silencing and tumour suppressive function of hsa-mir-124 in cervical cancer. Mol. Cancer 2010, 9, 167–180. [Google Scholar] [CrossRef]
- Chu, M.; Chang, Y.; Guo, Y.; Wang, N.; Cui, J.; Gao, W.-Q. Regulation and methylation of tumor suppressor mir-124 by androgen receptor in prostate cancer cells. PLoS ONE 2015, 10, e0116197. [Google Scholar] [CrossRef]
- Majid, A.; Wang, J.; Nawaz, M.; Abdul, S.; Ayesha, M.; Guo, C.; Liu, Q.; Liu, S.; Sun, M.-Z. Mir-124-3p suppresses the invasiveness and metastasis of hepatocarcinoma cells via targeting crkl. Front. Mol. Biosci. 2020, 7, 223–237. [Google Scholar] [CrossRef]
- Cai, Q.Q.; Dong, Y.W.; Wang, R.; Qi, B.; Guo, J.X.; Pan, J.; Liu, Y.Y.; Zhang, C.Y.; Wu, X.Z. Mir-124 inhibits the migration and invasion of human hepatocellular carcinoma cells by suppressing integrin αv expression. Sci. Rep. 2017, 7, 40733–40742. [Google Scholar] [CrossRef]
- Fu, W.; Wu, X.; Yang, Z.; Mi, H. The effect of mir-124-3p on cell proliferation and apoptosis in bladder cancer by targeting ednrb. Arch. Med Sci. 2019, 15, 1154–1162. [Google Scholar] [CrossRef]
- Lu, M.L.; Zhang, Y.; Li, J.; Fu, Y.; Li, W.H.; Zhao, G.F.; Li, X.H.; Wei, L.; Liu, G.B.; Huang, H. Microrna-124 inhibits colorectal cancer cell proliferation and suppresses tumor growth by interacting with plcb1 and regulating wnt/β-catenin signaling pathway. Eur. Rev. Med Pharmacol. Sci. 2019, 23, 121–136. [Google Scholar] [PubMed]
- Shahmohamadnejad, S.; Nouri Ghonbalani, Z.; Tahbazlahafi, B.; Panahi, G.; Meshkani, R.; Emami Razavi, A.; Shokri Afra, H.; Khalili, E. Aberrant methylation of mir-124 upregulates dnmt3b in colorectal cancer to accelerate invasion and migration. Arch. Physiol. Biochem. 2020, 1–7. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, Y.; Yue, X.; Li, H.; Luo, X.; Wang, Y.; Wang, K.; Wan, J. Mir-124 suppresses growth of human colorectal cancer by inhibiting stat3. PLoS ONE 2013, 8, e70300–e70310. [Google Scholar] [CrossRef]
- Kang, M.; Li, Y.; Liu, W.; Wang, R.; Tang, A.; Hao, H.; Liu, Z.; Ou, H. Mir-129-2 suppresses proliferation and migration of esophageal carcinoma cells through downregulation of sox4 expression. Int. J. Mol. Med. 2013, 32, 51–58. [Google Scholar] [CrossRef]
- Tang, X.; Tang, J.; Liu, X.; Zeng, L.; Cheng, C.; Luo, Y.; Li, L.; Qin, S.-L.; Sang, Y.; Deng, L.-M.; et al. Downregulation of mir-129-2 by promoter hypermethylation regulates breast cancer cell proliferation and apoptosis. Oncol. Rep. 2016, 35, 2963–2969. [Google Scholar] [CrossRef] [PubMed]
- Karaayvaz, M.; Zhai, H.; Ju, J. Mir-129 promotes apoptosis and enhances chemosensitivity to 5-fluorouracil in colorectal cancer. Cell Death Dis. 2013, 4, e659–e667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.-H.; Shan, T.; Aguilera-Barrantes, I.; Wang, L.-S.; Huang, T.H.-M.; Rader, J.S.; Sheng, X.; Huang, Y.-W. Mir-137 is a tumor suppressor in endometrial cancer and is repressed by DNA hypermethylation. Lab. Investig. 2018, 98, 1397–1407. [Google Scholar] [CrossRef]
- Bi, W.P.; Xia, M.; Wang, X.J. Mir-137 suppresses proliferation, migration and invasion of colon cancer cell lines by targeting tcf4. Oncol. Lett. 2018, 15, 8744–8748. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, J.; Feng, Z.; Tang, Q.; Zhou, X. Mir-137-3p inhibits colorectal cancer cell migration by regulating a kdm1a-dependent epithelial–mesenchymal transition. Dig. Dis. Sci. 2021, 66, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Guo, X.; Tian, S.; Zhu, C.; Chen, S.; Yu, C.; Jiang, J.; Sun, C. Microrna-137 reduces stemness features of pancreatic cancer cells by targeting klf12. J. Exp. Clin. Cancer Res. 2019, 38, 126–141. [Google Scholar] [CrossRef]
- Neault, M.; Mallette, F.A.; Richard, S. Mir-137 modulates a tumor suppressor network-inducing senescence in pancreatic cancer cells. Cell Rep. 2016, 14, 1966–1978. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Zhang, G.; Xie, C.; Zhou, Y. Mir-34b regulates cervical cancer cell proliferation and apoptosis. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Catuogno, S.; Cerchia, L.; Romano, G.; Pognonec, P.; Condorelli, G.; de Franciscis, V. Mir-34c may protect lung cancer cells from paclitaxel-induced apoptosis. Oncogene 2013, 32, 341–351. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, E.J.; Lee, S.; Tan, X.; Liu, X.; Park, S.; Kang, K.; Yoon, J.-S.; Ko, Y.H.; Kurie, J.M.; et al. Mir-34a and mir-34b/c have distinct effects on the suppression of lung adenocarcinomas. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Lee, J.-Y.; Ho, C.-C.; Hong, Q.-S.; Yu, S.-L.; Tzeng, C.-R.; Yang, P.-C.; Chen, H.-W. Mirna-34b as a tumor suppressor in estrogen-dependent growth of breast cancer cells. Breast Cancer Res. 2011, 13, R116–R131. [Google Scholar] [CrossRef]
- Hsieh, M.-J.; Lin, C.-W.; Su, S.-C.; Reiter, R.J.; Chen, A.W.-G.; Chen, M.-K.; Yang, S.-F. Effects of mir-34b/mir-892a upregulation and inhibition of abcb1/abcb4 on melatonin-induced apoptosis in vcr-resistant oral cancer cells. Mol. Ther. Nucleic Acids 2020, 19, 877–889. [Google Scholar] [CrossRef]
- Wang, L.-G.; Ni, Y.; Su, B.-H.; Mu, X.-R.; Shen, H.-C.; Du, J.-J. Microrna-34b functions as a tumor suppressor and acts as a nodal point in the feedback loop with met. Int. J. Oncol. 2013, 42, 957–962. [Google Scholar] [CrossRef]
- Li, Y.Q.; Ren, X.Y.; He, Q.M.; Xu, Y.F.; Tang, X.R.; Sun, Y.; Zeng, M.S.; Kang, T.B.; Liu, N.; Ma, J. Mir-34c suppresses tumor growth and metastasis in nasopharyngeal carcinoma by targeting met. Cell Death Dis. 2015, 6, e1618. [Google Scholar] [CrossRef][Green Version]
- Hagman, Z.; Haflidadottir, B.S.; Ansari, M.; Persson, M.; Bjartell, A.; Edsjö, A.; Ceder, Y. The tumour suppressor mir-34c targets met in prostate cancer cells. Br. J. Cancer 2013, 109, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Suzuki, H.; Sasaki, Y.; Maruyama, R.; Imai, K.; Shinomura, Y.; Tokino, T. Epigenetic silencing of microrna-34b/c and b-cell translocation gene 4 is associated with cpg island methylation in colorectal cancer. Cancer Res. 2008, 68, 4123–4132. [Google Scholar] [CrossRef]
- Li, Y.; Huang, R.; Wang, L.; Hao, J.; Zhang, Q.; Ling, R.; Yun, J. Microrna-762 promotes breast cancer cell proliferation and invasion by targeting irf7 expression. Cell Prolif. 2015, 48, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Ben Dhiab, M.; Ziadi, S.; Louhichi, T.; Ben Gacem, R.; Ksiaa, F.; Trimeche, M. Investigation of mir9-1, mir9-2 and mir9-3 methylation in hodgkin lymphoma. Pathobiology 2015, 82, 195–202. [Google Scholar] [CrossRef]
- Meng, Q.; Xiang, L.; Fu, J.; Chu, X.; Wang, C.; Yan, B. Transcriptome profiling reveals mir-9-3p as a novel tumor suppressor in gastric cancer. Oncotarget 2017, 8, 37321–37331. [Google Scholar] [CrossRef]
- Liu, X.; Bi, L.; Wang, Q.; Wen, M.; Li, C.; Ren, Y.; Jiao, Q.; Mao, J.-H.; Wang, C.; Wei, G.; et al. Mir-1204 targets vdr to promotes epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene 2018, 37, 3426–3439. [Google Scholar] [CrossRef]
- Zhao, X.; Shen, F.; Ma, J.; Zhao, S.; Meng, L.; Wang, X.; Liang, S.; Liang, J.; Hu, C.; Zhang, X. Creb1-induced mir-1204 promoted malignant phenotype of glioblastoma through targeting nr3c2. Cancer Cell Int. 2020, 20, 111–120. [Google Scholar] [CrossRef]
- Xu, J.; Meng, Q.; Li, X.; Yang, H.; Xu, J.; Gao, N.; Sun, H.; Wu, S.; Familiari, G.; Relucenti, M.; et al. Long noncoding rna mir17hg promotes colorectal cancer progression via mir-17-5p. Cancer Res. 2019, 79, 4882–4895. [Google Scholar] [CrossRef]
- Hsu, T.I.; Hsu, C.H.; Lee, K.H.; Lin, J.T.; Chen, C.S.; Chang, K.C.; Su, C.Y.; Hsiao, M.; Lu, P.J. Microrna-18a is elevated in prostate cancer and promotes tumorigenesis through suppressing stk4 in vitro and in vivo. Oncogenesis 2014, 3, e99. [Google Scholar] [CrossRef] [PubMed]
- Egeland, N.G.; Jonsdottir, K.; Aure, M.R.; Sahlberg, K.; Kristensen, V.N.; Cronin-Fenton, D.; Skaland, I.; Gudlaugsson, E.; Baak, J.P.A.; Janssen, E.A.M. Mir-18a and mir-18b are expressed in the stroma of oestrogen receptor alpha negative breast cancers. BMC Cancer 2020, 20, 377–390. [Google Scholar] [CrossRef]
- Lu, C.; Peng, K.; Guo, H.; Ren, X.; Hu, S.; Cai, Y.; Han, Y.; Ma, L.; Xu, P. Mir-18a-5p promotes cell invasion and migration of osteosarcoma by directly targeting irf2. Oncol. Lett. 2018, 16, 3150–3156. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Dai, Y.; Zhang, L.; Jiang, C.; Li, Z.; Yang, J.; McCarthy, J.B.; She, X.; Zhang, W.; Ma, J.; et al. Mir-18a promotes malignant progression by impairing microrna biogenesis in nasopharyngeal carcinoma. Carcinogenesis 2013, 34, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, X.; Wen, C.; Yang, X.; Song, M.; Chen, J.; Wang, C.; Zhang, B.; Wang, L.; Iwamoto, A.; et al. Hsa-mir-19a is associated with lymph metastasis and mediates the tnf-α induced epithelial-to-mesenchymal transition in colorectal cancer. Sci. Rep. 2015, 5, 13350–13361. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, R.; Yang, F.; Cheng, R.; Chen, X.; Cui, S.; Gu, Y.; Sun, W.; You, C.; Liu, Z.; et al. Mir-19a promotes colorectal cancer proliferation and migration by targeting tia1. Mol. Cancer 2017, 16, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, Z.; An, Y.; Hu, H.; Yin, J.; Zhang, P.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. Mir-19a/b modulate the metastasis of gastric cancer cells by targeting the tumour suppressor mxd1. Cell Death Dis. 2014, 5, e1144–e1154. [Google Scholar] [CrossRef]
- Jiang, X.-M.; Yu, X.-N.; Liu, T.-T.; Zhu, H.-R.; Shi, X.; Bilegsaikhan, E.; Guo, H.-Y.; Song, G.-Q.; Weng, S.-Q.; Huang, X.-X.; et al. Microrna-19a-3p promotes tumor metastasis and chemoresistance through the pten/akt pathway in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 105, 1147–1154. [Google Scholar] [CrossRef]
- Zhu, G.-F.; Xu, Y.-W.; Li, J.; Niu, H.-L.; Ma, W.-X.; Xu, J.; Zhou, P.-R.; Liu, X.; Ye, D.-L.; Liu, X.-R.; et al. Mir20a/106a-wtx axis regulates rhogdia/cdc42 signaling and colon cancer progression. Nat. Commun. 2019, 10, 112–125. [Google Scholar] [CrossRef]
- Torres-Martin, M.; Lassaletta, L.; de Campos, J.M.; Isla, A.; Pinto, G.R.; Burbano, R.R.; Melendez, B.; Castresana, J.S.; Rey, J.A. Genome-wide methylation analysis in vestibular schwannomas shows putative mechanisms of gene expression modulation and global hypomethylation at the hox gene cluster. Genes Chromosomes Cancer 2015, 54, 197–209. [Google Scholar] [CrossRef]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of axl receptor tyrosine kinase expression by mir-34a and mir-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef]
- Shi, Y.; Qiu, M.; Wu, Y.; Hai, L. Mir-548-3p functions as an anti-oncogenic regulator in breast cancer. Biomed. Pharmacother. Biomed. Pharmacother. 2015, 75, 111–116. [Google Scholar] [CrossRef]
- Laudato, S.; Patil, N.; Abba, M.L.; Leupold, J.H.; Benner, A.; Gaiser, T.; Marx, A.; Allgayer, H. P53-induced mir-30e-5p inhibits colorectal cancer invasion and metastasis by targeting itga6 and itgb1. Int. J. Cancer 2017, 141, 1879–1890. [Google Scholar] [CrossRef]
- Bentwich, I. Prediction and validation of micrornas and their targets. FEBS Lett. 2005, 579, 5904–5910. [Google Scholar] [CrossRef]
- Sproul, D.; Kitchen, R.R.; Nestor, C.E.; Dixon, J.M.; Sims, A.H.; Harrison, D.J.; Ramsahoye, B.H.; Meehan, R.R. Tissue of origin determines cancer-associated cpg island promoter hypermethylation patterns. Genome Biol. 2012, 13, R84–R99. [Google Scholar] [CrossRef]
- Bandres, E.; Agirre, X.; Bitarte, N.; Ramirez, N.; Zarate, R.; Roman-Gomez, J.; Prosper, F.; Garcia-Foncillas, J. Epigenetic regulation of microrna expression in colorectal cancer. Int. J. Cancer 2009, 125, 2737–2743. [Google Scholar] [CrossRef] [PubMed]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P. Defining driver DNA methylation changes in human cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [PubMed]
- Wajed, S.A.; Laird, P.W.; DeMeester, T.R. DNA methylation: An alternative pathway to cancer. Ann. Surg. 2001, 234, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.V.; Davies, R.; Down, T.A.; et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378–1385. [Google Scholar] [CrossRef]
- Hernando-Herraez, I.; Garcia-Perez, R.; Sharp, A.J.; Marques-Bonet, T. DNA methylation: Insights into human evolution. PLoS Genet. 2015, 11, e1005661–e1005672. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Li, J.; Zhong, Y.; Cai, S.; Zhou, P.; Yao, L. Microrna expression profiling in the colorectal normal-adenoma-carcinoma transition. Oncol. Lett. 2019, 18, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Ghorai, A.; Ghosh, U. Mirna gene counts in chromosomes vary widely in a species and biogenesis of mirna largely depends on transcription or post-transcriptional processing of coding genes. Front. Genet. 2014, 5, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.H.; Huang, H.R.; Lu, J.; Liu, X.; Zhao, F.P.; Zhang, B.; Lin, S.X.; Wang, L.; Chen, H.H.; Xu, X.; et al. Mir-124 suppresses tumor growth and metastasis by targeting foxq1 in nasopharyngeal carcinoma. Mol. Cancer 2014, 13, 186–198. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome (Chr) | Status | OpenSea | Island | Shelf | Shore |
|---|---|---|---|---|---|
| Chr1 | Hypomethylated | 786 | 39 | 142 | 179 |
| Hypermethylated | 57 | 636 | 10 | 210 | |
| Chr2 | Hypomethylated | 861 | 40 | 137 | 168 |
| Hypermethylated | 36 | 568 | 5 | 154 | |
| Chr3 | Hypomethylated | 532 | 17 | 73 | 70 |
| Hypermethylated | 30 | 430 | 15 | 155 | |
| Chr4 | Hypomethylated | 442 | 5 | 83 | 85 |
| Hypermethylated | 12 | 462 | 7 | 120 | |
| Chr5 | Hypomethylated | 588 | 28 | 91 | 118 |
| Hypermethylated | 62 | 541 | 4 | 116 | |
| Chr6 | Hypomethylated | 781 | 13 | 96 | 111 |
| Hypermethylated | 75 | 503 | 17 | 201 | |
| Chr7 | Hypomethylated | 728 | 55 | 151 | 195 |
| Hypermethylated | 50 | 533 | 21 | 162 | |
| Chr8 | Hypomethylated | 597 | 29 | 93 | 142 |
| Hypermethylated | 26 | 502 | 7 | 164 | |
| Chr9 | Hypomethylated | 106 | 13 | 26 | 69 |
| Hypermethylated | 12 | 177 | 5 | 28 | |
| Chr10 | Hypomethylated | 506 | 26 | 101 | 121 |
| Hypermethylated | 40 | 573 | 5 | 116 | |
| Chr11 | Hypomethylated | 686 | 19 | 84 | 131 |
| Hypermethylated | 41 | 448 | 4 | 147 | |
| Chr12 | Hypomethylated | 485 | 18 | 78 | 115 |
| Hypermethylated | 43 | 357 | 20 | 109 | |
| Chr13 | Hypomethylated | 517 | 17 | 85 | 97 |
| Hypermethylated | 29 | 372 | 11 | 170 | |
| Chr14 | Hypomethylated | 253 | 9 | 39 | 58 |
| Hypermethylated | 13 | 220 | 5 | 74 | |
| Chr15 | Hypomethylated | 313 | 9 | 35 | 39 |
| Hypermethylated | 9 | 219 | 4 | 34 | |
| Chr16 | Hypomethylated | 267 | 29 | 68 | 121 |
| Hypermethylated | 35 | 295 | 15 | 59 | |
| Chr17 | Hypomethylated | 242 | 7 | 75 | 80 |
| Hypermethylated | 27 | 229 | 4 | 46 | |
| Chr18 | Hypomethylated | 37 | 14 | 33 | 27 |
| Hypermethylated | 1 | 161 | 4 | 22 | |
| Chr19 | Hypomethylated | 262 | 40 | 114 | 119 |
| Hypermethylated | 37 | 524 | 18 | 147 | |
| Chr20 | Hypomethylated | 223 | 41 | 88 | 127 |
| Hypermethylated | 2 | 385 | 5 | 85 | |
| Chr21 | Hypomethylated | 67 | 4 | 18 | 23 |
| Hypermethylated | 5 | 88 | 0 | 20 | |
| Chr22 | Hypomethylated | 63 | 23 | 37 | 49 |
| Hypermethylated | 1 | 98 | 3 | 21 |
| Chromosome | Status | OpenSea | Island | Shelf | Shore |
|---|---|---|---|---|---|
| Chr1 | Hypomethylated | 5 | 0 | 0 | 0 |
| Hypermethylated | 0 | 1 | 0 | 2 | |
| Chr2 | Hypomethylated | 1 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr3 | Hypomethylated | 6 | 0 | 0 | 0 |
| Hypermethylated | 1 | 1 | 0 | 0 | |
| Chr4 | Hypomethylated | 0 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr5 | Hypomethylated | 1 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr6 | Hypomethylated | 5 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr7 | Hypomethylated | 12 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr8 | Hypomethylated | 1 | 0 | 0 | 1 |
| Hypermethylated | 0 | 3 | 0 | 7 | |
| Chr9 | Hypomethylated | 1 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr10 | Hypomethylated | 0 | 1 | 3 | 2 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr11 | Hypomethylated | 2 | 0 | 0 | 0 |
| Hypermethylated | 2 | 12 | 0 | 0 | |
| Chr12 | Hypomethylated | 0 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr13 | Hypomethylated | 12 | 0 | 3 | 1 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr14 | Hypomethylated | 25 | 1 | 1 | 2 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Chr15 | Hypomethylated | 1 | 1 | 0 | 1 |
| Hypermethylated | 0 | 5 | 0 | 0 | |
| Chr16 | Hypomethylated | 1 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 1 | |
| Chr17 | Hypomethylated | 0 | 0 | 0 | 0 |
| Hypermethylated | 0 | 0 | 0 | 0 | |
| Hypomethylated | 0 | 0 | 0 | 0 | |
| Chr18 | Hypermethylated | 0 | 0 | 0 | |
| Hypomethylated | 20 | 0 | 0 | 0 | |
| Chr19 | Hypermethylated | 0 | 0 | 0 | 0 |
| Hypomethylated | 5 | 0 | 0 | 0 | |
| Chr20 | Hypermethylated | 0 | 18 | 0 | 1 |
| Hypomethylated | 0 | 0 | 0 | 0 | |
| Chr21 | Hypermethylated | 0 | 0 | 0 | 0 |
| Hypomethylated | 0 | 0 | 0 | 0 | |
| Chr22 | Hypermethylated | 1 | 0 | 0 | 0 |
| Hypermethylated miRs Found in Our Study | miR Expression in Cancer | Regulation | Role | Cancer Types | Target Genes | References |
|---|---|---|---|---|---|---|
| hsa-miR-124-2 | Downregulated | Hypermethylated | Tumor suppressor | Cervical cancer | IGFBP7 | [30] |
| hsa-miR-124-3 | Downregulated | Hypermethylated | Tumor suppressor | Prostate cancer, Cervical cancer, HCC, Bladder cancer, CRC | IGFBP7, CRKL, Sp1, EDNRB, CCL20, DNMT3B, STAT3 | [30,31,32,33,34,35,36,37] |
| hsa-miR-129-2 | Downregulated | Hypermethylated | Tumor suppressor | Esophageal carcinoma, Breast cancer, CRC | SOX4, BCL2L2, BCL2 | [38,39,40] |
| hsa-miR-137 | Downregulated | Hypermethylated | Tumor suppressor | Endometrial cancer, CRC, Pancreatic cancer | EZH2, LSD1, TCF4, LSD1, KLF12, KDM4A | [14,41,42,43,44,45] |
| hsa-miR-34B | Downregulated | Hypermethylated | Tumor suppressor | Cervical cancer, Lung adenocarcinoma, Breast cancer, Oropharyngeal (oral) cancer, NSCLC | TGF-β1, BMF, Cyclin D1, JAG1 | [46,47,48,49,50,51] |
| hsa-miR-34C | Downregulated | Hyper- methylated | Tumor suppressor | Nasopharyngeal carcinoma, Prostate cancer | MET | [52,53] |
| hsa-miR-34b/c | Downregulated | Hypermethylated | Tumor suppressor | CRC | - | [54] |
| hsa-miR-762 | Upregulated | - | Tumor promoter | Breast cancer | IRF7 | [55] |
| hsa-miR-9-3 | Downregulated | Hypermethylated | Tumor suppressor | Hodgkin’s lymphoma, Gastric cancer | ITGB1 | [56,57] |
| Hypomethylated miRs Found in Our Study | miR Expression in Cancer | Regulation | Role | Cancer Types | Target Genes | References |
| hsa-miR-1204 | Upregulated | - | Tumor promoter | Breast cancer, Glioblastoma | VDR, CREB-1 | [58,59] |
| hsa-miR-17 | Upregulated | - | Tumor promoter | CRC | - | [60] |
| hsa-miR-18A | Upregulated | - | Tumor promoter | Prostate cancer, Breast cancers, Osteosarcoma, Nasopharyngeal carcinoma, CRC | STK4, IRF2, Dicer1 | [14,61,62,63,64] |
| hsa-miR-19A | Upregulated | - | Tumor promoter | CRC, Gastric cancer, HCC | TIA1, MXD1, PTEN | [14,65,66,67,68] |
| hsa-miR-19B1 | Upregulated | - | Tumor promoter | Gastric cancer | MXD1 | [67] |
| hsa-miR-20A | Upregulated | - | Tumor promoter | CRC | WTX | [69] |
| hsa-miR-548F5 | - | Hyper- methylated | - | Schwannomas | - | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, N.; Abba, M.L.; Zhou, C.; Chang, S.; Gaiser, T.; Leupold, J.H.; Allgayer, H. Changes in Methylation across Structural and MicroRNA Genes Relevant for Progression and Metastasis in Colorectal Cancer. Cancers 2021, 13, 5951. https://doi.org/10.3390/cancers13235951
Patil N, Abba ML, Zhou C, Chang S, Gaiser T, Leupold JH, Allgayer H. Changes in Methylation across Structural and MicroRNA Genes Relevant for Progression and Metastasis in Colorectal Cancer. Cancers. 2021; 13(23):5951. https://doi.org/10.3390/cancers13235951
Chicago/Turabian StylePatil, Nitin, Mohammed L. Abba, Chan Zhou, Shujian Chang, Timo Gaiser, Jörg H. Leupold, and Heike Allgayer. 2021. "Changes in Methylation across Structural and MicroRNA Genes Relevant for Progression and Metastasis in Colorectal Cancer" Cancers 13, no. 23: 5951. https://doi.org/10.3390/cancers13235951
APA StylePatil, N., Abba, M. L., Zhou, C., Chang, S., Gaiser, T., Leupold, J. H., & Allgayer, H. (2021). Changes in Methylation across Structural and MicroRNA Genes Relevant for Progression and Metastasis in Colorectal Cancer. Cancers, 13(23), 5951. https://doi.org/10.3390/cancers13235951