
Mesothelioma Malignancy and the Microenvironment: Molecular Mechanisms


 , , ,
, , ,  ,
,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Mesothelioma Stem Cells (MSCs) and Chemoresistance
3. Mesothelioma Stem Cells Malignancy and TME
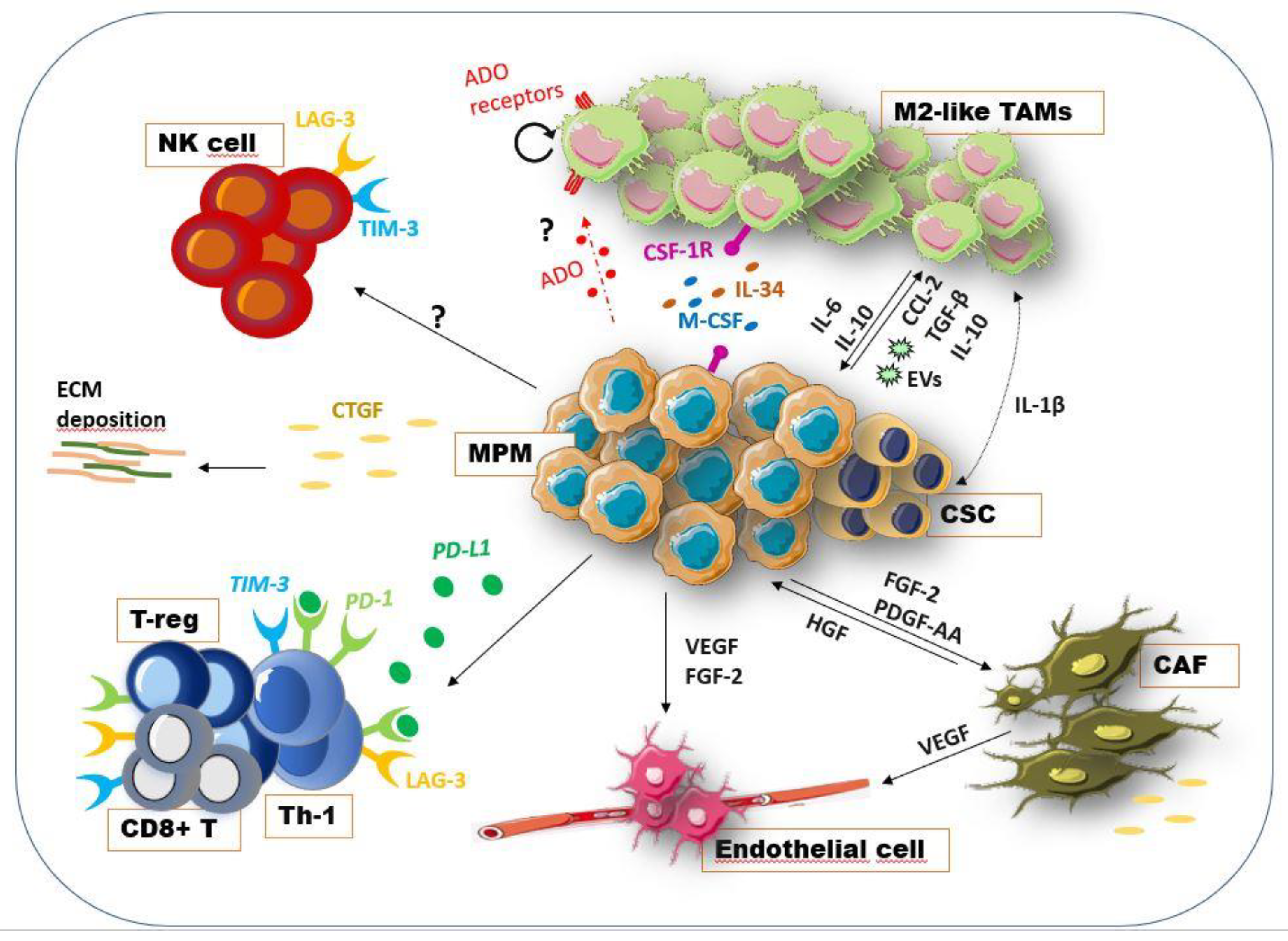
4. Mesothelioma Microenvironment Crosstalk: Molecular Mechanisms
4.1. Mesothelioma and Stroma
4.2. Macrophages in Mesothelioma
4.3. Adenosine Pathway and Mesothelioma Microenvironment

5. Intra-Tumor Heterogeneity within MPM Subtypes and TME
6. Therapeutic Approach Targeting TME
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thomas, A.; Chen, Y.; Yu, T.; Gill, A.; Prasad, V. Distinctive clinical characteristics of malignant mesothelioma in young patients. Oncotarget 2015, 6, 16766–16773. [Google Scholar] [CrossRef]
- FDA: U.S. Food and Drug Administration. FDA Approves Drug Combination For Treating Mesothelioma. Press release. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-drug-combination-treating-mesothelioma (accessed on 26 September 2021).
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef]
- Mangone, L.; Storchi, C.; Bisceglia, I.; Romanelli, A. Malignant Mesothelioma in the Italian Region Emilia-Romagna: Incidence and Asbestos exposure update to 2020. Ann. Res. Oncol. 2021, 1, 199–208. [Google Scholar] [CrossRef]
- Barbarino, M.; Giordano, A. Assessment of the Carcinogenicity of Carbon Nanotubes in the Respiratory System. Cancers 2021, 13, 1318. [Google Scholar] [CrossRef]
- Hillerdal, G.; Berg, J. Malignant Mesothelioma Secondary to Chronic Inflammation and Old Scars. Two New Cases and Review of the Literature. Cancer 1985, 55, 1968–1972. [Google Scholar] [CrossRef]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef]
- Testa, J.R.; Berns, A. Preclinical Models of Malignant Mesothelioma. Front. Oncol. 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.N.; Colby, T.V.; Ordóñez, N.G.; Allen, T.C.; Attanoos, R.L.; Beasley, M.B.; Butnor, K.J.; Chirieac, L.R.; Churg, A.M.; Dacic, S.; et al. Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2018, 142, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Illei, P.B.; Rusch, V.W.; Zakowski, M.F.; Ladanyi, M. Homozygous Deletion of CDKN2A and Codeletion of the Methylthioadenosine Phosphorylase Gene in the Majority of Pleural Mesotheliomas. Clin. Cancer Res. 2003, 9, 2108–2113. [Google Scholar] [PubMed]
- Bertino, J.R.; Waud, W.R.; Parker, W.B.; Lubin, M. Targeting Tumors That Lack Methylthioadenosine Phosphorylase (MTAP) Activity: Current Strategies. Cancer Biol. Ther. 2011, 11, 627–632. [Google Scholar] [CrossRef]
- Kirovski, G.; Stevens, A.P.; Czech, B.; Dettmer, K.; Weiss, T.S.; Wild, P.; Hartmann, A.; Bosserhoff, A.K.; Oefner, P.J.; Hellerbrand, C. Down-Regulation of Methylthioadenosine Phosphorylase (MTAP) Induces Progression of Hepatocellular Carcinoma via Accumulation of 5′-Deoxy-5′-Methylthioadenosine (MTA). Am. J. Pathol. 2011, 178, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Adusumilli, P.S.; Alexander, H.R.J.; Baas, P.; Bardelli, F.; Bononi, A.; Bueno, R.; Felley-Bosco, E.; Galateau-Salle, F.; Jablons, D.; et al. Mesothelioma: Scientific Clues for Prevention, Diagnosis, and Therapy. CA Cancer J. Clin. 2019, 69, 402–429. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Jhanwar, S.C.; Cheng, J.Q.; Testa, J.R. Deletion Mapping of the Short Arm of Chromosome 3 in Human Malignant Mesothelioma. Genes. Chromosomes Cancer 1994, 9, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef]
- Cheng, Y.Y.; Yuen, M.L.; Rath, E.M.; Johnson, B.; Zhuang, L.; Yu, T.K.; Aleksova, V.; Linton, A.; Kao, S.; Clarke, C.J.; et al. CDKN2A and MTAP Are Useful Biomarkers Detectable by Droplet Digital PCR in Malignant Pleural Mesothelioma: A Potential Alternative Method in Diagnosis Compared to Fluorescence In Situ Hybridisation. Front. Oncol. 2020, 10, 579327. [Google Scholar] [CrossRef]
- Thurneysen, C.; Opitz, I.; Kurtz, S.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Functional Inactivation of NF2/Merlin in Human Mesothelioma. Lung Cancer 2009, 64, 140–147. [Google Scholar] [CrossRef]
- Yang, H.; Hall, S.; Sun, B.; Zhao, L.; Gao, Y.; Schmid, R.A.; Tan, S.T.; Peng, R.W.; Yao, F. NF2 and Canonical Hippo-YAP Pathway Define Distinct Tumor Subsets Characterized by Different Immune Deficiency and Treatment Implications in Human Pleural Mesothelioma. Cancers 2021, 13, 1561. [Google Scholar] [CrossRef] [PubMed]
- Badhai, J.; Pandey, G.K.; Song, J.Y.; Krijgsman, O.; Bhaskaran, R.; Chandrasekaran, G.; Kwon, M.C.; Bombardelli, L.; Monkhorst, K.; Grasso, C.; et al. Combined deletion of Bap1, Nf2, and Cdkn2ab causes rapid onset of malignant mesothelioma in mice. J. Exp. Med. 2020, 217, e20191257. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Pezzuto, F.; Lunardi, F.; Vedovelli, L.; Fortarezza, F.; Urso, L.; Grosso, F.; Ceresoli, G.L.; Kern, I.; Vlacic, G.; Faccioli, E.; et al. P14/ARF-Positive Malignant Pleural Mesothelioma: A Phenotype with Distinct Immune Microenvironment. Front. Oncol. 2021, 11, 653497. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, J.L.; Sun, Q.; Harber, J.; Dawson, A.G.; Nakas, A.; Busacca, S.; Sharkey, A.J.; Waller, D.; Sheaff, M.T.; et al. Clonal architecture in mesothelioma is prognostic and shapes the tumour microenvironment. Nat. Commun. 2021, 12, 1751. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Cedrés, S.; Ponce-Aix, S.; Iranzo, P.; Callejo, A.; Pardo, N.; Navarro, A.; Martinez-Marti, A.; Gómez-Abecia, S.; Zucchiatti, A.C.; Sansano, I.; et al. Analysis of Mismatch Repair (MMR) Proteins Expression in a Series of Malignant Pleural Mesothelioma (MPM) Patients. Clin. Transl. Oncol. 2020, 22, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Sinn, K.; Mosleh, B.; Hoda, M.A. Malignant Pleural Mesothelioma: Recent Developments. Curr. Opin. Oncol. 2020, 33, 80–86. [Google Scholar] [CrossRef]
- Rozitis, E.; Johnson, B.; Cheng, Y.Y.; Lee, K. The Use of Immunohistochemistry, Fluorescence in Situ Hybridization, and Emerging Epigenetic Markers in the Diagnosis of Malignant Pleural Mesothelioma (MPM): A Review. Front. Oncol. 2020, 10, 1742. [Google Scholar] [CrossRef]
- Ferrari, L.; Carugno, M.; Mensi, C.; Pesatori, A.C. Circulating Epigenetic Biomarkers in Malignant Pleural Mesothelioma: State of the Art and Critical Evaluation. Front. Oncol. 2020, 10, 445. [Google Scholar] [CrossRef]
- McLoughlin, K.C.; Kaufman, A.S.; Schrump, D.S. Targeting the Epigenome in Malignant Pleural Mesothelioma. Transl. Lung Cancer Res. 2017, 6, 350–365. [Google Scholar] [CrossRef]
- Benedetti, S.; Nuvoli, B.; Catalani, S.; Galati, R. Reactive oxygen species a double-edged sword for mesothelioma. Oncotarget 2015, 6, 16848–16865. [Google Scholar] [CrossRef] [PubMed]
- Mossman, B.T.; Shukla, A.; Heintz, N.H.; Verschraegen, C.F.; Thomas, A.; Hassan, R. New insights into understanding the mechanisms, pathogenesis, and management of malignant mesotheliomas. Am. J. Pathol. 2013, 182, 1065–1077. [Google Scholar] [CrossRef]
- Minnema-Luiting, J.; Vroman, H.; Aerts, J.; Cornelissen, R. Heterogeneity in Immune Cell Content in Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2018, 19, 1041. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.S.; Righi, L.; Koeppen, H.; Zou, W.; Izzo, S.; Grosso, F.; Libener, R.; Loiacono, M.; Monica, V.; Buttigliero, C.; et al. Molecular and Histopathological Characterization of the Tumor Immune Microenvironment in Advanced Stage of Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2018, 13, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Pasello, G.; Zago, G.; Lunardi, F.; Urso, L.; Kern, I.; Vlacic, G.; Grosso, F.; Mencoboni, M.; Ceresoli, G.L.; Schiavon, M.; et al. Malignant pleural mesothelioma immune microenvironment and checkpoint expression: Correlation with clinical-pathological features and intratumor heterogeneity over time. Ann. Oncol. 2018, 29, 1258–1265. [Google Scholar] [CrossRef]
- Linton, A.; van Zandwijk, N.; Reid, G.; Clarke, S.; Cao, C.; Kao, S. Inflammation in malignant mesothelioma—Friend or foe? Annal. Cardiothorac. Surg. 2012, 1, 516–522. [Google Scholar] [CrossRef]
- Chu, G.J.; van Zandwijk, N.; Rasko, J. The Immune Microenvironment in Mesothelioma: Mechanisms of Resistance to Immunotherapy. Front. Oncol. 2019, 9, 1366. [Google Scholar] [CrossRef]
- Cornelissen, R.; Lievense, L.A.; Maat, A.P.; Hendriks, R.W.; Hoogsteden, H.C.; Bogers, A.J.; Hegmans, J.P.; Aerts, J.G. Ratio of intratumoral macrophage phenotypes is a prognostic factor in epithelioid malignant pleural mesothelioma. PLoS ONE 2014, 9, e106742. [Google Scholar] [CrossRef]
- Guo, W.; Lasky, J.L.; Wu, H. Cancer stem cells. Pediatr. Res. 2006, 59 Pt 2, 59R–64R. [Google Scholar] [CrossRef]
- Kuşoğlu, A.; Biray Avcı, Ç. Cancer stem cells: A brief review of the current status. Gene 2019, 681, 80–85. [Google Scholar] [CrossRef]
- Pasdar, E.A.; Smits, M.; Stapelberg, M.; Bajzikova, M.; Stantic, M.; Goodwin, J.; Yan, B.; Stursa, J.; Kovarova, J.; Sachaphibulkij, K.; et al. Characterisation of mesothelioma-initiating cells and their susceptibility to anti-cancer agents. PLoS ONE 2015, 10, e0119549. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Dericks, L.; Carboni, G.L.; Schmid, R.A.; Karoubi, G. Putative cancer stem cells in malignant pleural mesothelioma show resistance to cisplatin and pemetrexed. Int. J. Oncol. 2010, 37, 437–444. [Google Scholar] [CrossRef][Green Version]
- Oehl, K.; Vrugt, B.; Opitz, I.; Meerang, M. Heterogeneity in Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2018, 19, 1603. [Google Scholar] [CrossRef]
- Colak, S.; Medema, J.P. Cancer stem cells--important players in tumor therapy resistance. FEBS J. 2014, 281, 4779–4791. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; D’Costa, S.; Yoon, B.I.; Brody, A.R.; Sills, R.C.; Kim, Y. Characterization of side population cells in human malignant mesothelioma cell lines. Lung Cancer 2010, 70, 146–151. [Google Scholar] [CrossRef]
- Blum, W.; Pecze, L.; Felley-Bosco, E.; Wu, L.; de Perrot, M.; Schwaller, B. Stem Cell Factor-Based Identification and Functional Properties of In Vitro-Selected Subpopulations of Malignant Mesothelioma Cells. Stem Cell Rep. 2017, 8, 1005–1017. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Dericks, L.; Froment, L.; Boesch, R.; Schmid, R.A.; Karoubi, G. Cisplatin-resistant cells in malignant pleural mesothelioma cell lines show ALDH(high)CD44(+) phenotype and sphere-forming capacity. BMC Cancer 2014, 14, 304. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, V.; Kopecka, J.; Salaroglio, I.C.; Libener, R.; Napoli, F.; Izzo, S.; Orecchia, S.; Ananthanarayanan, P.; Bironzo, P.; Grosso, F.; et al. Wnt/IL-1β/IL-8 autocrine circuitries control chemoresistance in mesothelioma initiating cells by inducing ABCB5. Int. J. Cancer 2020, 146, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef]
- Menges, C.W.; Kadariya, Y.; Altomare, D.; Talarchek, J.; Neumann-Domer, E.; Wu, Y.; Xiao, G.H.; Shapiro, I.M.; Kolev, V.N.; Pachter, J.A.; et al. Tumor suppressor alterations cooperate to drive aggressive mesotheliomas with enriched cancer stem cells via a p53-miR-34a-c-Met axis. Cancer Res. 2014, 74, 1261–1271. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin deficiency predicts FAK inhibitor sensitivity: A synthetic lethal relationship. Sci. Transl. Med. 2014, 6, 237ra68. [Google Scholar] [CrossRef]
- Kolev, V.N.; Tam, W.F.; Wright, Q.G.; McDermott, S.P.; Vidal, C.M.; Shapiro, I.M.; Xu, Q.; Wicha, M.S.; Pachter, J.A.; Weaver, D.T. Inhibition of FAK kinase activity preferentially targets cancer stem cells. Oncotarget 2017, 8, 51733–51747. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biom. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Lau, B.W.; Kane, A.B. SDF1/CXCL12 is involved in recruitment of stem-like progenitor cells to orthotopic murine malignant mesothelioma spheroids. Anticancer Res. 2010, 30, 2153–2160. [Google Scholar]
- Adhikary, G.; Grun, D.; Alexander, H.R.; Friedberg, J.S.; Xu, W.; Keillor, J.W.; Kandasamy, S.; Eckert, R.L. Transglutaminase is a mesothelioma cancer stem cell survival protein that is required for tumor formation. Oncotarget 2018, 9, 34495–34505. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, S.; Adhikary, G.; Rorke, E.A.; Friedberg, J.S.; Mickle, M.B.; Alexander, H.R.; Eckert, R.L. The YAP1 Signaling Inhibitors, Verteporfin and CA3, Suppress the Mesothelioma Cancer Stem Cell Phenotype. Mol. Cancer Res. 2020, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Werb, Z.; Lu, P. The Role of Stroma in Tumor Development. Cancer J. 2015, 21, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef]
- Napoli, F.; Listì, A.; Zambelli, V.; Witel, G.; Bironzo, P.; Papotti, M.; Volante, M.; Scagliotti, G.; Righi, L. Pathological Characterization of Tumor Immune Microenvironment (TIME) in Malignant Pleural Mesothelioma. Cancers 2021, 13, 2564. [Google Scholar] [CrossRef]
- Lievense, L.A.; Cornelissen, R.; Bezemer, K.; Kaijen-Lambers, M.E.; Hegmans, J.P.; Aerts, J.G. Pleural Effusion of Patients with Malignant Mesothelioma Induces Macrophage-Mediated T Cell Suppression. J. Thorac. Onc. 2016, 11, 1755–1764. [Google Scholar] [CrossRef]
- Fujii, M.; Toyoda, T.; Nakanishi, H.; Yatabe, Y.; Sato, A.; Matsudaira, Y.; Ito, H.; Murakami, H.; Kondo, Y.; Kondo, E.; et al. TGF-β synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J. Exp. Med. 2012, 209, 479–494. [Google Scholar] [CrossRef]
- Chu, C.Y.; Chang, C.C.; Prakash, E.; Kuo, M.L. Connective tissue growth factor (CTGF) and cancer progression. J. Biomed. Sci. 2008, 15, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Jang, I.; Beningo, K.A. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers 2019, 11, 721. [Google Scholar] [CrossRef] [PubMed]
- Ohara, Y.; Enomoto, A.; Tsuyuki, Y.; Sato, K.; Iida, T.; Kobayashi, H.; Mizutani, Y.; Miyai, Y.; Hara, A.; Mii, S.; et al. Connective tissue growth factor produced by cancer-associated fibroblasts correlates with poor prognosis in epithelioid malignant pleural mesothelioma. Oncol. Rep. 2020, 44, 838–848. [Google Scholar] [CrossRef]
- Li, Q.; Wang, W.; Yamada, T.; Matsumoto, K.; Sakai, K.; Bando, Y.; Uehara, H.; Nishioka, Y.; Sone, S.; Iwakiri, S.; et al. Pleural mesothelioma instigates tumor-associated fibroblasts to promote progression via a malignant cytokine network. Am. J. Pathol. 2011, 179, 1483–1493. [Google Scholar] [CrossRef]
- Kumar-Singh, S.; Weyler, J.; Martin, M.J.; Vermeulen, P.B.; Van Marck, E. Angiogenic cytokines in mesothelioma: A study of VEGF, FGF-1 and -2, and TGF beta expression. J. Pathol. 1999, 189, 72–78. [Google Scholar] [CrossRef]
- Wang, F.T.; Sun, W.; Zhang, J.T.; Fan, Y.Z. Cancer-associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer. Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef]
- Chia, P.L.; Russell, P.; Asadi, K.; Thapa, B.; Gebski, V.; Murone, C.; Walkiewicz, M.; Eriksson, U.; Scott, A.M.; John, T. Analysis of angiogenic and stromal biomarkers in a large malignant mesothelioma cohort. Lung Cancer 2020, 150, 1–8. [Google Scholar] [CrossRef]
- Hegmans, J.P.; Hemmes, A.; Hammad, H.; Boon, L.; Hoogsteden, H.C.; Lambrecht, B.N. Mesothelioma environment comprises cytokines and T-regulatory cells that suppress immune responses. Eur. Resp. J. 2006, 27, 1086–1095. [Google Scholar] [CrossRef]
- Ujiie, H.; Kadota, K.; Nitadori, J.I.; Aerts, J.G.; Woo, K.M.; Sima, C.S.; Travis, W.D.; Jones, D.R.; Krug, L.M.; Adusumilli, P.S. The tumoral and stromal immune microenvironment in malignant pleural mesothelioma: A comprehensive analysis reveals prognostic immune markers. Oncoimmunology 2015, 4, e1009285. [Google Scholar] [CrossRef]
- Marcq, E.; Waele, J.; Audenaerde, J.V.; Lion, E.; Santermans, E.; Hens, N.; Pauwels, P.; van Meerbeeck, J.P.; Smits, E. Abundant expression of TIM-3, LAG-3, PD-1 and PD-L1 as immunotherapy checkpoint targets in effusions of mesothelioma patients. Oncotarget 2017, 8, 89722–89735. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci. Transl. Med. 2011, 3, 111ra120. [Google Scholar] [CrossRef]
- Klampatsa, A.; O’Brien, S.M.; Thompson, J.C.; Rao, A.S.; Stadanlick, J.E.; Martinez, M.C.; Liousia, M.; Cantu, E.; Cengel, K.; Moon, E.K.; et al. Phenotypic and functional analysis of malignant mesothelioma tumor-infiltrating lymphocytes. Oncoimmunology 2019, 8, e1638211. [Google Scholar] [CrossRef]
- Anraku, M.; Cunningham, K.S.; Yun, Z.; Tsao, M.S.; Zhang, L.; Keshavjee, S.; Johnston, M.R.; de Perrot, M. Impact of tumor-infiltrating T cells on survival in patients with malignant pleural mesothelioma. J. Thorac. Cardiovasc. Surg. 2008, 135, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Oizumi, S.; Kikuchi, E.; Shinagawa, N.; Konishi-Sakakibara, J.; Ishimine, A.; Aoe, K.; Gemba, K.; Kishimoto, T.; Torigoe, T.; et al. CD8+ tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol. Immunother. 2010, 59, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Hiltbrunner, S.; Mannarino, L.; Kirschner, M.B.; Opitz, I.; Rigutto, A.; Laure, A.; Lia, M.; Nozza, P.; Maconi, A.; Marchini, S.; et al. Tumor Immune Microenvironment and Genetic Alterations in Mesothelioma. Front. Oncol. 2021, 11, 660039. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Bedrossian, C.W. The pathogenesis of mesothelioma. Semin. Diagn. Path. 2006, 23, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Désage, A.L.; Karpathiou, G.; Peoc’h, M.; Froudarakis, M.E. The Immune Microenvironment of Malignant Pleural Mesothelioma: A Literature Review. Cancers 2021, 13, 3205. [Google Scholar] [CrossRef]
- Wang, M.; Gauthier, A.; Daley, L.; Dial, K.; Wu, J.; Woo, J.; Lin, M.; Ashby, C.; Mantell, L.L. The Role of HMGB1, a Nuclear Damage-Associated Molecular Pattern Molecule, in the Pathogenesis of Lung Diseases. Antiox. Redox Signal. 2019, 31, 954–993. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H. Mesothelioma: Recent highlights. Ann. Transl. Med. 2017, 5, 238. [Google Scholar] [CrossRef] [PubMed]
- Hiraku, Y.; Guo, F.; Ma, N.; Yamada, T.; Wang, S.; Kawanishi, S.; Murata, M. Multi-walled carbon nanotube induces nitrative DNA damage in human lung epithelial cells via HMGB1-RAGE interaction and Toll-like receptor 9 activation. Part. Fibre Toxicol. 2016, 13, 16. [Google Scholar] [CrossRef]
- Boyles, M.S.; Young, L.; Brown, D.M.; MacCalman, L.; Cowie, H.; Moisala, A.; Smail, F.; Smith, P.J.; Proudfoot, L.; Windle, A.H.; et al. Multi-walled carbon nanotube induced frustrated phagocytosis, cytotoxicity and pro-inflammatory conditions in macrophages are length dependent and greater than that of asbestos. Toxicol. In Vitro 2015, 29, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H. Molecular pathways: Targeting mechanisms of asbestos and erionite carcinogenesis in mesothelioma. Clin. Cancer Res. 2012, 18, 598–604. [Google Scholar] [CrossRef]
- Schürch, C.M.; Forster, S.; Brühl, F.; Yang, S.H.; Felley-Bosco, E.; Hewer, E. The “don’t eat me” signal CD47 is a novel diagnostic biomarker and potential therapeutic target for diffuse malignant mesothelioma. Oncoimmunology 2017, 7, e1373235. [Google Scholar] [CrossRef]
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. Int. J. Mol. Sci. 2020, 21, 5207. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O. Regulators of macrophage activation. Eur. J. Immunol. 2011, 41, 1531–1534. [Google Scholar] [CrossRef]
- Xu, F.; Wei, Y.; Tang, Z.; Liu, B.; Dong, J. Tumor-associated macrophages in lung cancer: Friend or foe? (Review). Mol. Med. Rep. 2020, 22, 4107–4115. [Google Scholar] [CrossRef]
- Yang, Q.; Guo, N.; Zhou, Y.; Chen, J.; Wei, Q.; Han, M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm. Sin. B 2020, 10, 2156–2170. [Google Scholar] [CrossRef] [PubMed]
- Colin, D.J.; Cottet-Dumoulin, D.; Faivre, A.; Germain, S.; Triponez, F.; Serre-Beinier, V. Experimental Model of Human Malignant Mesothelioma in Athymic Mice. Int. J. Mol. Sci. 2018, 19, 1881. [Google Scholar] [CrossRef]
- Chéné, A.L.; d’Almeida, S.; Blondy, T.; Tabiasco, J.; Deshayes, S.; Fonteneau, J.F.; Cellerin, L.; Delneste, Y.; Grégoire, M.; Blanquart, C. Pleural Effusions from Patients with Mesothelioma Induce Recruitment of Monocytes and Their Differentiation into M2 Macrophages. J. Thorac. Oncol. 2016, 11, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Sarode, P.; Schaefer, M.B.; Grimminger, F.; Seeger, W.; Savai, R. Macrophage and Tumor Cell Cross-Talk Is Fundamental for Lung Tumor Progression: We Need to Talk. Front. Oncol. 2020. 10, 324. [CrossRef]
- Izzi, V.; Chiurchiù, V.; D’Aquilio, F.; Palumbo, C.; Tresoldi, I.; Modesti, A.; Baldini, P.M. Differential effects of malignant mesothelioma cells on THP-1 monocytes and macrophages. Int. J. Oncol. 2009, 34, 543–550. [Google Scholar] [CrossRef][Green Version]
- Burt, B.M.; Rodig, S.J.; Tilleman, T.R.; Elbardissi, A.W.; Bueno, R.; Sugarbaker, D.J. Circulating and tumor-infiltrating myeloid cells predict survival in human pleural mesothelioma. Cancer 2011, 117, 5234–5244. [Google Scholar] [CrossRef]
- Horio, D.; Minami, T.; Kitai, H.; Ishigaki, H.; Higashiguchi, Y.; Kondo, N.; Hirota, S.; Kitajima, K.; Nakajima, Y.; Koda, Y.; et al. Tumor-associated macrophage-derived inflammatory cytokine enhances malignant potential of malignant pleural mesothelioma. Cancer Sci. 2020, 111, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Blondy, T.; d’Almeida, S.M.; Briolay, T.; Tabiasco, J.; Meiller, C.; Chéné, A.L.; Cellerin, L.; Deshayes, S.; Delneste, Y.; Fonteneau, J.F.; et al. Involvement of the M-CSF/IL-34/CSF-1R pathway in malignant pleural mesothelioma. J. Immunother. Cancer 2020, 8, e000182. [Google Scholar] [CrossRef]
- Cioce, M.; Canino, C.; Goparaju, C.; Yang, H.; Carbone, M.; Pass, H.I. Autocrine CSF-1R signaling drives mesothelioma chemoresistance via AKT activation. Cell Death Dis. 2014, 5, e1167. [Google Scholar] [CrossRef] [PubMed]
- Dammeijer, F.; Lievense, L.A.; Kaijen-Lambers, M.E.; van Nimwegen, M.; Bezemer, K.; Hegmans, J.P.; van Hall, T.; Hendriks, R.W.; Aerts, J.G. Depletion of Tumor-Associated Macrophages with a CSF-1R Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by DC Immunotherapy. Cancer Immunol. Res. 2017, 5, 535–546. [Google Scholar] [CrossRef]
- Magkouta, S.F.; Vaitsi, P.C.; Pappas, A.G.; Iliopoulou, M.; Kosti, C.N.; Psarra, K.; Kalomenidis, I.T. CSF1/CSF1R Axis Blockade Limits Mesothelioma and Enhances Efficiency of Anti-PDL1 Immunotherapy. Cancers 2021, 13, 2546. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Miselis, N.R.; Wu, Z.J.; Van Rooijen, N.; Kane, A.B. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol. Cancer Ther. 2008, 7, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Veltman, J.D.; Lambers, M.E.; van Nimwegen, M.; Hendriks, R.W.; Hoogsteden, H.C.; Hegmans, J.P.; Aerts, J.G. Zoledronic acid impairs myeloid differentiation to tumour-associated macrophages in mesothelioma. Br. J. Cancer 2010, 103, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Andón, F.T.; Digifico, E.; Maeda, A.; Erreni, M.; Mantovani, A.; Alonso, M.J.; Allavena, P. Targeting tumor associated macrophages: The new challenge for nanomedicine. Semin. Immunol. 2017, 34, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Giurisato, E.; Lonardi, S.; Telfer, B.; Lussoso, S.; Risa-Ebrí, B.; Zhang, J.; Russo, I.; Wang, J.; Santucci, A.; Finegan, K.G.; et al. Extracellular-Regulated Protein Kinase 5-Mediated Control of p21 Expression Promotes Macrophage Proliferation Associated with Tumor Growth and Metastasis. Cancer Res. 2020, 80, 3319–3330. [Google Scholar] [CrossRef]
- Allard, B.; Beavis, P.A.; Darcy, P.K.; Stagg, J. Immunosuppressive activities of adenosine in cancer. Curr. Opin. Pharmacol. 2016, 29, 7–16. [Google Scholar] [CrossRef]
- Whiteside, T.L. The potential of tumor-derived exosomes for noninvasive cancer monitoring. Expert. Rev. Mol. Diagn. 2015, 15, 1293–1310. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Ghalamfarsa, G.; Kazemi, M.H.; Raoofi Mohseni, S.; Masjedi, A.; Hojjat-Farsangi, M.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. CD73 as a potential opportunity for cancer immunotherapy. Expert. Opin. Ther. Targets 2019, 23, 127–142. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef]
- Reyna-Jeldes, M.; Díaz-Muñoz, M.; Madariaga, J.A.; Coddou, C.; Vázquez-Cuevas, F.G. Autocrine and paracrine purinergic signaling in the most lethal types of cancer. Purinergic Signal. 2021, 1–26. [Google Scholar] [CrossRef]
- Yang, H.; Yao, F.; Davis, P.F.; Tan, S.T.; Hall, S. CD73, Tumor Plasticity and Immune Evasion in Solid Cancers. Cancers 2021, 13, 177. [Google Scholar] [CrossRef]
- Al-Taei, S.; Salimu, J.; Spary, L.K.; Clayton, A.; Lester, J.F.; Tabi, Z. Prostaglandin E2-mediated adenosinergic effects on CD14+ cells: Self-amplifying immunosuppression in cancer. Oncoimmunology 2016, 6, e1268308. [Google Scholar] [CrossRef]
- Haskó, G.; Pacher, P. Regulation of macrophage function by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Ernens, I.; Léonard, F.; Vausort, M.; Rolland-Turner, M.; Devaux, Y.; Wagner, D.R. Adenosine up-regulates vascular endothelial growth factor in human macrophages. Biochem. Biophys. Res. Commun. 2010, 392, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef]
- Pinhal-Enfield, G.; Ramanathan, M.; Hasko, G.; Vogel, S.N.; Salzman, A.L.; Boons, G.J.; Leibovich, S.J. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am. J. Pathol. 2003, 163, 711–721. [Google Scholar] [CrossRef]
- Csóka, B.; Németh, Z.H.; Virág, L.; Gergely, P.; Leibovich, S.J.; Pacher, P.; Sun, C.X.; Blackburn, M.R.; Vizi, E.S.; Deitch, E.A.; et al. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood 2007, 110, 2685–2695. [Google Scholar] [CrossRef]
- Haskó, G.; Kuhel, D.G.; Chen, J.F.; Schwarzschild, M.A.; Deitch, E.A.; Mabley, J.G.; Marton, A.; Szabó, C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J. 2000, 14, 2065–2074. [Google Scholar] [CrossRef]
- Kreckler, L.M.; Wan, T.C.; Ge, Z.D.; Auchampach, J.A. Adenosine inhibits tumor necrosis factor-alpha release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor. J. Pharmacol. Exp. Ther. 2006, 317, 172–180. [Google Scholar] [CrossRef]
- Németh, Z.H.; Lutz, C.S.; Csóka, B.; Deitch, E.A.; Leibovich, S.J.; Gause, W.C.; Tone, M.; Pacher, P.; Vizi, E.S.; Haskó, G. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J. Immunol. 2005, 175, 8260–8270. [Google Scholar] [CrossRef]
- Csóka, B.; Selmeczy, Z.; Koscsó, B.; Németh, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Chu, Y.; Li, Z.; Yu, X.; Huang, Z.; Xu, J.; Zheng, L. Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J. Hepatol. 2021, 74, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Blum, Y.; Meiller, C.; Quetel, L.; Elarouci, N.; Ayadi, M.; Tashtanbaeva, D.; Armenoult, L.; Montagne, F.; Tranchant, R.; Renier, A.; et al. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat. Commun. 2019, 10, 1333. [Google Scholar] [CrossRef]
- Lee, H.S.; Jang, H.J.; Choi, J.M.; Zhang, J.; de Rosen, V.L.; Wheeler, T.M.; Lee, J.S.; Tu, T.; Jindra, P.T.; Kerman, R.H.; et al. Comprehensive immunoproteogenomic analyses of malignant pleural mesothelioma. JCI Insight 2018, 3, e98575. [Google Scholar] [CrossRef]
- Wadowski, B.; Bueno, R.; De Rienzo, A. Immune Microenvironment and Genetics in Malignant Pleural Mesothelioma. Front. Oncol. 2021, 11, 684025. [Google Scholar] [CrossRef]
- Thapa, B.; Salcedo, A.; Lin, X.; Walkiewicz, M.; Murone, C.; Ameratunga, M.; Asadi, K.; Deb, S.; Barnett, S.A.; Knight, S.; et al. The Immune Microenvironment, Genome-wide Copy Number Aberrations, and Survival in Mesothelioma. J. Thorac. Oncol. 2017, 12, 850–859. [Google Scholar] [CrossRef]
- Muller, S.; Victoria Lai, W.; Adusumilli, P.S.; Desmeules, P.; Frosina, D.; Jungbluth, A.; Ni, A.; Eguchi, T.; Travis, W.D.; Ladanyi, M.; et al. V-domain Ig-containing suppressor of T-cell activation (VISTA), a potentially targetable immune checkpoint molecule, is highly expressed in epithelioid malignant pleural mesothelioma. Mod. Pathol. 2020, 33, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xu, D.; Schmid, R.A.; Peng, R.W. Biomarker-guided targeted and immunotherapies in malignant pleural mesothelioma. Ther. Adv. Med. Oncol. 2020, 12, 1758835920971421. [Google Scholar] [CrossRef]
- Alcala, N.; Mangiante, L.; Le-Stang, N.; Gustafson, C.E.; Boyault, S.; Damiola, F.; Alcala, K.; Brevet, M.; Thivolet-Bejui, F.; Blanc-Fournier, C.; et al. Redefining malignant pleural mesothelioma types as a continuum uncovers immune-vascular interactions. EBioMedicine 2019, 48, 191–202. [Google Scholar] [CrossRef]
- Tazzari, M.; Brich, S.; Tuccitto, A.; Bozzi, F.; Beretta, V.; Spagnuolo, R.D.; Negri, T.; Stacchiotti, S.; Deraco, M.; Baratti, D.; et al. Complex Immune Contextures Characterise Malignant Peritoneal Mesothelioma: Loss of Adaptive Immunological Signature in the More Aggressive Histological Types. J. Immunol. Res. 2018, 5804230. [Google Scholar] [CrossRef] [PubMed]
- Alay, A.; Cordero, D.; Hijazo-Pechero, S.; Aliagas, E.; Lopez-Doriga, A.; Marín, R.; Palmero, R.; Llatjós, R.; Escobar, I.; Ramos, R.; et al. Integrative transcriptome analysis of malignant pleural mesothelioma reveals a clinically relevant immune-based classification. J. Immunother. Cancer 2021, 9, e001601. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Kotova, S.; Wong, R.M.; Cameron, R.B. New and emerging therapeutic options for malignant pleural mesothelioma: Review of early clinical trials. Cancer Manag. Res. 2015, 7, 51–63. [Google Scholar] [CrossRef][Green Version]
- Bronte, G.; Incorvaia, L.; Rizzo, S.; Passiglia, F.; Galvano, A.; Rizzo, F.; Rolfo, C.; Fanale, D.; Listì, A.; Natoli, C.; et al. The resistance related to targeted therapy in malignant pleural mesothelioma: Why has not the target been hit yet? Crit. Rev. Oncol. Hematol. 2016, 107, 20–32. [Google Scholar] [CrossRef]
- Bograd, A.J.; Suzuki, K.; Vertes, E.; Colovos, C.; Morales, E.A.; Sadelain, M.; Adusumilli, P.S. Immune responses and immunotherapeutic interventions in malignant pleural mesothelioma. Cancer Immunol. Immunother. 2011, 60, 1509–1527. [Google Scholar] [CrossRef]
- Chia, P.L.; Russell, P.A.; Scott, A.M.; John, T. Targeting the vasculature: Anti-angiogenic agents for malignant mesothelioma. Expert Rev. Anticancer Ther. 2016, 16, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; Lindwasser, O.W.; Adjei, A.A.; Adusumilli, P.S.; Beyers, M.L.; Blumenthal, G.M.; Bueno, R.; Burt, B.M.; Carbone, M.; Dahlberg, S.E.; et al. Current and Future Management of Malignant Mesothelioma: A Consensus Report from the National Cancer Institute Thoracic Malignancy Steering Committee, International Association for the Study of Lung Cancer, and Mesothelioma Applied Research Foundation. J. Thorac. Oncol. 2018, 13, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Grosso, F.; Steele, N.; Novello, S.; Nowak, A.K.; Popat, S.; Greillier, L.; John, T.; Leighl, N.B.; Reck, M.; Taylor, P.; et al. Nintedanib Plus Pemetrexed/Cisplatin in Patients With Malignant Pleural Mesothelioma: Phase II Results From the Randomized, Placebo-Controlled LUME-Meso Trial. J. Clin. Oncol. 2017, 35, 3591–3600. [Google Scholar] [CrossRef] [PubMed]
- Berneman, Z.N.; Germonpre, P.; Huizing, T.M.; Van de Velde, A.; Nijs, G.; Stein, B.; Van Tendeloo, V.F.; Lion, E.; Smits, E.L.; Anguille, S. Dendritic cell vaccination in malignant pleural mesothelioma: A phase I/II study. J. Clin. Oncol. 2014, 32 (Suppl. 15), 7583. [Google Scholar] [CrossRef]
- Noordam, L.; Kaijen, M.; Bezemer, K.; Cornelissen, R.; Maat, L.; Hoogsteden, H.C.; Aerts, J.; Hendriks, R.W.; Hegmans, J.; Vroman, H. Low-dose cyclophosphamide depletes circulating naïve and activated regulatory T cells in malignant pleural mesothelioma patients synergistically treated with dendritic cell-based immunotherapy. Oncoimmunology 2018, 7, e1474318. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.; Bocchini, M.; Bronte, G.; Delmonte, A.; Guidoboni, M.; Crinò, L.; Mazza, M. Malignant Pleural Mesothelioma: State-of-the-Art on Current Therapies and Promises for the Future. Front. Oncol. 2020, 9, 1519. [Google Scholar] [CrossRef]
- Obacz, J.; Yung, H.; Shamseddin, M.; Linnane, E.; Liu, X.; Azad, A.A.; Rassl, D.M.; Fairen-Jimenez, D.; Rintoul, R.C.; Nikolić, M.Z.; et al. Biological basis for novel mesothelioma therapies. Br. J. Cancer 2021, 10, 1–17. [Google Scholar] [CrossRef]
- Cinausero, M.; Rihawi, K.; Cortiula, F.; Follador, A.; Fasola, G.; Ardizzoni, A. Emerging therapies in malignant pleural mesothelioma. Crit. Rev. Oncol. Hematol. 2019, 144, 102815. [Google Scholar] [CrossRef]
- Gray, S.G.; Mutti, L. Immunotherapy for mesothelioma: A critical review of current clinical trials and future perspectives. Trans. Lung Cancer Res. 2020, 9 (Suppl. 1), S100–S119. [Google Scholar] [CrossRef]
- Tan, Z.; Chiu, M.S.; Yan, C.W.; Wong, Y.C.; Huang, H.; Man, K.; Chen, Z. Antimesothelioma Immunotherapy by CTLA-4 Blockade Depends on Active PD1-Based TWIST1 Vaccination. Mol. Ther. Oncolytics 2020, 16, 302–317. [Google Scholar] [CrossRef]
- Klampatsa, A.; Albelda, S.M. Current Advances in CAR T Cell Therapy for Malignant Mesothelioma. J. Cell. Immunol. 2020, 2, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, T.; Achard, C.; Boisgerault, N.; Grard, M.; Petithomme, T.; Chatelain, C.; Dutoit, S.; Blanquart, C.; Royer, P.J.; Minvielle, S.; et al. Frequent Homozygous Deletions of Type I Interferon Genes in Pleural Mesothelioma Confer Sensitivity to Oncolytic Measles Virus. J. Thorac. Oncol. 2020, 15, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xu, D.; Gao, Y.; Schmid, R.A.; Peng, R.W. Oncolytic Viral Therapy for Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2020, 15, e111–e113. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug Name | Therapeutic Strategy | Molecular Target | Phase | Clinical Trials | Ref |
|---|---|---|---|---|---|
| Mesothelin-targeted CAR-T | CAR-T | Mesothelin | Phase I Phase II | NCT02414269 NCT03054298 | [152] |
| Nivolumab | mAb | PD-1 | Phase I Phase II Phase III | NCT02497508 JapicCTI163247 NCT03063450 | [148] |
| Pembrolizumab | mAb | PD-1 | Phase I Phase II Phase III | NCT02054806 NCT02399371 NCT02991482 | [147] |
| Tremelimumab | mAb | CTLA-4 | Phase II | NCT01843374 | [147] |
| Bevacizumab | mAb | VEGF | NCT00651456 | [142,143] | |
| FAP-targeted CAR-T | CAR-T | Fibroblast-activating protein (FAP) | Phase I | NCT01722149 | [152] |
| WT1- DCV | DC vaccination | Wilms tumor protein 1 (WT1) | Phase I/II | NCT02649829 | [145] |
| Adenoviral-mediated IFN-α/β | Cytokine-based gene therapy | Interferon | Phase I Phase I | NCT01212367 NCT01119664 | [140,141] |
| Nintedanib | Tyrosine kinase Inhibitor | VEGF, PDGF, FGF receptors | Phase II | NCT01907100 | [144] |
| Emactuzumab | mAb | CSF-1 receptor | Phase I | NCT01494688 | [153] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cersosimo, F.; Barbarino, M.; Lonardi, S.; Vermi, W.; Giordano, A.; Bellan, C.; Giurisato, E. Mesothelioma Malignancy and the Microenvironment: Molecular Mechanisms. Cancers 2021, 13, 5664. https://doi.org/10.3390/cancers13225664
Cersosimo F, Barbarino M, Lonardi S, Vermi W, Giordano A, Bellan C, Giurisato E. Mesothelioma Malignancy and the Microenvironment: Molecular Mechanisms. Cancers. 2021; 13(22):5664. https://doi.org/10.3390/cancers13225664
Chicago/Turabian StyleCersosimo, Francesca, Marcella Barbarino, Silvia Lonardi, William Vermi, Antonio Giordano, Cristiana Bellan, and Emanuele Giurisato. 2021. "Mesothelioma Malignancy and the Microenvironment: Molecular Mechanisms" Cancers 13, no. 22: 5664. https://doi.org/10.3390/cancers13225664
APA StyleCersosimo, F., Barbarino, M., Lonardi, S., Vermi, W., Giordano, A., Bellan, C., & Giurisato, E. (2021). Mesothelioma Malignancy and the Microenvironment: Molecular Mechanisms. Cancers, 13(22), 5664. https://doi.org/10.3390/cancers13225664