
Transcriptional and Mutational Profiling of B-Other Acute Lymphoblastic Leukemia for Improved Diagnostics

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort, Sample Preparation, DNA and RNA Extraction
2.2. Whole-Transcriptome RNA Sequencing
2.3. Karyotyping, FISH Analysis and Array CGH
2.4. Immunophenotyping
2.5. Fusion Detection
2.6. Mutation Analysis
2.7. Gene Expression Profiling
2.8. Sample Classification
2.9. Differential Gene Expression
2.10. Differential Splicing Analysis
3. Results
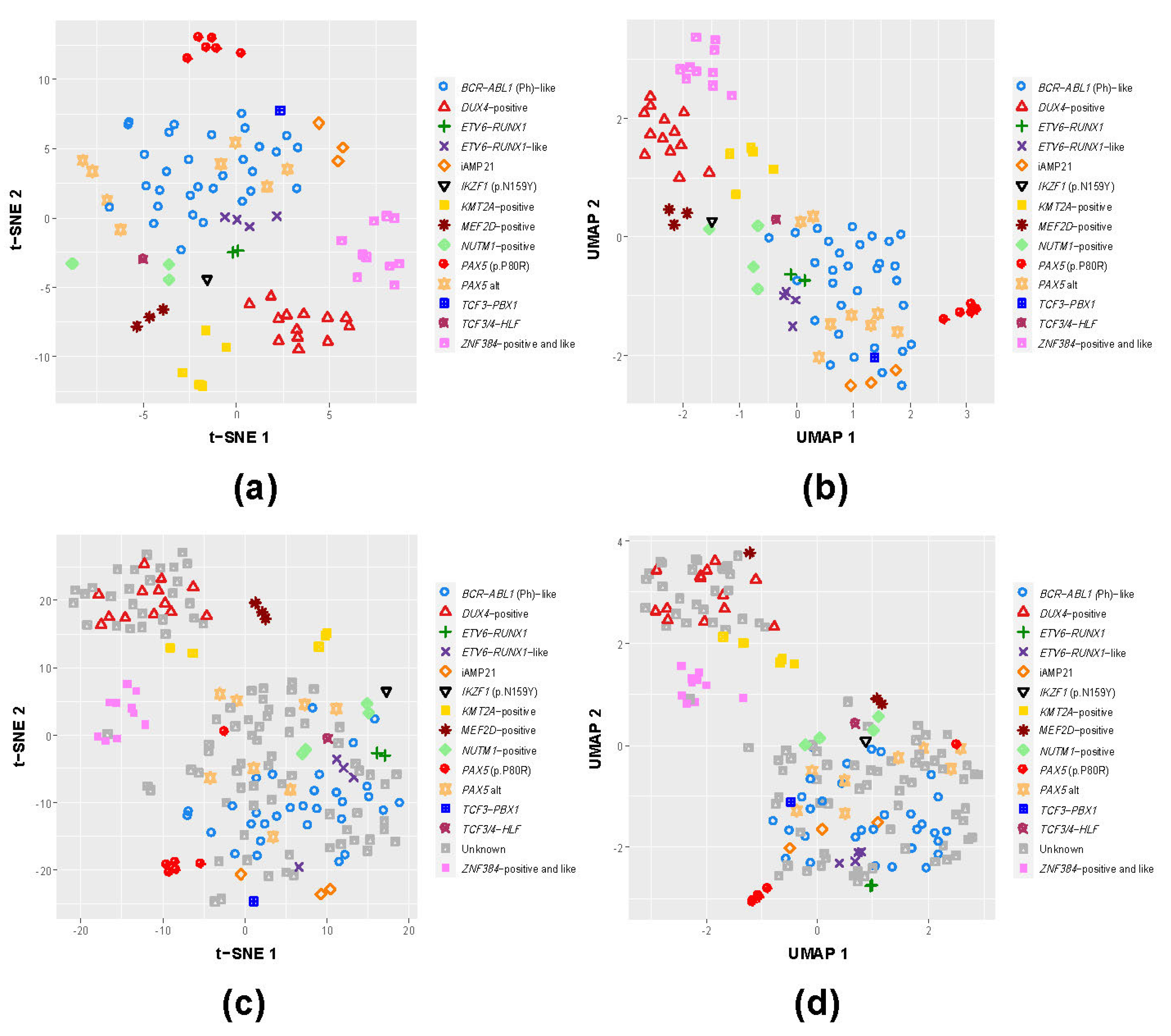
3.1. B-Other Samples Cluster with Known B-ALL Subgroups Based on Gene Expression
3.2. Splicing Profiles of the Known BCP-ALL Subgroups Provide Additional Information for Potential Improvement in Diagnostics of B-Other Samples
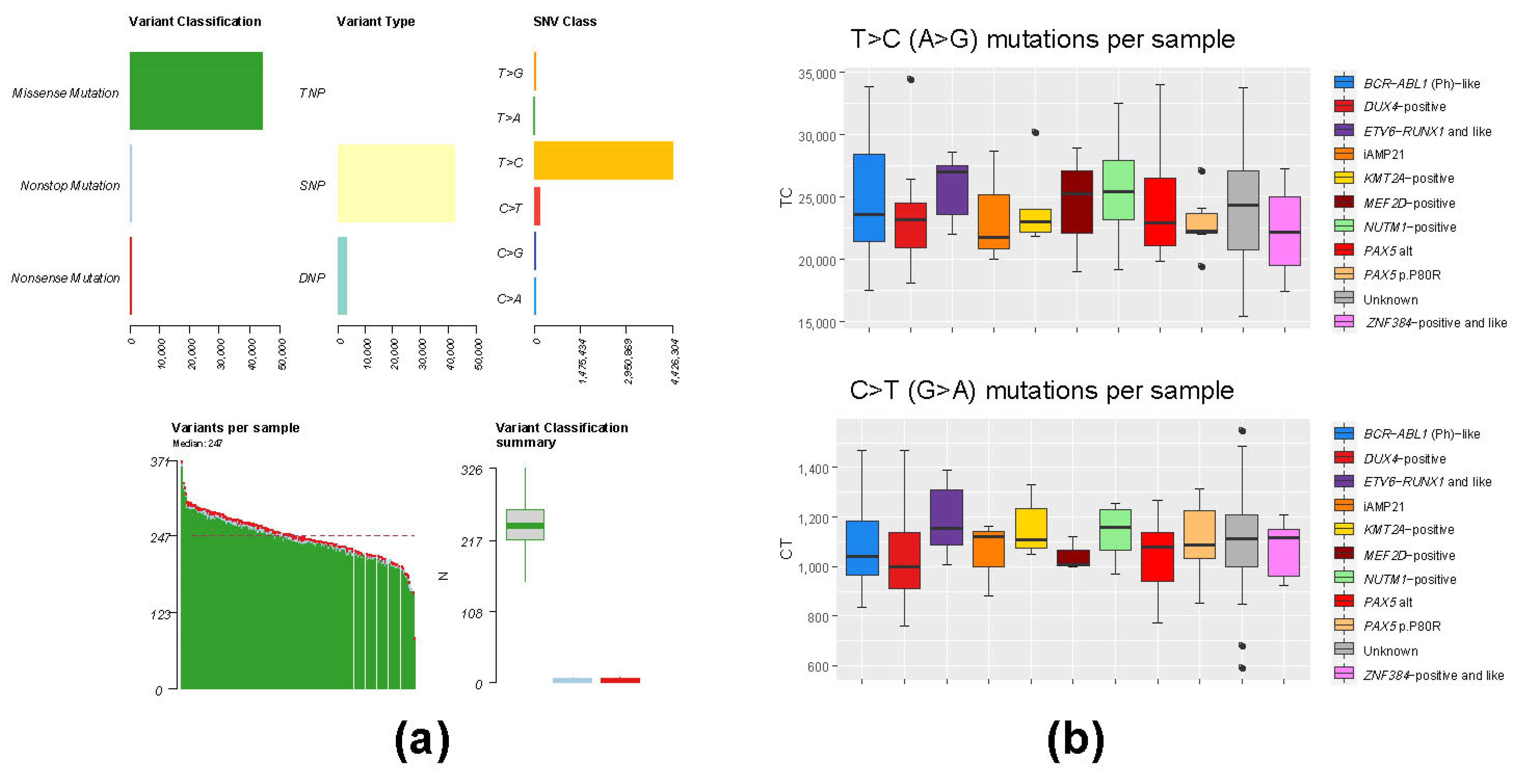
3.3. Mutation Analysis of Whole-Transcriptome BCP-ALL Data Has Limited Applicability Due to High Level of RNA Editing Events
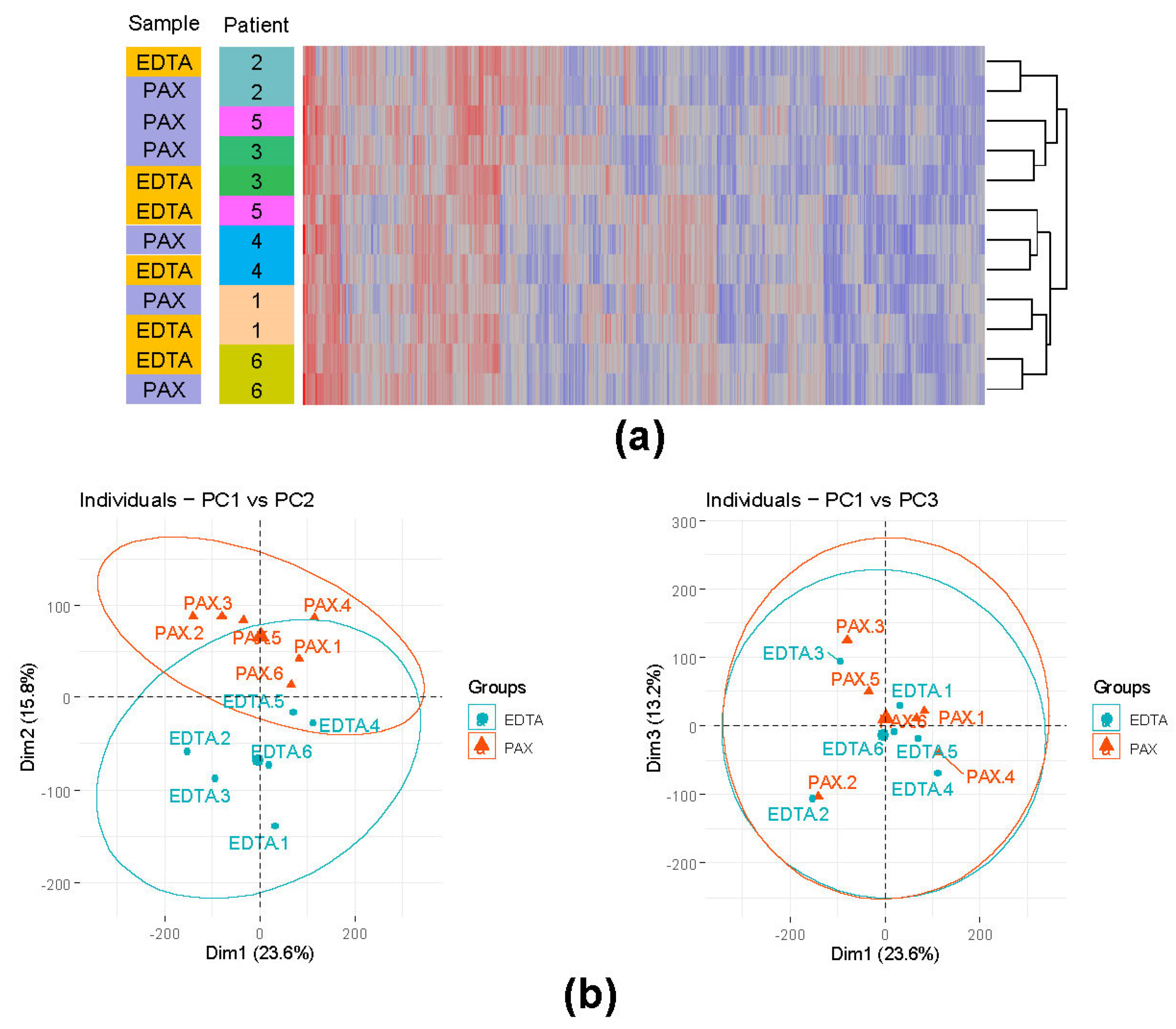
3.4. EDTA Tubes Are a Viable Alternative to PAXgene RNA Stabilizing Tubes
3.4.1. Comparison of Sequencing Output (PAXgene vs. EDTA) Showed Slightly Higher Duplication Rate in PAXgene Samples
3.4.2. Influence of the Different Storage Tubes on the Overall Gene Expression
3.4.3. No Appreciable Correlation between Expression and Decay Constants Exists in Either EDTA or PAXgene Stored Samples
3.4.4. Fusion Detection Assessment (PAXgene vs. EDTA)
3.5. Low Blast Count Samples Can Be Reliably Profiled by Whole-Transcriptome RNA-seq Given Sufficient Sequencing Depth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barr, R.D.; Ferrari, A.; Ries, L.; Whelan, J.; Archie Bleyer, W. Cancer in Adolescents and Young Adults: A Narrative Review of the Current Status and a View of the Future. JAMA Pediatr. 2016, 170, 495–501. [Google Scholar] [CrossRef]
- Katanoda, K.; Shibata, A.; Matsuda, T.; Hori, M.; Nakata, K.; Narita, Y.; Ogawa, C.; Munakata, W.; Kawai, A.; Nishimoto, H. Childhood, Adolescent and Young Adult Cancer Incidence in Japan in 2009–2011. Jpn. J. Clin. Oncol. 2017, 47, 762–771. [Google Scholar] [CrossRef] [Green Version]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef]
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; Bouzbid, S.; et al. International incidence of childhood cancer, 2001–10: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved Survival for Children and Adolescents with Acute Lymphoblastic Leukemia Between 1990 and 2005: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef]
- Schrappe, M.; Bleckmann, K.; Zimmermann, M.; Biondi, A.; Möricke, A.; Locatelli, F.; Cario, G.; Rizzari, C.; Attarbaschi, A.; Valsecchi, M.G.; et al. Reduced-Intensity Delayed Intensification in Standard-Risk Pediatric Acute Lymphoblastic Leukemia Defined by Undetectable Minimal Residual Disease: Results of an International Randomized Trial (AIEOP-BFM ALL 2000). J. Clin. Oncol. 2018, 36, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieters, R.; de Groot-Kruseman, H.; Van der Velden, V.; Fiocco, M.; Berg, H.V.D.; de Bont, E.; Egeler, R.M.; Hoogerbrugge, P.; Kaspers, G.; Van der Schoot, E.; et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J. Clin. Oncol. 2016, 34, 2591–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef]
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Liao, E.; Li, C.; Yen, C.; Yu, S. Pathogenesis of pediatric B-cell acute lymphoblastic leukemia: Molecular pathways and disease treatments (Review). Oncol. Lett. 2020, 20, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Boer, M.L.D.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion ofIKZF1and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Cario, G.; Leoni, V.; Conter, V.; Baruchel, A.; Schrappe, M.; Biondi, A. BCR-ABL1-like acute lymphoblastic leukemia in childhood and targeted therapy. Haematologica 2020, 105, 2200–2204. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Reshmi, S.C.; Harvey, R.; Chen, I.-M.; Patel, K.; Stonerock, E.; Jenkins, H.; Dai, Y.; Valentine, M.; Gu, Z.; et al. Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: A report from the Children’s Oncology Group. Blood 2018, 132, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Carpiuc, K.T.; Stephens, J.M.; Botteman, M.F.; Feng, W.; Hay, J.W. A review of the clinical and economic outcomes of imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia. Expert Opin. Pharmacother. 2007, 8, 2775–2787. [Google Scholar] [CrossRef]
- Shen, S.; Chen, X.; Cai, J.; Yu, J.; Gao, J.; Hu, S.; Zhai, X.; Liang, C.; Ju, X.; Jiang, H.; et al. Effect of Dasatinib vs Imatinib in the Treatment of Pediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 358–366. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef] [Green Version]
- Hormann, F.M.; Hoogkamer, A.Q.; Beverloo, H.B.; Boeree, A.; Dingjan, I.; Wattel, M.M.; Stam, R.W.; Escherich, G.; Pieters, R.; den Boer, M.L.; et al. NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute Lymphoblastic Leukemia As-sociated with Increased Expression of Genes on Chromosome Band 10p12.31-12.2. Haematologica 2019, 104, e455–e459. [Google Scholar] [CrossRef] [Green Version]
- Boer, J.M.; Valsecchi, M.G.; Hormann, F.M.; Antić, Z.; Zaliova, M.; Schwab, C.; Cazzaniga, G.; Arfeuille, C.; Cavé, H.; Attarbaschi, A.; et al. Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 2021, 35, 2978–2982. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirabayashi, S.; Butler, E.R.; Ohki, K.; Kiyokawa, N.; Bergmann, A.K.; Möricke, A.; Boer, J.M.; Cavé, H.; Cazzaniga, G.; Yeoh, A.E.J.; et al. Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: A retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 2021, 35, 3272–3277. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-Rearranged Subtypes in Paediatric B-Cell Pre-cursor Acute Lymphoblastic Leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Waanders, E.; Van Der Velden, V.H.J.; Van Reijmersdal, S.V.; Venkatachalam, R.; Scheijen, B.; Sonneveld, E.; Van Dongen, J.J.M.; Veerman, A.J.P.; Van Leeuwen, F.N.; et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia 2010, 24, 1258–1264. [Google Scholar] [CrossRef]
- Antić, Ž.; Yu, J.; Van Reijmersdal, S.V.; Van Dijk, A.; Dekker, L.; Segerink, W.H.; Sonneveld, E.; Fiocco, M.; Pieters, R.; Hoogerbrugge, P.M.; et al. Multiclonal complexity of pediatric acute lymphoblastic leukemia and the prognostic relevance of subclonal mutations. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.M.; Walton, M.A.; Carter, P.M. The Major Causes of Death in Children and Adolescents in the United States. N. Engl. J. Med. 2018, 379, 2468–2475. [Google Scholar] [CrossRef]
- Schlegelberger, B.; Metzke, S.; Harder, S.; Zühlke-Jenisch, R.; Zhang, Y.; Siebert, R. Classical and Molecular Cytogenetics of Tumor Cells. In Diagnostic Cytogenetics; Wegner, R.-D., Ed.; Springer: Berlin/Heidelberg, Germany, 1999; pp. 151–185. [Google Scholar]
- Schieck, M.; Lentes, J.; Thomay, K.; Hofmann, W.; Behrens, Y.L.; Hagedorn, M.; Ebersold, J.; Davenport, C.F.; Fazio, G.; Möricke, A.; et al. Implementation of RNA sequencing and array CGH in the diagnostic workflow of the AIEOP-BFM ALL 2017 trial on acute lymphoblastic leukemia. Ann. Hematol. 2020, 99, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Dworzak, M.N.; Buldini, B.; Gaipa, G.; Ratei, R.; Hrusak, O.; Luria, D.; Rosenthal, E.; Bourquin, J.-P.; Sartor, M.; Schumich, A.; et al. AIEOP-BFM Consensus Guidelines 2016 for Flow Cytometric Immunophenotyping of Pediatric Acute Lymphoblastic Leukemia. Cytom. Part B Clin. Cytom. 2017, 94, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bene, M.C.; Castoldi, G.; Knapp, W.; Ludwig, W.D.; Matutes, E.; Orfao, A.; Veer, M.B.V. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 1995, 9, 1783–1786. [Google Scholar]
- Schinnerl, D.; Mejstrikova, E.; Schumich, A.; Zaliova, M.; Fortschegger, K.; Nebral, K.; Attarbaschi, A.; Fiser, K.; Kauer, M.O.; Popitsch, N.; et al. CD371 cell surface expression: A unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica 2019, 104, e352–e355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Kubota, Y.; Kiyoi, H.; et al. Tran-scriptional Landscape of B Cell Precursor Acute Lymphoblastic Leukemia Based on an International Study of 1,223 Cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef] [Green Version]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:e-prints/1802.03426. [Google Scholar]
- Risso, D.; Schwartz, K.; Sherlock, G.; Dudoit, S. GC-Content Normalization for RNA-Seq Data. BMC Bioinform. 2011, 12, 480. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Col-laborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Merino, G.A.; Fernández, E.A. Differential splicing analysis based on isoforms expression with NBSplice. J. Biomed. Inform. 2020, 103, 103378. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Passet, M.; Boissel, N.; Sigaux, F.; Saillard, C.; Bargetzi, M.; Ba, I.; Thomas, X.; Graux, C.; Chalandon, Y.; Leguay, T.; et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 2019, 133, 280–284. [Google Scholar] [CrossRef]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van der Vorst, M.; Jongmans, M.C.J.; et al. Mutational Landscape and Patterns of Clonal Evolution in Relapsed Pediatric Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef]
- Ma, X.; Edmonson, M.; Yergeau, D.; Muzny, D.M.; Hampton, O.A.; Rusch, M.; Song, G.; Easton, J.; Harvey, R.; Wheeler, D.A.; et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat. Commun. 2015, 6, 6604. [Google Scholar] [CrossRef] [PubMed]
- Coudray, A.; Battenhouse, A.; Bucher, P.; Iyer, V.R. Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data. PeerJ 2018, 6, e5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picardi, E.; Manzari, C.; Mastropasqua, F.; Aiello, I.; D’Erchia, A.M.; Pesole, G. Profiling RNA editing in human tissues: Towards the inosinome Atlas. Sci. Rep. 2015, 5, 14941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingett, S.W.; Andrews, S. FastQ Screen: A Tool for Multi-Genome Mapping and Quality Control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 28 July 2021).
- Rainen, L.; Oelmueller, U.; Jurgensen, S.; Wyrich, R.; Ballas, C.; Schram, J.; Herdman, C.; Bankaitis-Davis, D.; Nicholls, N.; Trollinger, D.; et al. Stabilization of mRNA Expression in Whole Blood Samples. Clin. Chem. 2002, 48, 1883–1890. [Google Scholar] [CrossRef] [Green Version]
- Romero, I.G.; Pai, A.A.; Tung, J.; Gilad, Y. RNA-seq: Impact of RNA degradation on transcript quantification. BMC Biol. 2014, 12, 42. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.M.; Lonsdale, A.; Zhu, A.; Davidson, N.M.; Schmidt, B.; Hawkins, A.; Wallach, E.; Martin, M.; Mechinaud, F.M.; Khaw, S.L.; et al. The Application of RNA Sequencing for the Diagnosis and Genomic Classification of Pediatric Acute Lym-phoblastic Leukemia. Blood Adv. 2020, 4, 930–942. [Google Scholar] [CrossRef]
- Clappier, E.; Auclerc, M.-F.; Rapion, J.; Bakkus, M.; Caye, A.; Khemiri, A.; Giroux, C.; Hernandez, L.; Kabongo, E.; Savola, S.; et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 2013, 28, 70–77. [Google Scholar] [CrossRef]
- Zaliova, M.; Potuckova, E.; Hovorkova, L.; Musilova, A.; Winkowska, L.; Fiser, K.; Stuchly, J.; Mejstrikova, E.; Starkova, J.; Zuna, J.; et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 2019, 104, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Stanulla, M.; Cavé, H.; Moorman, A.V. IKZF1 Deletions in Pediatric Acute Lymphoblastic Leukemia: Still a Poor Prognostic Marker? Blood 2020, 135, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chouvarine, P.; Antić, Ž.; Lentes, J.; Schröder, C.; Alten, J.; Brüggemann, M.; Carrillo-de Santa Pau, E.; Illig, T.; Laguna, T.; Schewe, D.; et al. Transcriptional and Mutational Profiling of B-Other Acute Lymphoblastic Leukemia for Improved Diagnostics. Cancers 2021, 13, 5653. https://doi.org/10.3390/cancers13225653
Chouvarine P, Antić Ž, Lentes J, Schröder C, Alten J, Brüggemann M, Carrillo-de Santa Pau E, Illig T, Laguna T, Schewe D, et al. Transcriptional and Mutational Profiling of B-Other Acute Lymphoblastic Leukemia for Improved Diagnostics. Cancers. 2021; 13(22):5653. https://doi.org/10.3390/cancers13225653
Chicago/Turabian StyleChouvarine, Philippe, Željko Antić, Jana Lentes, Charlotte Schröder, Julia Alten, Monika Brüggemann, Enrique Carrillo-de Santa Pau, Thomas Illig, Teresa Laguna, Denis Schewe, and et al. 2021. "Transcriptional and Mutational Profiling of B-Other Acute Lymphoblastic Leukemia for Improved Diagnostics" Cancers 13, no. 22: 5653. https://doi.org/10.3390/cancers13225653
APA StyleChouvarine, P., Antić, Ž., Lentes, J., Schröder, C., Alten, J., Brüggemann, M., Carrillo-de Santa Pau, E., Illig, T., Laguna, T., Schewe, D., Stanulla, M., Tang, M., Zimmermann, M., Schrappe, M., Schlegelberger, B., Cario, G., & Bergmann, A. K. (2021). Transcriptional and Mutational Profiling of B-Other Acute Lymphoblastic Leukemia for Improved Diagnostics. Cancers, 13(22), 5653. https://doi.org/10.3390/cancers13225653