
Liquid Biopsies beyond Mutation Calling: Genomic and Epigenomic Features of Cell-Free DNA in Cancer

 , , and
, , and 
Simple Summary
Abstract
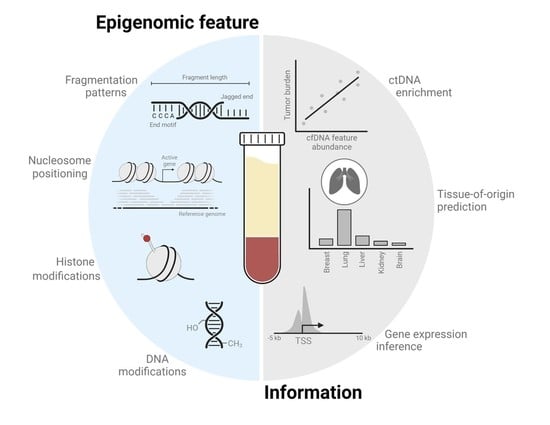
1. Introduction
2. Fragmentation Patterns of cfDNA
2.1. Shortening of cfDNA Fragments in Cancer
2.2. Cancer-Associated Alterations of cfDNA Fragment End Sequences
3. Nucleosome Positioning and Chromatin Modifications in cfDNA
3.1. Deducing Gene Expression from Nucleosome Positioning and Occupancy
3.2. Inference of Transcription Factor Binding
3.3. Histone Modification of cfDNA Nucleosomes as a Measure of Transcriptional Activity
4. Epigenomic Modifications of cfDNA
4.1. Methylation Profiling of cfDNA
4.2. 5-hydroxymethylation Profiling of cfDNA
5. Combinatorial Biomarkers for cfDNA Analysis
6. Challenges and Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Vlaminck, I.; Valantine, H.A.; Snyder, T.M.; Strehl, C.; Cohen, G.; Luikart, H.; Neff, N.F.; Okamoto, J.; Bernstein, D.; Weisshaar, D. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci. Transl. Med. 2014, 6, 241ra77. [Google Scholar] [CrossRef]
- De Vlaminck, I.; Martin, L.; Kertesz, M.; Patel, K.; Kowarsky, M.; Strehl, C.; Cohen, G.; Luikart, H.; Neff, N.F.; Okamoto, J. Noninvasive monitoring of infection and rejection after lung transplantation. Proc. Natl. Acad. Sci. USA 2015, 112, 13336–13341. [Google Scholar] [CrossRef]
- Snyder, T.M.; Khush, K.K.; Valantine, H.A.; Quake, S.R. Universal noninvasive detection of solid organ transplant rejection. Proc. Natl. Acad. Sci. USA 2011, 108, 6229–6234. [Google Scholar] [CrossRef]
- Chiu, R.W.; Chan, K.C.; Gao, Y.; Lau, V.Y.; Zheng, W.; Leung, T.Y.; Foo, C.H.; Xie, B.; Tsui, N.B.; Lun, F.M.; et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc. Natl. Acad. Sci. USA 2008, 105, 20458–20463. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Nabet, B.Y.; Rizvi, H.; Chaudhuri, A.A.; Wells, D.K.; Dunphy, M.P.S.; Chabon, J.J.; Liu, C.L.; Hui, A.B.; Arbour, K.C.; et al. Circulating Tumor DNA Analysis to Assess Risk of Progression after Long-term Response to PD-(L)1 Blockade in NSCLC. Clin. Cancer Res. 2020, 26, 2849–2858. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Van der Pol, Y.; Mouliere, F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell 2019, 36, 350–368. [Google Scholar] [CrossRef]
- Ottaviano, M.; Giuliano, M.; Tortora, M.; La Civita, E.; Liotti, A.; Longo, M.; Bruzzese, D.; Cennamo, M.; Riccio, V.; De Placido, P.; et al. A New Horizon of Liquid Biopsy in Thymic Epithelial Tumors: The Potential Utility of Circulating Cell-Free DNA. Front. Oncol. 2020, 10, 602153. [Google Scholar] [CrossRef]
- Christopoulos, P.; Dietz, S.; Angeles, A.K.; Rheinheimer, S.; Kazdal, D.; Volckmar, A.L.; Janke, F.; Endris, V.; Meister, M.; Kriegsmann, M.; et al. Earlier extracranial progression and shorter survival in ALK-rearranged lung cancer with positive liquid rebiopsies. Transl. Lung Cancer Res. 2021, 10, 2118–2131. [Google Scholar] [CrossRef]
- Dietz, S.; Christopoulos, P.; Yuan, Z.; Angeles, A.K.; Gu, L.; Volckmar, A.-L.; Ogrodnik, S.J.; Janke, F.; Dalle Fratte, C.; Zemojtel, T. Longitudinal therapy monitoring of ALK-positive lung cancer by combined copy number and targeted mutation profiling of cell-free DNA. EBioMedicine 2020, 62, 103103. [Google Scholar] [CrossRef] [PubMed]
- Angeles, A.K.; Christopoulos, P.; Yuan, Z.; Bauer, S.; Janke, F.; Ogrodnik, S.J.; Schlesner, M.; Reck, M.; Meister, M.; Schneider, M.; et al. Early identification of disease progression in ALK-rearranged lung cancer using circulating tumor DNA analysis. NPJ Precis. Oncol. in press. 2021. [Google Scholar]
- Le Guin, C.H.D.; Bornfeld, N.; Bechrakis, N.E.; Jabbarli, L.; Richly, H.; Lohmann, D.R.; Zeschnigk, M. Early detection of metastatic uveal melanoma by the analysis of tumor-specific mutations in cell-free plasma DNA. Cancer Med. 2021, 10, 5974–5982. [Google Scholar] [CrossRef]
- Kim, Y.W.; Kim, Y.H.; Song, Y.; Kim, H.S.; Sim, H.W.; Poojan, S.; Eom, B.W.; Kook, M.C.; Joo, J.; Hong, K.M. Monitoring circulating tumor DNA by analyzing personalized cancer-specific rearrangements to detect recurrence in gastric cancer. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Märtens, K.; Mägi, R.; Kolde, R.; Koltšina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, 3248. [Google Scholar] [CrossRef]
- Ponnaluri, V.C.; Ehrlich, K.C.; Zhang, G.; Lacey, M.; Johnston, D.; Pradhan, S.; Ehrlich, M. Association of 5-hydroxymethylation and 5-methylation of DNA cytosine with tissue-specific gene expression. Epigenetics 2017, 12, 123–138. [Google Scholar] [CrossRef]
- De Gobbi, M.; Anguita, E.; Hughes, J.; Sloane-Stanley, J.A.; Sharpe, J.A.; Koch, C.M.; Dunham, I.; Gibbons, R.J.; Wood, W.G.; Higgs, D.R. Tissue-specific histone modification and transcription factor binding in α globin gene expression. Blood 2007, 110, 4503–4510. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; He, X.; Bar-Joseph, Z. Predicting tissue specific transcription factor binding sites. BMC Genom. 2013, 14, 796. [Google Scholar] [CrossRef]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W. CancerLocator: Non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef]
- Sadeh, R.; Sharkia, I.; Fialkoff, G.; Rahat, A.; Gutin, J.; Chappleboim, A.; Nitzan, M.; Fox-Fisher, I.; Neiman, D.; Meler, G.; et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat. Biotechnol. 2021, 39, 586–598. [Google Scholar] [CrossRef]
- Church, T.R.; Wandell, M.; Lofton-Day, C.; Mongin, S.J.; Burger, M.; Payne, S.R.; Castanos-Velez, E.; Blumenstein, B.A.; Rosch, T.; Osborn, N.; et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 2014, 63, 317–325. [Google Scholar] [CrossRef]
- Powrózek, T.; Krawczyk, P.; Kucharczyk, T.; Milanowski, J. Septin 9 promoter region methylation in free circulating DNA—Potential role in noninvasive diagnosis of lung cancer: Preliminary report. Med Oncol. 2014, 31, 917. [Google Scholar] [CrossRef]
- Hennigan, S.T.; Trostel, S.Y.; Terrigino, N.T.; Voznesensky, O.S.; Schaefer, R.J.; Whitlock, N.C.; Wilkinson, S.; Carrabba, N.V.; Atway, R.; Shema, S.; et al. Low Abundance of Circulating Tumor DNA in Localized Prostate Cancer. JCO Precis. Oncol. 2019, 3, 1–13. [Google Scholar] [CrossRef]
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.C.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef]
- Smith, C.G.; Moser, T.; Mouliere, F.; Field-Rayner, J.; Eldridge, M.; Riediger, A.L.; Chandrananda, D.; Heider, K.; Wan, J.C.M.; Warren, A.Y.; et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med. 2020, 12, 23. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef]
- Hellwig, S.; Nix, D.A.; Gligorich, K.M.; O’Shea, J.M.; Thomas, A.; Fuertes, C.L.; Bhetariya, P.J.; Marth, G.T.; Bronner, M.P.; Underhill, H.R. Automated size selection for short cell-free DNA fragments enriches for circulating tumor DNA and improves error correction during next generation sequencing. PLoS ONE 2018, 13, e0197333. [Google Scholar] [CrossRef]
- Chan, R.W.Y.; Serpas, L.; Ni, M.; Volpi, S.; Hiraki, L.T.; Tam, L.S.; Rashidfarrokhi, A.; Wong, P.C.H.; Tam, L.H.P.; Wang, Y.; et al. Plasma DNA Profile Associated with DNASE1L3 Gene Mutations: Clinical Observations, Relationships to Nuclease Substrate Preference, and In Vivo Correction. Am. J. Hum. Genet. 2020, 107, 882–894. [Google Scholar] [CrossRef]
- Cheng, T.H.T.; Lui, K.O.; Peng, X.L.; Cheng, S.H.; Jiang, P.; Chan, K.C.A.; Chiu, R.W.K.; Lo, Y.M.D. DNase1 Does Not Appear to Play a Major Role in the Fragmentation of Plasma DNA in a Knockout Mouse Model. Clin. Chem. 2018, 64, 406–408. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef]
- Jiang, P.; Chan, C.W.M.; Chan, K.C.A.; Cheng, S.H.; Wong, J.; Wong, V.W.-S.; Wong, G.L.H.; Chan, S.L.; Mok, T.S.K.; Chan, H.L.Y.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef]
- Chan, K.C.; Zhang, J.; Hui, A.B.; Wong, N.; Lau, T.K.; Leung, T.N.; Lo, K.W.; Huang, D.W.; Lo, Y.M. Size distributions of maternal and fetal DNA in maternal plasma. Clin. Chem. 2004, 50, 88–92. [Google Scholar] [CrossRef]
- Lo, Y.M.; Chan, K.C.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.R.; et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef]
- Zheng, Y.W.; Chan, K.C.; Sun, H.; Jiang, P.; Su, X.; Chen, E.Z.; Lun, F.M.; Hung, E.C.; Lee, V.; Wong, J.; et al. Nonhematopoietically derived DNA is shorter than hematopoietically derived DNA in plasma: A transplantation model. Clin. Chem. 2012, 58, 549–558. [Google Scholar] [CrossRef]
- Jensen, T.J.; Kim, S.K.; Zhu, Z.; Chin, C.; Gebhard, C.; Lu, T.; Deciu, C.; van den Boom, D.; Ehrich, M. Whole genome bisulfite sequencing of cell-free DNA and its cellular contributors uncovers placenta hypomethylated domains. Genome Biol. 2015, 16, 78. [Google Scholar] [CrossRef]
- Lun, F.M.; Chiu, R.W.; Sun, K.; Leung, T.Y.; Jiang, P.; Chan, K.C.; Sun, H.; Lo, Y.M. Noninvasive prenatal methylomic analysis by genomewide bisulfite sequencing of maternal plasma DNA. Clin. Chem. 2013, 59, 1583–1594. [Google Scholar] [CrossRef]
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [CrossRef]
- Mouliere, F.; Piskorz, A.M.; Chandrananda, D.; Moore, E.; Morris, J.; Smith, C.G.; Goranova, T.; Heider, K.; Mair, R.; Supernat, A.; et al. Selecting short DNA fragments in plasma improves detection of circulating tumour DNA. bioRxiv 2017, 134437. [Google Scholar] [CrossRef]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Mair, R.; Chandrananda, D.; Marass, F.; Smith, C.G.; Su, J.; Morris, J.; Watts, C.; Brindle, K.M.; Rosenfeld, N. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol. Med. 2018, 10, e9323. [Google Scholar] [CrossRef]
- Burnham, P.; Kim, M.S.; Agbor-Enoh, S.; Luikart, H.; Valantine, H.A.; Khush, K.K.; De Vlaminck, I. Single-stranded DNA library preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci. Rep. 2016, 6, 27859. [Google Scholar] [CrossRef]
- Sanchez, C.; Roch, B.; Mazard, T.; Blache, P.; Al Amir Dache, Z.; Pastor, B.; Pisareva, E.; Tanos, R.; Thierry, A.R. Circulating nuclear DNA structural features, origins, and complete size profile revealed by fragmentomics. JCI Insight 2021, 6, e144561. [Google Scholar] [CrossRef]
- Sanchez, C.; Snyder, M.W.; Tanos, R.; Shendure, J.; Thierry, A.R. New insights into structural features and optimal detection of circulating tumor DNA determined by single-strand DNA analysis. NPJ Genom. Med. 2018, 3, 31. [Google Scholar] [CrossRef]
- Chan, K.C.; Jiang, P.; Sun, K.; Cheng, Y.K.; Tong, Y.K.; Cheng, S.H.; Wong, A.I.; Hudecova, I.; Leung, T.Y.; Chiu, R.W.; et al. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc. Natl. Acad. Sci. USA 2016, 113, E8159–E8168. [Google Scholar] [CrossRef]
- Jiang, P.; Sun, K.; Tong, Y.K.; Cheng, S.H.; Cheng, T.H.T.; Heung, M.M.S.; Wong, J.; Wong, V.W.S.; Chan, H.L.Y.; Chan, K.C.A.; et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E10925–E10933. [Google Scholar] [CrossRef]
- Chandrananda, D.; Thorne, N.P.; Bahlo, M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med. Genom. 2015, 8, 29. [Google Scholar] [CrossRef]
- Serpas, L.; Chan, R.W.Y.; Jiang, P.; Ni, M.; Sun, K.; Rashidfarrokhi, A.; Soni, C.; Sisirak, V.; Lee, W.-S.; Cheng, S.H.; et al. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc. Natl. Acad. Sci. USA 2019, 116, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Han, D.S.C.; Ni, M.; Chan, R.W.Y.; Chan, V.W.H.; Lui, K.O.; Chiu, R.W.K.; Lo, Y.M.D. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020, 106, 202–214. [Google Scholar] [CrossRef]
- Jiang, P.; Xie, T.; Ding, S.C.; Zhou, Z.; Cheng, S.H.; Chan, R.W.Y.; Lee, W.S.; Peng, W.; Wong, J.; Wong, V.W.S.; et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res. 2020, 30, 1144–1153. [Google Scholar] [CrossRef]
- Zhou, Z.; Cheng, S.H.; Ding, S.C.; Heung, M.M.S.; Xie, T.; Cheng, T.H.T.; Lam, W.K.J.; Peng, W.; Teoh, J.Y.C.; Chiu, P.K.F.; et al. Jagged Ends of Urinary Cell-Free DNA: Characterization and Feasibility Assessment in Bladder Cancer Detection. Clin. Chem. 2021, 67, 621–630. [Google Scholar] [CrossRef]
- Sun, K.; Jiang, P.; Wong, A.I.C.; Cheng, Y.K.Y.; Cheng, S.H.; Zhang, H.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc. Natl. Acad. Sci. USA 2018, 115, E5106–E5114. [Google Scholar] [CrossRef]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jiang, P.; Chan, K.C.; Wong, J.; Cheng, Y.K.; Liang, R.H.; Chan, W.K.; Ma, E.S.; Chan, S.L.; Cheng, S.H.; et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef]
- Napirei, M.; Ludwig, S.; Mezrhab, J.; Klöckl, T.; Mannherz, H.G. Murine serum nucleases—Contrasting effects of plasmin and heparin on the activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J. 2009, 276, 1059–1073. [Google Scholar] [CrossRef]
- Jiang, P.; Sun, K.; Peng, W.; Cheng, S.H.; Ni, M.; Yeung, P.C.; Heung, M.M.S.; Xie, T.; Shang, H.; Zhou, Z.; et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discov. 2020, 10, 664–673. [Google Scholar] [CrossRef]
- Valouev, A.; Johnson, S.M.; Boyd, S.D.; Smith, C.L.; Fire, A.Z.; Sidow, A. Determinants of nucleosome organization in primary human cells. Nature 2011, 474, 516–520. [Google Scholar] [CrossRef]
- Teif, V.B.; Vainshtein, Y.; Caudron-Herger, M.; Mallm, J.P.; Marth, C.; Hofer, T.; Rippe, K. Genome-wide nucleosome positioning during embryonic stem cell development. Nat. Struct. Mol. Biol. 2012, 19, 1185–1192. [Google Scholar] [CrossRef]
- Murtaza, M.; Caldas, C. Nucleosome mapping in plasma DNA predicts cancer gene expression. Nat. Genet. 2016, 48, 1105–1106. [Google Scholar] [CrossRef]
- Sun, K.; Jiang, P.; Cheng, S.H.; Cheng, T.H.T.; Wong, J.; Wong, V.W.S.; Ng, S.S.M.; Ma, B.B.Y.; Leung, T.Y.; Chan, S.L.; et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 2019, 29, 418–427. [Google Scholar] [CrossRef]
- Ulz, P.; Perakis, S.; Zhou, Q.; Moser, T.; Belic, J.; Lazzeri, I.; Wolfler, A.; Zebisch, A.; Gerger, A.; Pristauz, G.; et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat. Commun. 2019, 10, 4666. [Google Scholar] [CrossRef]
- Lam, W.K.J.; Gai, W.; Sun, K.; Wong, R.S.M.; Chan, R.W.Y.; Jiang, P.; Chan, N.P.H.; Hui, W.W.I.; Chan, A.W.H.; Szeto, C.C.; et al. DNA of Erythroid Origin Is Present in Human Plasma and Informs the Types of Anemia. Clin. Chem. 2017, 63, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Vad-Nielsen, J.; Meldgaard, P.; Sorensen, B.S.; Nielsen, A.L. Cell-free Chromatin Immunoprecipitation (cfChIP) from blood plasma can determine gene-expression in tumors from non-small-cell lung cancer patients. Lung Cancer 2020, 147, 244–251. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Pradhan, K.; Campbell, N.; Mazdo, J.; Vasantkumar, A.; Maqbool, S.; Bhagat, T.D.; Gupta, S.; Suzuki, M.; Yu, Y.; et al. Altered hydroxymethylation is seen at regulatory regions in pancreatic cancer and regulates oncogenic pathways. Genome Res. 2017, 27, 1830–1842. [Google Scholar] [CrossRef]
- Saghafinia, S.; Mina, M.; Riggi, N.; Hanahan, D.; Ciriello, G. Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep. 2018, 25, 1066–1080. [Google Scholar] [CrossRef]
- Li, M.; Gao, F.; Xia, Y.; Tang, Y.; Zhao, W.; Jin, C.; Luo, H.; Wang, J.; Li, Q.; Wang, Y. Filtrating colorectal cancer associated genes by integrated analyses of global DNA methylation and hydroxymethylation in cancer and normal tissue. Sci. Rep. 2016, 6, 31826. [Google Scholar] [CrossRef]
- Dor, Y.; Cedar, H. Principles of DNA methylation and their implications for biology and medicine. Lancet 2018, 392, 777–786. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.L.; Zhang, K.; Zhang, K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, H.; Huang, Y.; Xu, S.; Zhou, Y.; Zhang, W.; Li, J.; Ming, Y.; Wang, X.; Zhao, S.; et al. Genome-wide cell-free DNA methylation analyses improve accuracy of non-invasive diagnostic imaging for early-stage breast cancer. Mol. Cancer 2021, 20, 36. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Moss, J.; Zick, A.; Grinshpun, A.; Carmon, E.; Maoz, M.; Ochana, B.L.; Abraham, O.; Arieli, O.; Germansky, L.; Meir, K.; et al. Circulating breast-derived DNA allows universal detection and monitoring of localized breast cancer. Ann. Oncol. 2020, 31, 395–403. [Google Scholar] [CrossRef]
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 2020, 26, 1044–1047. [Google Scholar] [CrossRef]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Abou Alaiwi, S.; Chakravarthy, A.; Shen, S.Y.; Bakouny, Z.; Boccardo, F.; et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef]
- Togneri, F.S.; Ward, D.G.; Foster, J.M.; Devall, A.J.; Wojtowicz, P.; Alyas, S.; Vasques, F.R.; Oumie, A.; James, N.D.; Cheng, K.K.; et al. Genomic complexity of urothelial bladder cancer revealed in urinary cfDNA. Eur. J. Hum. Genet. 2016, 24, 1167–1174. [Google Scholar] [CrossRef]
- Xia, Y.; Huang, C.C.; Dittmar, R.; Du, M.; Wang, Y.; Liu, H.; Shenoy, N.; Wang, L.; Kohli, M. Copy number variations in urine cell free DNA as biomarkers in advanced prostate cancer. Oncotarget 2016, 7, 35818–35831. [Google Scholar] [CrossRef]
- Lu, T.; Li, J. Clinical applications of urinary cell-free DNA in cancer: Current insights and promising future. Am. J. Cancer Res. 2017, 7, 2318–2332. [Google Scholar] [PubMed]
- Andersson, E.; Steven, K.; Guldberg, P. Size-based enrichment of exfoliated tumor cells in urine increases the sensitivity for DNA-based detection of bladder cancer. PLoS ONE 2014, 9, e94023. [Google Scholar] [CrossRef]
- Van den Helder, R.; Wever, B.M.M.; van Trommel, N.E.; van Splunter, A.P.; Mom, C.H.; Kasius, J.C.; Bleeker, M.C.G.; Steenbergen, R.D.M. Non-invasive detection of endometrial cancer by DNA methylation analysis in urine. Clin. Epigenet. 2020, 12, 165. [Google Scholar] [CrossRef]
- Larsen, L.K.; Jakobsen, J.S.; Abdul-Al, A.; Guldberg, P. Noninvasive Detection of High Grade Prostate Cancer by DNA Methylation Analysis of Urine Cells Captured by Microfiltration. J. Urol. 2018, 200, 749–757. [Google Scholar] [CrossRef]
- Bach, S.; Paulis, I.; Sluiter, N.R.; Tibbesma, M.; Martin, I.; van de Wiel, M.A.; Tuynman, J.B.; Bahce, I.; Kazemier, G.; Steenbergen, R.D.M. Detection of colorectal cancer in urine using DNA methylation analysis. Sci. Rep. 2021, 11, 2363. [Google Scholar] [CrossRef]
- Cheng, T.H.T.; Jiang, P.; Teoh, J.Y.C.; Heung, M.M.S.; Tam, J.C.W.; Sun, X.; Lee, W.S.; Ni, M.; Chan, R.C.K.; Ng, C.F.; et al. Noninvasive Detection of Bladder Cancer by Shallow-Depth Genome-Wide Bisulfite Sequencing of Urinary Cell-Free DNA for Methylation and Copy Number Profiling. Clin. Chem. 2019, 65, 927–936. [Google Scholar] [CrossRef]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef]
- Liu, M.; Klein, E.; Hubbell, E.; Maddala, T.; Aravanis, A.; Beausang, J.; Filippova, D.; Gross, S.; Jamshidi, A.; Kurtzman, K. Plasma cell-free DNA (cfDNA) assays for early multi-cancer detection: The circulating cell-free genome atlas (CCGA) study. Ann. Oncol. 2018, 29, viii14. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Klein, E.A.; Seiden, M.; Hubbell, E.; Venn, O.; Jamshidi, A.; Zhang, N.; Beausang, J.F.; Gross, S.; Kurtzman, K.N. Simultaneous multi-cancer detection and tissue of origin (TOO) localization using targeted bisulfite sequencing of plasma cell-free DNA (cfDNA). Ann. Oncol. 2019, 30, v912. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Nadauld, L.D.; McDonnell, C.H.; Beer, T.M.; Liu, M.C.; Klein, E.A.; Hudnut, A.; Whittington, R.A.; Taylor, B.; Oxnard, G.R.; Lipson, J.; et al. The PATHFINDER Study: Assessment of the Implementation of an Investigational Multi-Cancer Early Detection Test Into Clinical Practice. Cancers 2021, 13, 3501. [Google Scholar] [CrossRef]
- U.S. National Library of the Medicine. The SUMMIT Study: A Cancer Screening Study. Available online: https://clinicaltrials.gov/ct2/show/NCT03934866 (accessed on 20 August 2021).
- U.S. National Library of the Medicine. The STRIVE Study: Development of a Blood Test for Early Detection of Multiple Cancer Types. Available online: https://clinicaltrials.gov/ct2/show/NCT03085888 (accessed on 20 August 2021).
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, X.; Lu, J.; Liang, H.; Dai, Q.; Xu, G.L.; Luo, C.; Jiang, H.; He, C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012, 8, 328–330. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Brazauskas, P.; Kriaucionis, S. DNA modifications: Another stable base in DNA. Nat. Chem. 2014, 6, 1031–1033. [Google Scholar] [CrossRef] [PubMed]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Williams, M.; Murrell, A.; Balasubramanian, S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014, 6, 1049–1055. [Google Scholar] [CrossRef]
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lu, Y.; Jelinek, J.; Liang, S.; Estecio, M.R.; Barton, M.C.; Issa, J.P. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014, 42, 6956–6971. [Google Scholar] [CrossRef]
- Jeong, M.; Sun, D.; Luo, M.; Huang, Y.; Challen, G.A.; Rodriguez, B.; Zhang, X.; Chavez, L.; Wang, H.; Hannah, R.; et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014, 46, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Manzo, M.; Wirz, J.; Ambrosi, C.; Villasenor, R.; Roschitzki, B.; Baubec, T. Isoform-specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands. EMBO J. 2017, 36, 3421–3434. [Google Scholar] [CrossRef]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef]
- Thomson, J.P.; Lempiainen, H.; Hackett, J.A.; Nestor, C.E.; Muller, A.; Bolognani, F.; Oakeley, E.J.; Schubeler, D.; Terranova, R.; Reinhardt, D.; et al. Non-genotoxic carcinogen exposure induces defined changes in the 5-hydroxymethylome. Genome Biol. 2012, 13, R93. [Google Scholar] [CrossRef]
- Li, W.; Liu, M. Distribution of 5-Hydroxymethylcytosine in Different Human Tissues. J. Nucleic Acids 2011, 2011, 870726. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [PubMed]
- Song, C.X.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.H.; Zhang, W.; Jian, X.; et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. [Google Scholar] [CrossRef]
- Bergamaschi, A.; Ning, Y.; Ku, C.-J.; Ellison, C.; Collin, F.; Guler, G.; Phillips, T.; McCarthy, E.; Wang, W.; Antoine, M.; et al. Pilot study demonstrating changes in DNA hydroxymethylation enable detection of multiple cancers in plasma cell-free DNA. medRxiv 2020. [Google Scholar] [CrossRef]
- Cai, J.; Chen, L.; Zhang, Z.; Zhang, X.; Lu, X.; Liu, W.; Shi, G.; Ge, Y.; Gao, P.; Yang, Y.; et al. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut 2019, 68, 2195–2205. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, J.; He, Y.; Xia, L.; Dong, X.; Chen, G.; Zhou, Y.; Hu, X.; Zhong, S.; Wang, Y.; et al. Liquid biopsy by combining 5-hydroxymethylcytosine signatures of plasma cell-free DNA and protein biomarkers for diagnosis and prognosis of hepatocellular carcinoma. ESMO Open 2021, 6, 100021. [Google Scholar] [CrossRef]
- Cao, F.; Wei, A.; Hu, X.; He, Y.; Zhang, J.; Xia, L.; Tu, K.; Yuan, J.; Guo, Z.; Liu, H.; et al. Integrated epigenetic biomarkers in circulating cell-free DNA as a robust classifier for pancreatic cancer. Clin. Epigenet. 2020, 12, 112. [Google Scholar] [CrossRef]
- Chiu, B.C.; Zhang, Z.; You, Q.; Zeng, C.; Stepniak, E.; Bracci, P.M.; Yu, K.; Venkataraman, G.; Smith, S.M.; He, C.; et al. Prognostic implications of 5-hydroxymethylcytosines from circulating cell-free DNA in diffuse large B-cell lymphoma. Blood Adv. 2019, 3, 2790–2799. [Google Scholar] [CrossRef]
- Guler, G.D.; Ning, Y.; Ku, C.J.; Phillips, T.; McCarthy, E.; Ellison, C.K.; Bergamaschi, A.; Collin, F.; Lloyd, P.; Scott, A.; et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat. Commun. 2020, 11, 5270. [Google Scholar] [CrossRef]
- Song, C.X.; Yin, S.; Ma, L.; Wheeler, A.; Chen, Y.; Zhang, Y.; Liu, B.; Xiong, J.; Zhang, W.; Hu, J.; et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 2017, 27, 1231–1242. [Google Scholar] [CrossRef]
- Tian, X.; Sun, B.; Chen, C.; Gao, C.; Zhang, J.; Lu, X.; Wang, L.; Li, X.; Xing, Y.; Liu, R.; et al. Circulating tumor DNA 5-hydroxymethylcytosine as a novel diagnostic biomarker for esophageal cancer. Cell Res. 2018, 28, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, X.; Gao, C.; Xing, Y.; Qi, Z.; Liu, R.; Wang, Y.; Zhang, X.; Yang, Y.G.; Li, X.; et al. 5-Hydroxymethylome in Circulating Cell-free DNA as A Potential Biomarker for Non-small-cell Lung Cancer. Genom. Proteom. Bioinform. 2018, 16, 187–199. [Google Scholar] [CrossRef]
- Pedersen, S.K.; Baker, R.T.; McEvoy, A.; Murray, D.H.; Thomas, M.; Molloy, P.L.; Mitchell, S.; Lockett, T.; Young, G.P.; LaPointe, L.C. A two-gene blood test for methylated DNA sensitive for colorectal cancer. PLoS ONE 2015, 10, e0125041. [Google Scholar] [CrossRef]
- Peneder, P.; Stutz, A.M.; Surdez, D.; Krumbholz, M.; Semper, S.; Chicard, M.; Sheffield, N.C.; Pierron, G.; Lapouble, E.; Totzl, M.; et al. Multimodal analysis of cell-free DNA whole-genome sequencing for pediatric cancers with low mutational burden. Nat. Commun. 2021, 12, 3230. [Google Scholar] [CrossRef]
- De Rubis, G.; Rajeev Krishnan, S.; Bebawy, M. Liquid Biopsies in Cancer Diagnosis, Monitoring, and Prognosis. Trends Pharmacol. Sci. 2019, 40, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.C.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; McLaughlin, B.; Jendrisak, A.; Wang, Y.; Lee, J.; Greene, S.; Krupa, R.; Lu, D.; et al. Phenotypic Heterogeneity of Circulating Tumor Cells Informs Clinical Decisions between AR Signaling Inhibitors and Taxanes in Metastatic Prostate Cancer. Cancer Res. 2017, 77, 5687–5698. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Azizi, M.; Eslami, S.Z.; Cortes-Hernandez, L.E.; Heidarifard, M.; Nouri, M.; Alix-Panabieres, C. The Role of Circulating Tumor Cells in the Metastatic Cascade: Biology, Technical Challenges, and Clinical Relevance. Cancers 2020, 12, 867. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.T.; Cui, X.; Chen, Q.; Li, Y.F.; Cui, Y.H.; Wang, Y.; Jiang, J. Circulating tumor cell status monitors the treatment responses in breast cancer patients: A meta-analysis. Sci. Rep. 2017, 7, 43464. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; McCormack, R.; Kheoh, T.; Molina, A.; Smith, M.R.; Dreicer, R.; Saad, F.; de Wit, R.; Aftab, D.T.; Hirmand, M.; et al. Circulating Tumor Cell Number as a Response Measure of Prolonged Survival for Metastatic Castration-Resistant Prostate Cancer: A Comparison With Prostate-Specific Antigen Across Five Randomized Phase III Clinical Trials. J. Clin. Oncol. 2018, 36, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Alva, A.; Friedlander, T.; Clark, M.; Huebner, T.; Daignault, S.; Hussain, M.; Lee, C.; Hafez, K.; Hollenbeck, B.; Weizer, A.; et al. Circulating Tumor Cells as Potential Biomarkers in Bladder Cancer. J. Urol. 2015, 194, 790–798. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| cfDNA Fragment Feature | Method | Analyte | Control Cohort | Cancer Entity Tested | Result | Reference |
|---|---|---|---|---|---|---|
| Fragment length shortening | WGS | Plasma | Healthy controls (n = 32), HBV (n = 67), liver cirrhosis (n = 36) | HCC (n = 90) | Short cfDNA fragments preferentially carry tumor-associated CNVs | [42] |
| Fragment length shortening | sWGS | CSF | n/a | Glioma (n = 13) | Detectable CNVs in CSF correlates with higher abundance of short (<145 bp) cfDNA fragments | [51] |
| Fragment length shortening | tNGS; ddPCR | Plasma | n/a | Melanoma (n = 8), CRC (n = 3), and pancreatic (n = 2) cancer | 2-fold enrichment of SNVs in short (<142 bp) cfDNA fragments | [38] |
| Fragment length shortening | sWGS; tNGS | Plasma | n/a | High-grade serous ovarian cancer (n = 13) | Up to 11-fold enrichment of the mutated cfDNA fraction in short fragments (90 to 150 bp) | [49] |
| Multiple fragment size features | sWGS; tNGS | Plasma | Healthy controls (n = 144) | Multiple cancerentities (n = 200) | Enhanced CNV/SNV detection by short fragments; fragmentation feature-based cancer classification (AUC = 0.99) | [41] |
| Position-specific fragment length variations | WGS | Plasma | Healthy controls (n = 245) | Multiple cancer entities (n = 236) | Cancer detection based on genome-wide cfDNA fragmentation profiles (AUC = 0.94) | [37] |
| Preferred end coordinates | WGS | Plasma | Healthy controls (n = 32), HBV (n = 67), liver cirrhosis (n = 36) | HCC (n = 90) | Cancer classification based on fragments at tumor-associated preferred end coordinates (AUC = 0.88) | [56] |
| Fragment end motifs | WGS | Plasma | Healthy controls (n = 38), HBV (n = 17) | HCC (n = 34) | Increased fragment end motif diversity in patients compared with controls | [67] |
| Jagged ends | WGBS | Plasma | Healthy controls (n = 8), HBV (n = 17) | HCC (n = 34) | Cancer classification based on the increased jaggedness in cancer patients | [60] |
| Jagged ends | WGBS | Urine | Healthy controls (n = 39) | Bladder cancer (n = 43) | Cancer classification based on the decreased jaggedness in cancer patients | [61] |
| Cf Nucleosome Feature | Method | Control Cohort Used for Method Establishment | Cancer Entity Tested | Result | Reference |
|---|---|---|---|---|---|
| Nucleosome positioning | Evaluation of windowed protection score (WPS) using deep WGS | Pooled (n = 1) and individual cfDNA (n = 2) from healthy controls | Small and squamous cell lung cancer, colorectal adenocarcinoma, hepatocellular carcinoma, ductal carcinoma in situ breast cancer | Matched 3 out of 5 cancer test samples to a reference cell-of-origin model | [8] |
| Nucleosome depleted regions | Analysis of promoter read depth using deep WGS | cfDNA from healthy controls (n = 104) | Colon (n = 128), prostate (n = 139), breast (n = 125), lung (n = 31) cancers | Identified expressed cancer driver genes from cfDNA in regions with copy number gains | [63] |
| Nucleosome phasing | Analysis of differential phasing of upstream and downstream cfDNA fragments using WGS | Pooled cfDNA (n = 32) from healthy controls | Hepatocellular carcinoma (n = 90), colorectal (n = 11), and lung (n = 9) cancers | Positive correlation of nucleosome phasing between cell-of-origin and patient cfDNA | [71] |
| Nucleosome footprinting | Assessment of transcription factor accessibility score using WGS | cfDNA from healthy controls (n = 24) | Prostate (n = 8), breast (n = 2), colon (n = 1) cancers | Identified cell lineage reprogramming in prostate cancer; identified increased accessibility by tumor entity specific TFs in breast and colon cancer samples | [72] |
| Histone modifications | cfDNA ChIP-seq | cfDNA from healthy controls (n = 61) | Metastatic colorectal cancer (n = 56) | Colorectal cancer classifier was established based on histone modification occupancy-inferred expression; longitudinal monitoring reflected the clinical status of patients | [29] |
| Histone modifications | cfDNA ChIP-qPCR | n/a | Non-small cell lung cancer (n = 14) | Correlation of H3K36me3 occupancy and gene expression in lung cancer associated genes | [74] |
| cfDNA Modification | Method | Analyte | Control Cohort | Cancer Entity Tested | Result | Reference |
|---|---|---|---|---|---|---|
| 5mC | Illumina 450k methylation array | Plasma | Healthy control cfDNA (n = 105) combined to 8 pools | Colon (n = 4), lung (n = 4), breast (n = 3) cancer, and CUP (n = 4) | Tissue-of-origin deconvolution from cfDNA agrees with clinical findings | [64] |
| 5mC | Affinity-based profiling (cfMeDIP) | Plasma | Healthy controls (n = 62) | Multiple cancer entities (n = 189) | Robust detection and classification across various cancer types (AUC = 0.91–0.98 depending on entity and stage) | [87] |
| 5mC | Affinity-based profiling (cfMeDIP) | Plasma | Various extracranial tumors and healthy controls (n = 387) | Multiple intracranial tumor types (n = 220) | Accurate detection (AUC = 0.99) and discrimination of common intracranial tumor types (AUC = 0.71–0.95 depending on the type of brain tumor) | [85] |
| 5mC | Affinity-based profiling (cfMeDIP) | Plasma; urine | Healthy controls (n = 133 plasma samples; n = 15 urine samples) | RCC (n = 69 plasma samples; n = 30 urine samples), UBC (n = 15) | Detection of RCC patients (plasma AUC = 0.99; urine AUC = 0.86) differentiation between RCC and UBC (plasma AUC = 0.98) | [86] |
| 5mC | Targeted bisulfite sequencing | Plasma | Healthy controls (n = 64) | Localized (n = 30) and metastatic (n = 17) breast cancer patients | Cancer detection and therapy surveillance based on a breast-unique 3-marker methylation panel | [84] |
| 5mC | Targeted bisulfite sequencing (PanSeer) | Plasma | Asymptotic individuals (n = 414) | Cancer patients (n = 223) and asymptotic individuals that were later diagnosed with cancer (n = 191) | Cancer detection (up to four years prior to diagnosis) across five cancer entities using a methylation panel covering 447 genomic loci | [88] |
| 5mC | Targeted bisulfite sequencing | Plasma | Healthy controls (n = 4207) | >50 cancer entities (n = 2482) | Cancer detection in >50 entities with a specificity of 99.3% and accurate tissue-of-origin prediction in 93% of cases | [83] |
| 5hmC | Affinity-based profiling (hMeSEAL) | Plasma | Healthy controls (n = 8), HBV (n = 7) | Lung (n = 15), pancreatic (n = 7), gastric (n = 5), breast cancer (n = 4), HCC (n = 10), CRC (n = 4), and glioblastoma (n = 4) | Detection of disease/stage specific 5hmC changes capable of differentiating lung, liver, and pancreatic cancers | [130] |
| 5hmC | Affinity-based profiling (hMeSEAL) | Plasma | Healthy controls (n = 243) | Pancreatic cancer (n = 64) | Classification of pancreatic cancer patients based on 5hmC features (AUC = 0.88) | [129] |
| 5hmC | Affinity-based profiling (hMeSEAL) | Plasma | Healthy controls (n = 177) | Esophageal cancer (n = 150) | 5hmC signature-based detection of esophageal cancer (AUC = 0.96) | [131] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeles, A.K.; Janke, F.; Bauer, S.; Christopoulos, P.; Riediger, A.L.; Sültmann, H. Liquid Biopsies beyond Mutation Calling: Genomic and Epigenomic Features of Cell-Free DNA in Cancer. Cancers 2021, 13, 5615. https://doi.org/10.3390/cancers13225615
Angeles AK, Janke F, Bauer S, Christopoulos P, Riediger AL, Sültmann H. Liquid Biopsies beyond Mutation Calling: Genomic and Epigenomic Features of Cell-Free DNA in Cancer. Cancers. 2021; 13(22):5615. https://doi.org/10.3390/cancers13225615
Chicago/Turabian StyleAngeles, Arlou Kristina, Florian Janke, Simone Bauer, Petros Christopoulos, Anja Lisa Riediger, and Holger Sültmann. 2021. "Liquid Biopsies beyond Mutation Calling: Genomic and Epigenomic Features of Cell-Free DNA in Cancer" Cancers 13, no. 22: 5615. https://doi.org/10.3390/cancers13225615
APA StyleAngeles, A. K., Janke, F., Bauer, S., Christopoulos, P., Riediger, A. L., & Sültmann, H. (2021). Liquid Biopsies beyond Mutation Calling: Genomic and Epigenomic Features of Cell-Free DNA in Cancer. Cancers, 13(22), 5615. https://doi.org/10.3390/cancers13225615