
The Role of HGF/MET Signaling in Metastatic Uveal Melanoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Structure and Biological Function of Hepatocyte Growth Factor
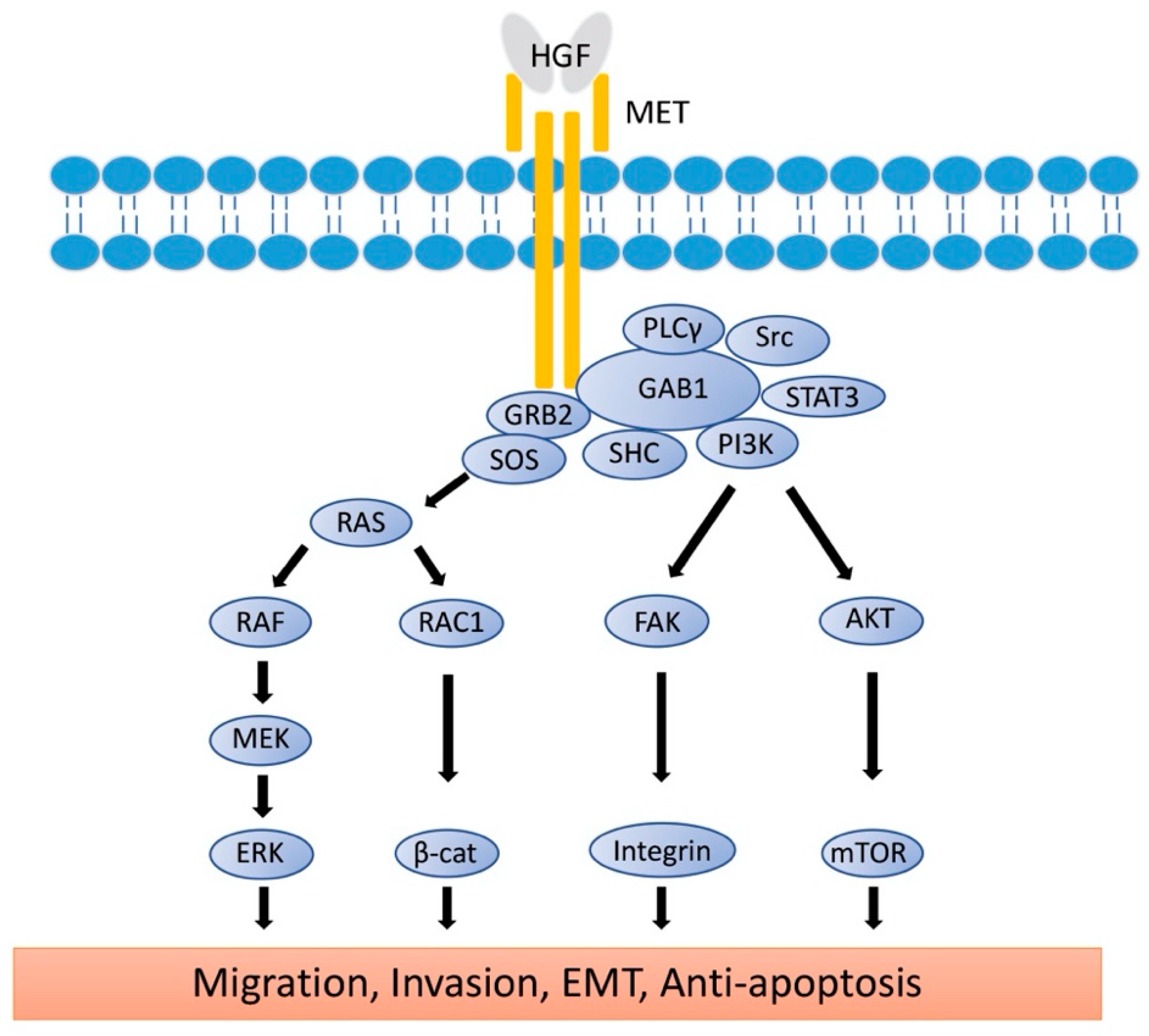
3. Structure and Biological Function of MET
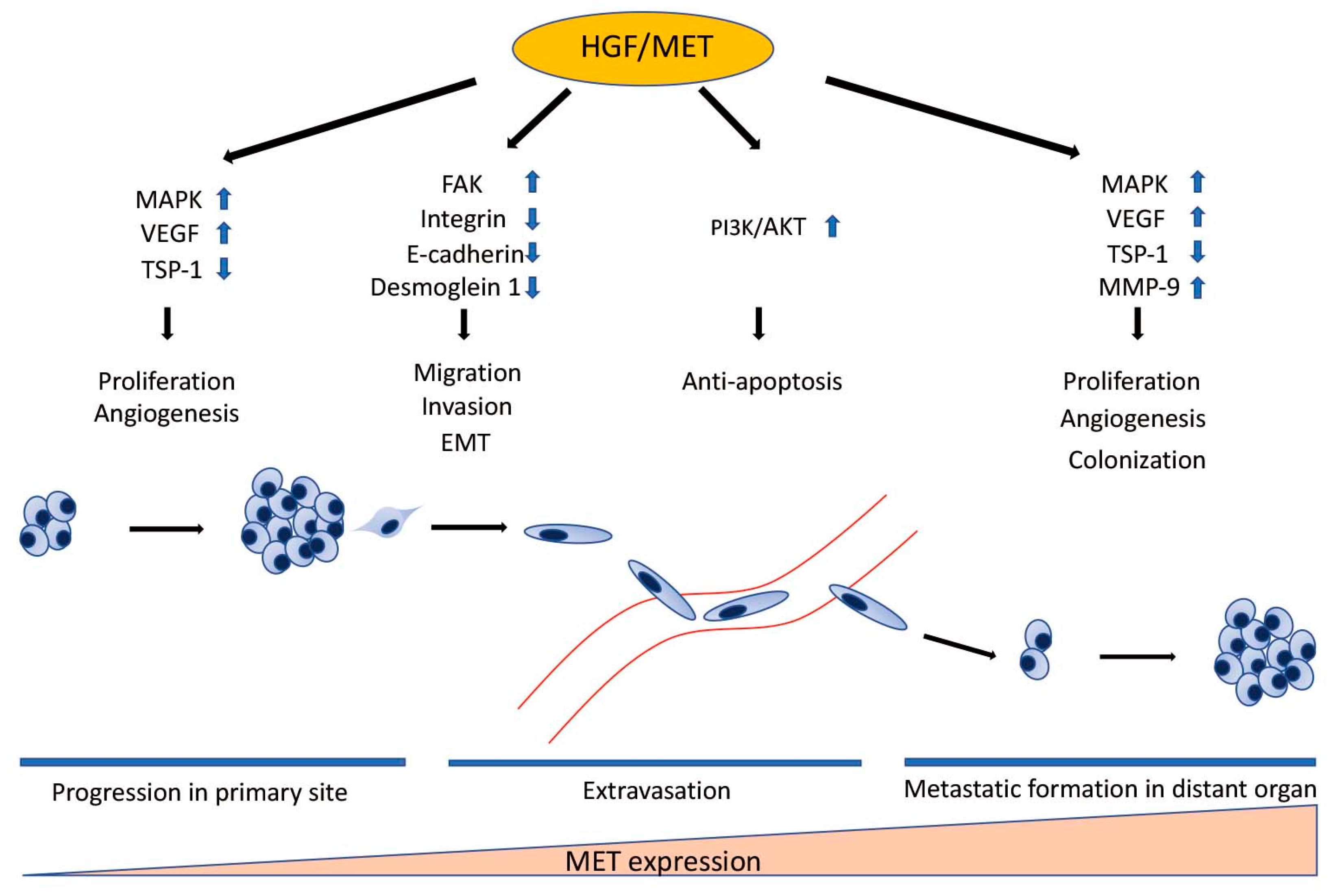
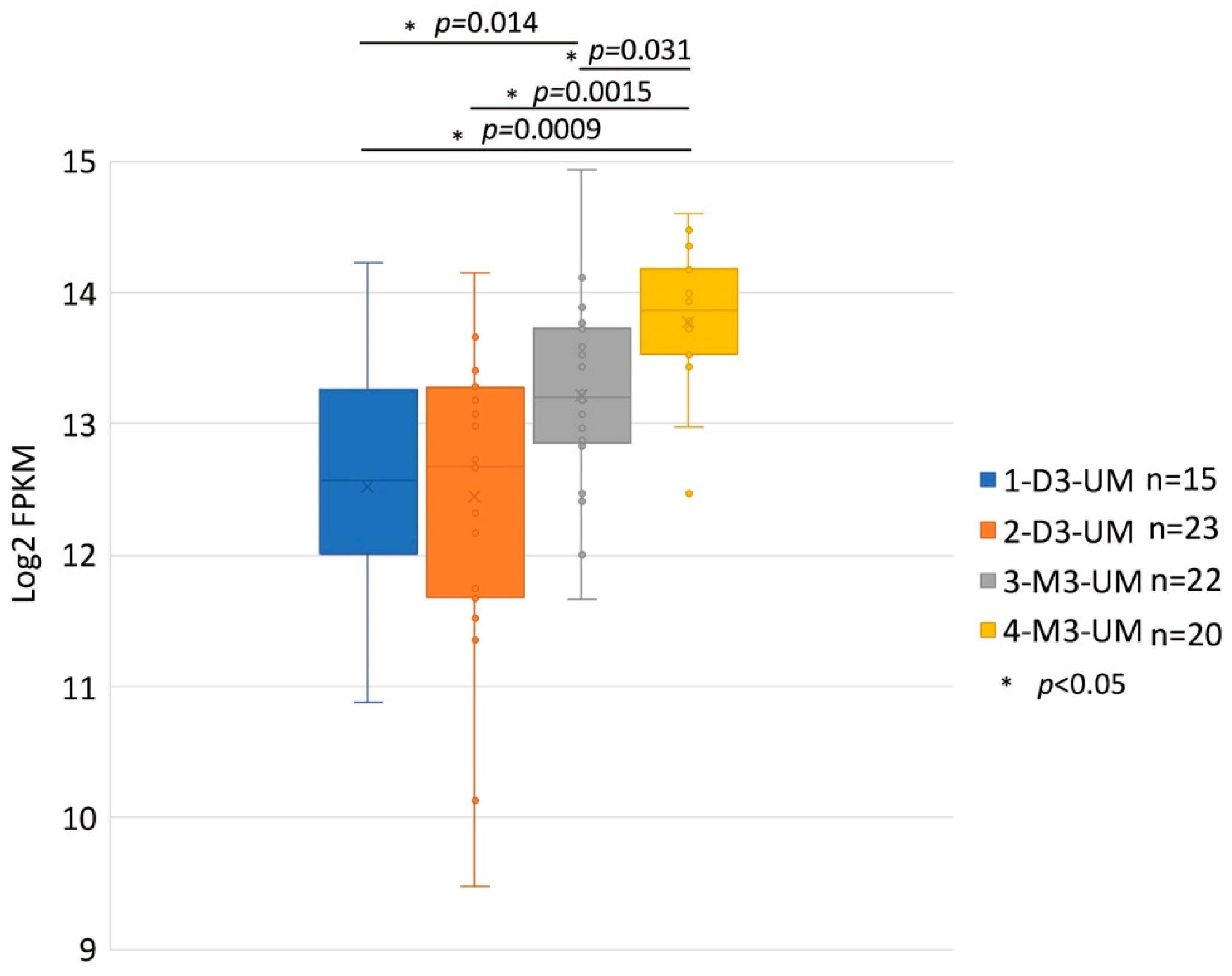
4. The Role of HGF/MET Signaling in Metastatic Uveal Melanoma
4.1. The Role of HGF/MET Signaling in Cell Migration and Invasion
4.2. The Role of HGF/MET Signaling in Epithelial-Mesenchymal Transition
4.3. The Role of HGF/MET Signaling in Survival in the Bloodstream
4.4. The Role of HGF/MET Signaling in Angiogenesis
4.5. The Role of HGF/MET Signaling in Production of Metalloproteinase
5. Targeting HGF/MET Signaling in Metastatic Uveal Melanoma
6. The Role of HGF/MET Signaling in Therapeutic Resistance
6.1. Resistance to Chemotherapy
6.2. Resistance to Targeted Therapy
6.3. Resistance to Radiation Therapy
6.4. Resistance to Immunotherapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krantz, B.A.; Dave, N.; Komatsubara, K.M.; Marr, B.P.; Carvajal, R.D. Uveal melanoma: Epidemiology, etiology, and treatment of primary disease. Clin. Opthalmol. 2017, 11, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Sussman, T.A.; Fuchain, P.; Singh, A. Clinical Trial in Metastatic Uveal Melanoma: Current Status. Ocul. Oncol. Pathol. 2020, 6, 381–387. [Google Scholar] [CrossRef]
- Aronow, M.E.; Topham, A.K.; Singh, A.D. Uveal Melanoma: 5-year Update on Incidence, Treatment, and Survival (SEER 1973-2013). Ocul. Oncol. Pathol. 2018, 4, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of Metastatic Disease After Enrollment in the COMS Trials for Treatment of Choroidal Melanoma. Arch. Opthalmol. 2005, 123, 1639–1643. [Google Scholar]
- Gonsalves, C.F.; Eschelman, D.J.; Adamo, R.D.; Anne, P.R.; Orloff, M.M.; Terai, M.; Hage, A.N.; Yi, M.; Chevoneva, I.; Sato, T. A Prospective Phase II Trial of Radioembolization for Treatment of Uveal Melanoma Hepatic Metastasis. Radiology 2019, 293, 223–231. [Google Scholar] [CrossRef]
- Bustamante, P.; Piquet, L.; Landreville, S.; Burnier, J.V. Uveal melanoma pathobiology: Metastasis to the liver. Semin. Cancer Biol. 2021, 71, 65–85. [Google Scholar] [CrossRef]
- Seedor, R.S.; Eschelman, D.J.; Gonsalves, C.F.; Adamo, R.D.; Orloff, M.; Amjad, A.; Sharpe-Mills, E.; Chervoneva, I.; Shields, C.L.; Shields, J.A.; et al. An Outcome Assessment of a Single Institution’s Longitudinal Experience with Uveal Melanoma Patients with Liver Metastasis. Cancers 2020, 12, 117. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue nevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, K.N.; Jager, M.J.; Klein, A.; Kilic, E. Uveal melanoma: Towards a molecular understanding. Prog. Retin. Eye Res. 2020, 75, 100800. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLBC4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [Green Version]
- Shain, A.H.; Bagger, M.M.; Yu, R.; Chang, D.; Liu, S.; Vemula, S.; Weier, J.F.; Wadt, K.; Heegaard, S.; Bastian, B.C.; et al. The genetic evolution of metastatic uveal melanoma. Nat. Genet. 2019, 51, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanoma. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220. [Google Scholar] [CrossRef] [Green Version]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; Bodegom, A.; Paridaens, D.; Kilic, E.; Klein, A. Uveal Melanomas with SF3B1 Mutations. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Szalai, E.; Jiang, Y.; Poppelen, N.M.; Jager, M.J.; Klein, A.; Kilic, E.; Grossniklaus, H.E. Association of uveal melanoma metastatic rate with stochastic mutation rate and type of mutation. JAMA Opthalmol. 2018, 136, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Poppelen, N.M.; Drabarek, W.; Smit, K.N.; Vaarwater, J.; Brands, T.; Paridaens, D.; Kilic, E.; Klein, A. SRSF2 mutation in uveal melanoma: A preference for in-frame deletions? Cancers 2019, 11, 1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Mizuno, S. The discovery of Hepatocyte Growth Factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, E.; Birchemeier, W.; Birchemeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Miranda, O.; Farooqui, M.; Siegfried, J.M. Status of Agents Targeting the HGF/c-Met Axis in Lung Cancer. Cancers 2018, 10, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minuti, G.; Landi, L. MET deregulation in breast cancer. Ann. Transl. Med. 2015, 3, 181. [Google Scholar] [PubMed]
- Shao, Z.; Pan, H.; Tu, S.; Zhang, J.; Yan, S.; Shao, A. HGF/c-Met Axis: The Advanced Development in Digestive System Cancer. Front. Cell. Dev. Biol. 2020, 8, 801. [Google Scholar] [CrossRef] [PubMed]
- Gardner, F.P.; Serie, D.J.; Salomao, D.R.; Wu, K.J.; Markovic, S.N.; Pulido, J.S.; Joseph, R.W. c-MET expression in primary and liver metastases in uveal melanoma. Melanoma Res. 2014, 24, 617–620. [Google Scholar] [CrossRef]
- Barisione, G.; Fabbi, M.; Gino, A.; Queirolo, P.; Orgiano, L.; Spano, L.; Picasso, V.; Pfeffer, U.; Mosci, C.; Jager, M.J. Potential role of soluble c-Met as a new candidate biomarker of metastatic uveal melanoma. JAMA Opthalmol. 2015, 133, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Surriga, O.; Rajasekhar, V.K.; Ambrosini, G.; Dogan, Y.; Huang, R.; Schwartz, G.K. Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol. Cancer Ther. 2013, 12, 2817–2826. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Jing, Y.; Kou, X.; Ye, F.; Gao, L.; Fan, Q.; Yang, Y.; Zhao, Q.; Li, R.; Wu, M.; et al. Hepatic stellate cells secreted hepatocyte growth factor contributes to the chemoresistance of hepatocellular carcinoma. PLoS ONE 2013, 8, e73312. [Google Scholar] [CrossRef]
- Czyz, M. HGF/c-MET signaling in melanocytes and melanoma. Int. J. Mol. Sci. 2018, 19, 3844. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, E.; Sandin, S.; Petoukhov, M.V.; Finch, J.; Youles, M.E.; Ofverstedt, L.G.; Miguel, R.N.; Blundell, T.L.; Vande Woude, G.F.; Skoglund, U.; et al. Structural basis of hepatocyte growth factor/scatter factor and MET signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4046–4051. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J. Gastroenterol. Hepatol. 2011, 26, 188–202. [Google Scholar] [CrossRef] [Green Version]
- Owen, K.A.; Qiu, D.; Alves, J.; Schumacher, A.M.; Kilpatrick, L.M.; Li, J.; Harris, J.L.; Ellis, V. Pericellular activation of hepatocyte growth factor by the transmembrane serine proteases matriptase and hepsin, but not by the membrane-associated protease uPA. Biochem. J. 2010, 426, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Kirchhofer, D.; Yao, X.; Peek, M.; Eigenbrot, C.; Lipari, M.T.; Billeci, K.L.; Maun, H.R.; Moran, P.; Santell, L.; Wiesmann, C.; et al. Structural and functional basis of the serine protease-like hepatocyte growth factor β-chain in Met binding and signaling. J. Bio. Chem. 2004, 279, 39915–39924. [Google Scholar] [CrossRef] [Green Version]
- Jakubczak, J.L.; LaRochelle, W.J.; Merlino, G. NK1, a natural splice variant of hepatocyte growth factor/scatter factor, is a partial agonist in vivo. Mol. Cell Biol. 1998, 18, 1275–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, H.H. Structural basis of MET receptor dimerization by the bacterial invasion protein InlB and the HGF/SF splice variant NK1. Biochim. Biophys. Acta 2013, 1834, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Young Song, K.; Giubellino, A. The role of MET in melanoma and melanocytic lesion. Am. J. Pathol. 2019, 189, 2138–2148. [Google Scholar] [CrossRef] [Green Version]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic. Dis. Transl. Med. 2017, 3, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Kong-Beltran, M.; Stamos, J.; Wickramasinghe, D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell 2004, 6, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017, 108, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Mizuuchi, H.; Tomida, S.; Terashima, M.; Hayashi, H.; Nishio, K.; Mitsudomi, T. MET gene exon 14 deletion created using the CRISPR/Cas9 system enhances cellular growth and sensitivity to a MET inhibitor. Lung Cancer 2015, 90, 590–597. [Google Scholar] [CrossRef]
- Darsa, H.E.; Sayed, R.E.; Abdel-Rahman, O. MET Inhibitors for the Treatment of Gastric Cancer: What’s Their Potential? J. Exp. Pharmacol. 2020, 12, 349–361. [Google Scholar] [CrossRef]
- Amemiya, H.; Kono, K.; Itakura, J.; Feng Tang, R.; Takahashi, A.; An, F.Q.; Kamei, S.; Iizuka, H.; Fujii, H.; Matsumoto, Y. c-Met Expression in Gastric Cancer with Liver Metastasis. Oncology 2002, 63, 286–296. [Google Scholar] [CrossRef]
- Lai, A.Z.; Abella, J.V.; Park, M. Crosstalk in Met receptor oncogenesis. Trends. Cell Biol. 2009, 19, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhou, J.; Rogers, A.M.; Janne, P.A.; Benedettini, E.; Loda, M.; Hodi, F.S. c-Met, epidermal growth factor receptor, and insulin-like growth factor-1 receptor are important for growth in uveal melanoma and independently contribute to migration and metastatic potential. Melanoma Res. 2012, 22, 123–132. [Google Scholar] [CrossRef]
- Varkaris, A.; Gaur, S.; Parikh, N.U.; Song, J.H.; Dayyani, F.; Jin, J.K.; Logothetis, C.J.; Gallick, G.E. Ligand-independent Activation of MET Through IGF-1/IGF-1R Signaling. Int. J. Cancer 2013, 133, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Guan, X. Cancer metastases: Challenges and opportumities. Acta Pharm. Sin. B. 2015, 5, 402–418. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, A.M.; Shiri, S.; Farsinejad, S. Metastasis review: From bench to bedside. Tumour Biol. 2014, 35, 8483–8523. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Hu, D.; Tu, L.; Zbou, X.; Lu, F.; Wen, B.; Wu, W.; Lin, Y.; Zbou, Z.; Qu, J. Involvement of PI3K/Akt Pathway in Hepatocyte Growth Factor-Induced Migration of Uveal Melanoma Cells. IVOS 2008, 49, 497–504. [Google Scholar] [CrossRef]
- Beviglia, L.; Kramer, R.H. HGF induces FAK activation and integrin-mediated adhesion in MTLn3 breast carcinoma cells. Int. J. Cancer 1999, 83, 640–649. [Google Scholar] [CrossRef]
- Lai, J.F.; Kao, S.C.; Jiang, S.T.; Tang, M.J.; Chan, P.C.; Chen, H.C. Involvement of focal adhesion kinase in hepatocyte growth factor-induced scatter of Madin-Darby canine kidney cells. J. Biol. Chem. 2000, 275, 7474–7480. [Google Scholar] [CrossRef] [Green Version]
- Guarino, M. Epithelial-mesenchymal transition and tumour invasion. Int. J. Biochem. Cell Biol. 2007, 39, 2153–2160. [Google Scholar] [CrossRef]
- Koefinger, P.; Wels, C.; Joshi, S.; Damm, S.; Steinbauer, E.; Beham-Schmid, C.; Frank, S.; Bergler, H.; Schaider, H. The cadherin switch in melanoma instigated by HGF is mediated through epithelial-mesenchymal transition regulators. Pigment Cell Melanoma Res. 2011, 24, 382–385. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Schaider, H.; Satyamoorthy, K.; Hanakawa, Y.; Hashimoto, K.; Herlyn, M. Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene 2001, 20, 8125–8135. [Google Scholar] [CrossRef] [Green Version]
- Venza, M.; Visalli, M.; Catalano, T.; Biondo, C.; Beninati, C.; Teti, D.; Venza, I. DNA methylation-induced E-cadherin silencing is correlated with the clinicopathological features of melanoma. Oncol. Rep. 2016, 35, 2451–2460. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Chen, R. The DecisionDx-UM Gene Expression Profile Test Provides Risk Stratification and Individualized Patient Care in Veal Melanoma. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Chang, S.H.; Worley, L.A.; Onken, M.D.; Harbour, J.W. Prognostic biomarkers in uveal melanoma: Evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008, 18, 191–200. [Google Scholar] [CrossRef]
- Kaliki, S.; Shields, C.L.; Shields, J.A. Uveal melanoma: Estimating prognosis. Indian J. Ophthalmol. 2015, 63, 93–102. [Google Scholar] [CrossRef]
- Onken, M.D.; Ehlers, J.P.; Worley, L.A.; Makita, J.; Yokota, Y.; Harbour, J.W. Functional Gene Expression Analysis Uncovers Phenotypic Switch in Aggressive Uveal Melanoma. Cancer Res. 2006, 66, 4602–4609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, E.; Sugihara, H.; Bamba, M.; Hattori, T. Dynamic alteration of the E-cadherin/catenin complex during cell differentiation and invasion of undifferentiated-type gastric carcinomas. J. Pathol. 2005, 205, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Chen, S.; You, Z.; Yang, F.; Carey, T.E.; Saims, D.; Wang, C.Y. Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFkappa B. J. Biol. Chem. 2002, 277, 25203–25208. [Google Scholar] [CrossRef] [Green Version]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abounader, R.; Laterra, J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro-Oncology 2005, 7, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Su, Y.; Volpert, O.V.; Vande Woude, G.F. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.A.; Lee, K.H. HGF mediated upregulation of lipocalin 2 regulates MMP9 through nuclear factor-κB activation. Oncol. Rep. 2015, 34, 2179–2187. [Google Scholar] [CrossRef] [Green Version]
- El-Shabrawi, Y.; Ardjomand, N.; Radner, H.; Ardjomand, N. MMP-9 is predominantly expressed in epithelioid melanoma and not spindle cell uveal melanoma. J. Pathol. 2001, 194, 201–206. [Google Scholar] [CrossRef]
- Sato, T.; Han, F.; Yamamoto, A. The biology and management of uveal melanoma. Curr. Oncol. Rep. 2008, 10, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, B.; Moran, P.; Lipari, T.; Ganesan, R.; Corpuz, R.; Ludlam, M.J.C.; Gogineni, A.; Koeppen, H.; Bunting, S.; et al. Pegylated kunitz domain inhibitor suppresses hepsin-mediated invasive tumor growth and metastasis. Cancer Res. 2009, 69, 8395–8402. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Yang, R.; Zheng, Z.; Romero, M.; Ross, J.; Bou-Reslan, H.; Carano, R.A.D.; Kasman, I.; Mai, E.; Young, J.; et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008, 68, 4360–4368. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Nakamura, T. NK4 (HGF-antagonist/angiogenesis inhibitor) in cancer biology and therapeutics. Cancer Sci. 2003, 94, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-dependent solid tumours—Molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zou, Q.; Liu, H.; Qiu, B.; Li, Q.; Lin, Y.; Liang, Y. Management of Non-small Cell Lung Cancer Patients with MET Exon 14 Skipping Mutations. Curr. Treat. Options Oncol. 2020, 21, 33. [Google Scholar] [CrossRef]
- Yan, S.B.; Peek, V.L.; Ajamie, R.; Buchanan, S.G.; Graff, J.R.; Heidler, S.A.; Hui, Y.H.; Huss, K.L.; Konicek, B.W.; Manro, J.R.; et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Investig. New Drugs 2013, 31, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Bi, C.; Credille, K.M.; Manro, J.R.; Peek, V.L.; Donoho, G.P.; Yan, L.; Wijsman, J.A.; Yan, S.B.; Walgren, R.A. Inhibition of tumor growth and metastasis in non-small cell lung cancer by LY2801653, an inhibitor of several oncokinases, including MET. Clin. Cancer Res. 2013, 19, 5699–5710. [Google Scholar] [CrossRef] [Green Version]
- Ohara, M.; Saito, K.; Kageyama, K.; Terai, M.; Cheg, H.; Aplin, A.E.; Sato, T. Dual Targeting of CDK4/6 and cMET in Metastatic Uveal Melanoma. Cancers 2021, 13, 1104. [Google Scholar] [CrossRef] [PubMed]
- He, A.R.; Cohen, R.B.; Denlinger, C.S.; Sama, A.; Birnbaum, A.; Hwang, J.; Sato, T.; Lewis, N.; Mynderse, M.; Niland, M.; et al. First-in-Human Phase I Study of Merestinib, an Oral Multikinase Inhibitor, in Patients with Advanced Cancer. Oncologist 2019, 24, e930–e942. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Olson, D.J.; Allred, J.B.; Strand, C.A.; Bao, R.; Zha, Y.; Carll, T.; Labadie, B.W.; Bastos, B.R.; Butler, M.; et al. Randomized Phase II Trial and Tumor Mutational Spectrum Analysis from Cabozantinib versus Chemotherapy in Metastatic Uveal Melanoma (Alliance A091201). Clin. Cancer Res. 2020, 26, 804–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennacchietti, S.; Cazzanti, M.; Bertotti, A.; Rideout III, W.M.; Han, M.; Gyuris, J.; Perera, T.; Comoglio, P.M.; Trusolino, L.; Michieli, P. Microenvironment-Derived HGF Overcome Genetically Determined Sensitivity to Anti-MET Drugs. Cancer Res. 2014, 74, 6598–6609. [Google Scholar] [CrossRef] [Green Version]
- Wood, G.E.; Hockings, H.; Hilton, D.M.; Kermorgant, S. The role of MET in chemotherapy resistance. Oncogene 2021, 40, 1927–1942. [Google Scholar] [CrossRef]
- Xu, J.; Liu, S.; Yang, X.; Cao, S.; Zhou, Y. Paracrine HGF promotes EMT and mediates the effects of PSC on chemoresistance by activating c-Met/PI3K/Akt signaling in pancreatic cancer in vivo. Life Sci. 2020, 263, 118523. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Huang, C.Y.; Chiang, Y.Y.; Chen, W.H.; Chiou, S.H.; Chen, C.Y.; Chow, K.C. HGF increases cisplatin resistance via down-regulation of AIF in lung cancer cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Raghav, K.P.S.; Gonzalez-Augulo, A.M.; Blumenschein, G.R. Role of HGF/MET axis in resistance of lung cancer to contemporary management. Transl. Lung Cancer Res. 2012, 1, 179–193. [Google Scholar] [PubMed]
- Canadas, I.; Rojo, F.; Taus, A.; Arpi, O.; Arumi-Uria, M.; Pijuan, L.; Menendez, S.; Zazo, S.; Domine, M.; Salido, M.; et al. Targeting Epithelial-to-Transition with Met Inhibitors Reverts Chemoresistance in Small Cell Lung Cancer. Clin. Cancer Res. 2014, 20, 938–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapitejin, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination with Dacarbazine in Patients with Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoushtari, A.N.; Kudchadkar, R.R.; Panageas, K.; Murthy, R.K.; Jung, M.; Shah, R.; O’Donnell, B.; Khawaja, T.T.; Shames, Y.; Prempeh-Keteku, N.A.; et al. A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J. Clin. Oncol. 2016, 34, 9511. [Google Scholar] [CrossRef]
- Cheng, H.; Terai, M.; Kageyama, K.; Ozaki, S.; McCue, P.A.; Sato, T.; Aplin, A.E. Paracrine Effect of NRG1 and HGF Drives Resistance to MEK Inhibitors in Metastatic Uveal Melanoma. Cancer Res. 2015, 75, 2737–2748. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Chua, V.; Liao, C.; Purwin, T.J.; Terai, M.; Kageyama, K.; Davies, M.A.; Sato, T.; Aplin, A.E. Co-targeting HGF-cMET signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol. Cancer Ther. 2017, 16, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Tohyama, O.; Yamaguchi, A.; Matsushima, T.; Takahashi, K.; Funasaka, S.; Shirotori, S.; Asada, M.; Obaishi, H. E7050: A dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010, 101, 210–215. [Google Scholar] [CrossRef]
- Huang, X.; Li, E.; Shen, H.; Wang, X.; Tang, T.; Zhang, X.; Xu, J.; Tang, Z.; Guo, C.; Bai, X.; et al. Targeting the HGF/MET Axis in Cancer Therapy: Challenges in Resistance and Opportunities for Improvement. Front. Cell Dev. Biol. 2020, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Hamada, N.; Matsumoto, H.; Hara, T.; Kobayashi, Y. Intercellular and intracellular signaling pathways mediating ionizing radiation-induced bystander effects. J. Radiat. Res. 2007, 48, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweigerer, L.; Rave-Frank, M.; Schmidberger, H.; Hecht, M. Sublethal irradiation promotes invasiveness of neuroblastoma cells. Biochem. Biophys. Res. Commun. 2005, 330, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Albitar, M.; Sudarsanam, S.; Ma, W.; Jiang, S.; Chen, W.; Funari, V.; Blocker, F.; Agersborg, S. Correlation of MET gene amplification and TP53 mutation with PD-L1 expression in non-small cell lung cancer. Oncotarget 2018, 9, 13682–13693. [Google Scholar] [CrossRef] [PubMed]
- Saigi, M.; Alburquerque-Bejar, J.J.; Leer-Florin, A.M.; Pereira, C.; Pros, E.; Romero, O.A.; Baixeras, N.; Esteve-Codina, A.; Nadal, E.; Brambilla, E.; et al. MET-Oncogenic and JAK2-Inactivating Alterations Are Independent Factors That Affect Regulation of PD-L1 Expression in Lung Cancer. Clin. Cancer Res. 2018, 24, 4579–4587. [Google Scholar] [CrossRef] [Green Version]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.R.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Terai, M.; Londin, E.; Rochani, A.; Link, E.; Lam, B.; Kaushal, G.; Bhushan, A.; Orloff, M.; Sato, T. Expression of Tryptophan 2,3-Dioxygenase in Metastatic Uveal Melanoma. Cancers 2020, 12, 405. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
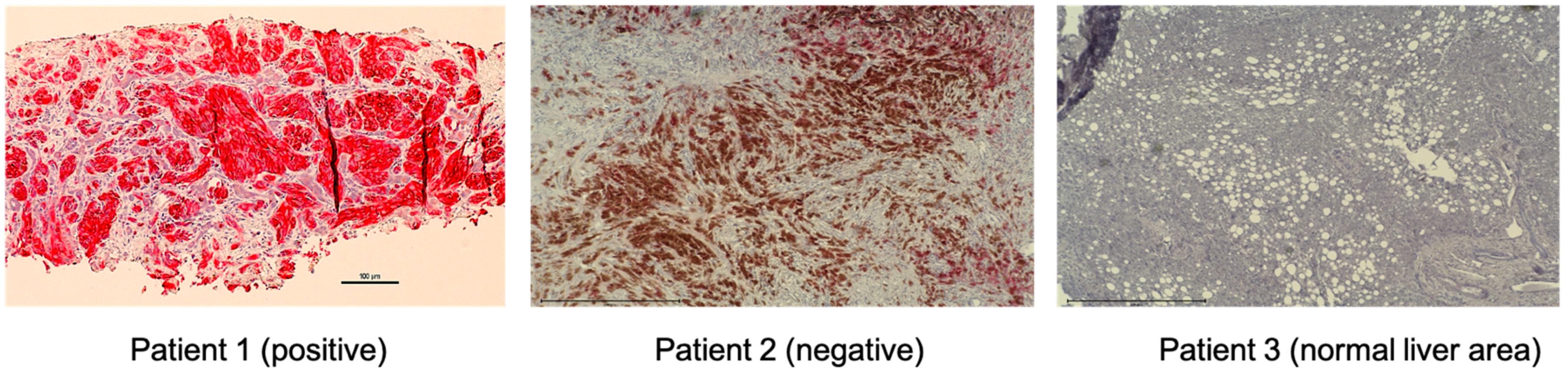
| MET Positive | MET Negative | ||
|---|---|---|---|
| Location | Number | Location | Number |
| Liver | 21 | Liver | 2 |
| Lung | 1 | Omentum | 1 |
| Abdomen | 1 | Mediastium | 1 |
| Cervical Lymph node | 1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, R.; Terai, M.; Londin, E.; Sato, T. The Role of HGF/MET Signaling in Metastatic Uveal Melanoma. Cancers 2021, 13, 5457. https://doi.org/10.3390/cancers13215457
Tanaka R, Terai M, Londin E, Sato T. The Role of HGF/MET Signaling in Metastatic Uveal Melanoma. Cancers. 2021; 13(21):5457. https://doi.org/10.3390/cancers13215457
Chicago/Turabian StyleTanaka, Ryota, Mizue Terai, Eric Londin, and Takami Sato. 2021. "The Role of HGF/MET Signaling in Metastatic Uveal Melanoma" Cancers 13, no. 21: 5457. https://doi.org/10.3390/cancers13215457
APA StyleTanaka, R., Terai, M., Londin, E., & Sato, T. (2021). The Role of HGF/MET Signaling in Metastatic Uveal Melanoma. Cancers, 13(21), 5457. https://doi.org/10.3390/cancers13215457