
New Therapeutic Strategy for Overcoming Multidrug Resistance in Cancer Cells with Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors

 ,
,  , , ,
, , ,  , ,
, , 

Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. SFK Inhibitors Decrease Cell Viability
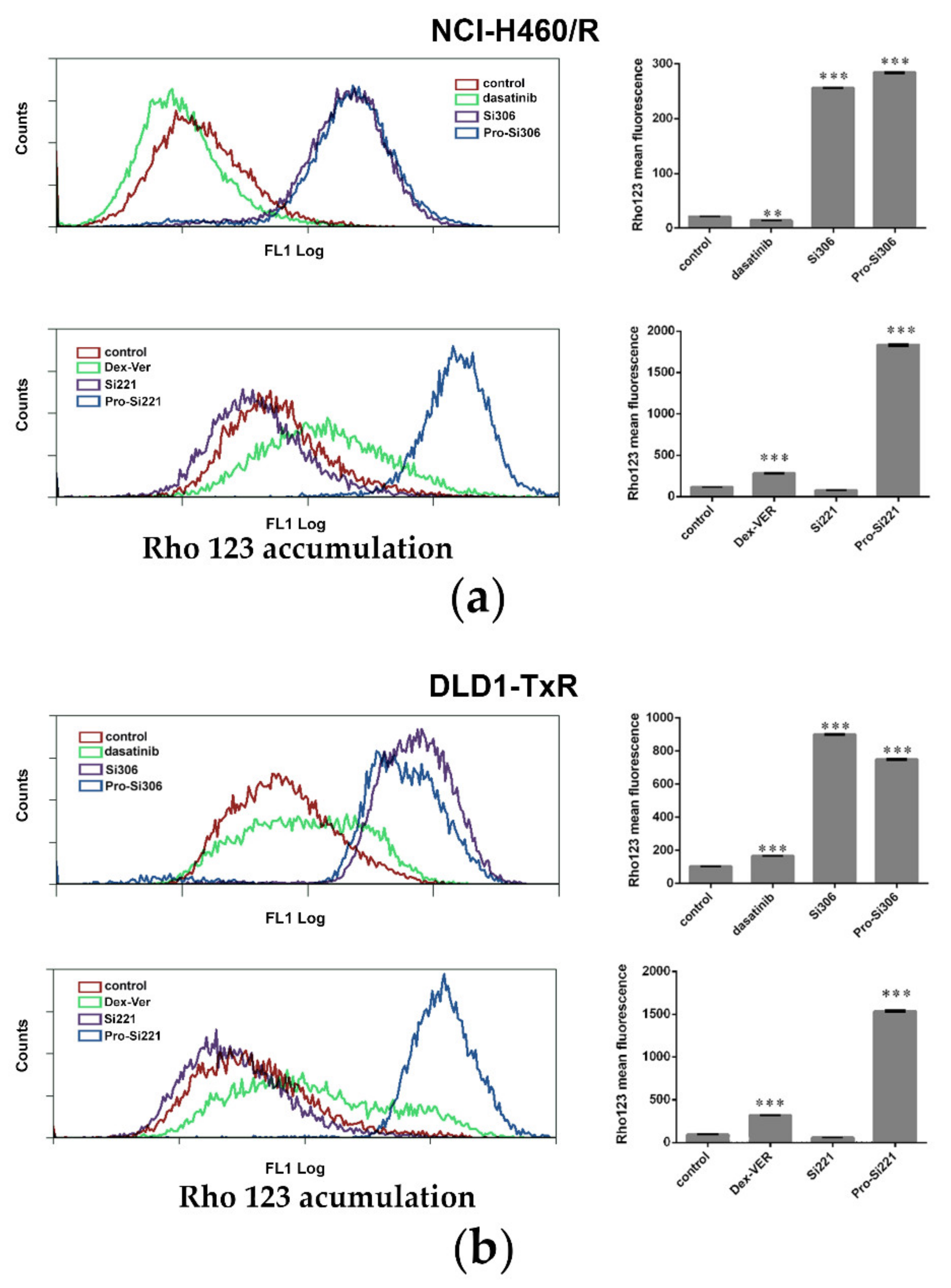
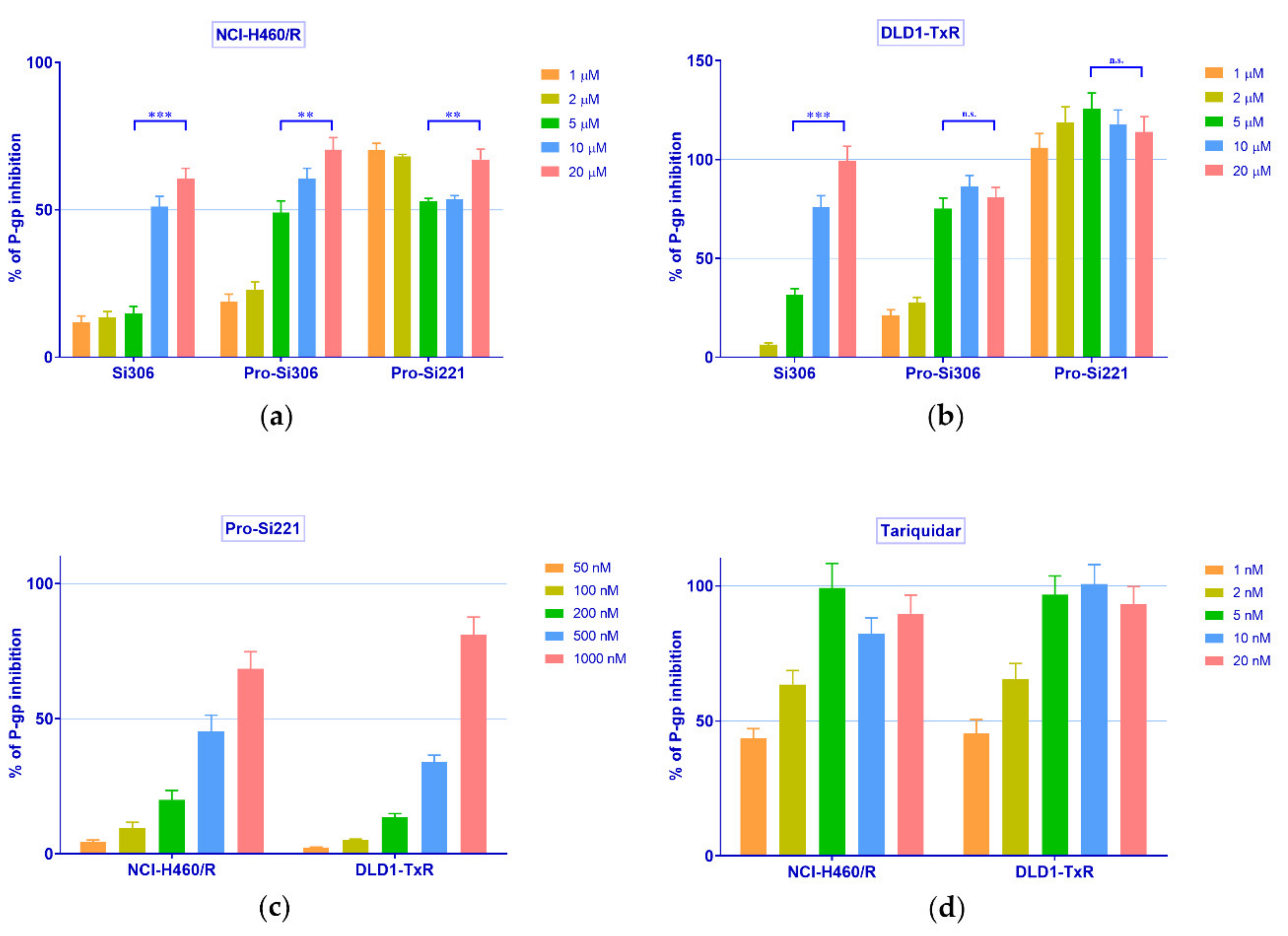
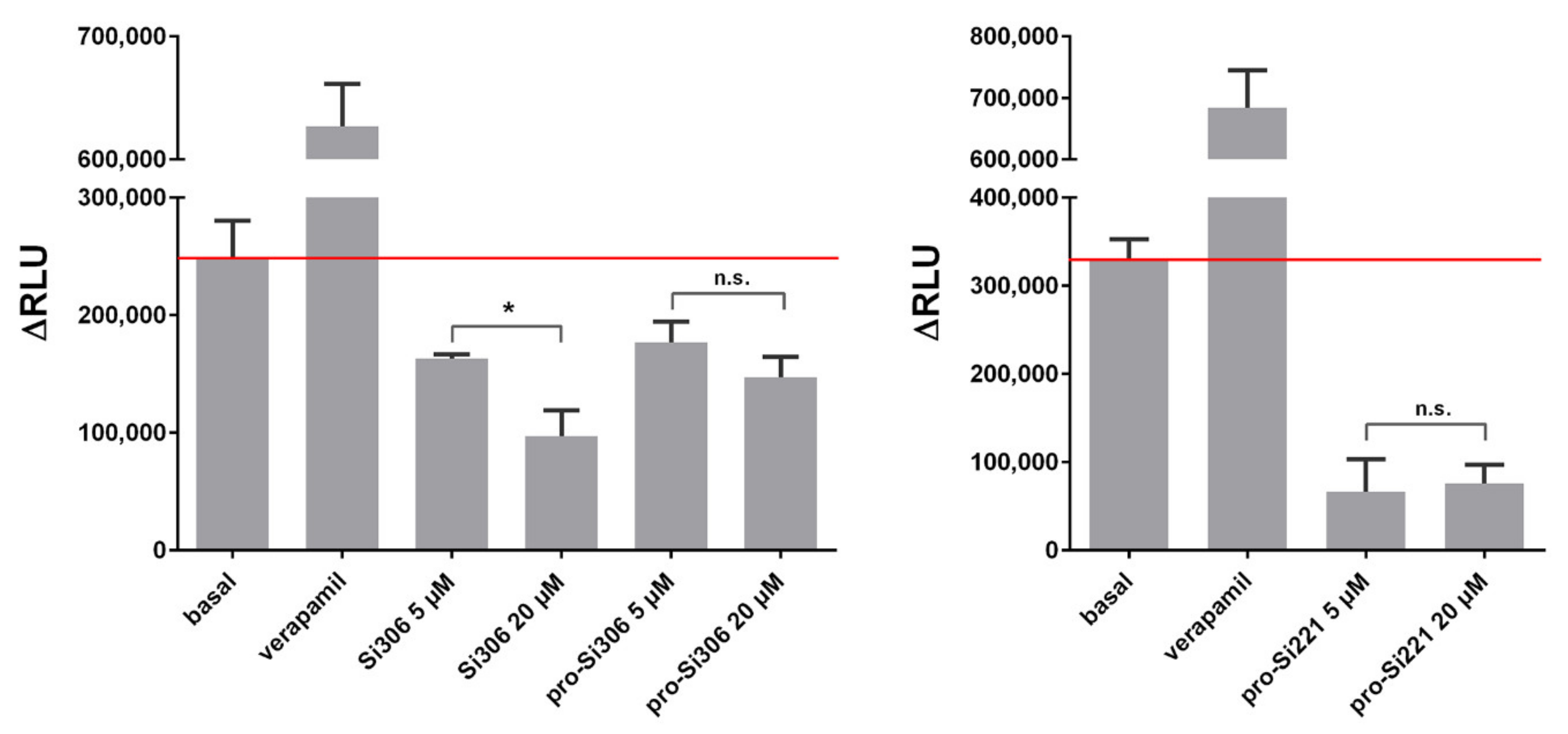
2.2. SFK Inhibitors Modify P-gp Activity
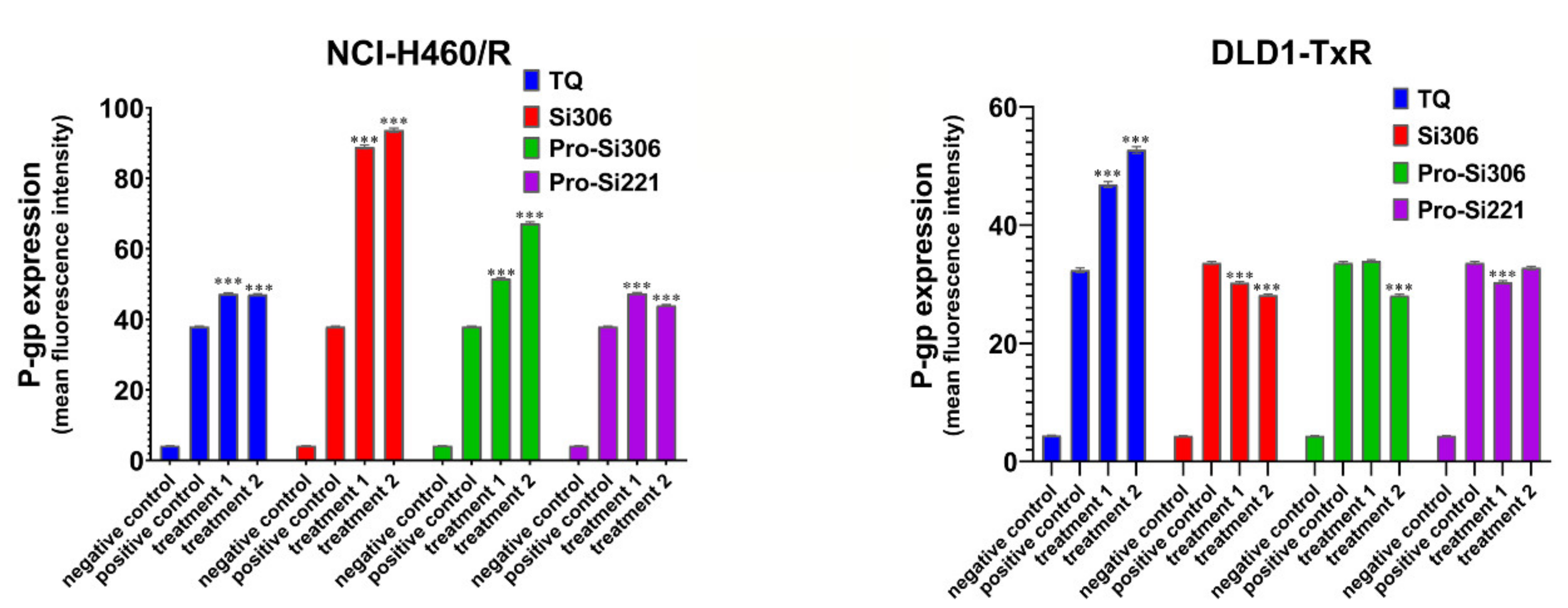
2.3. The Effect of SFK Inhibitors on P-gp Expression
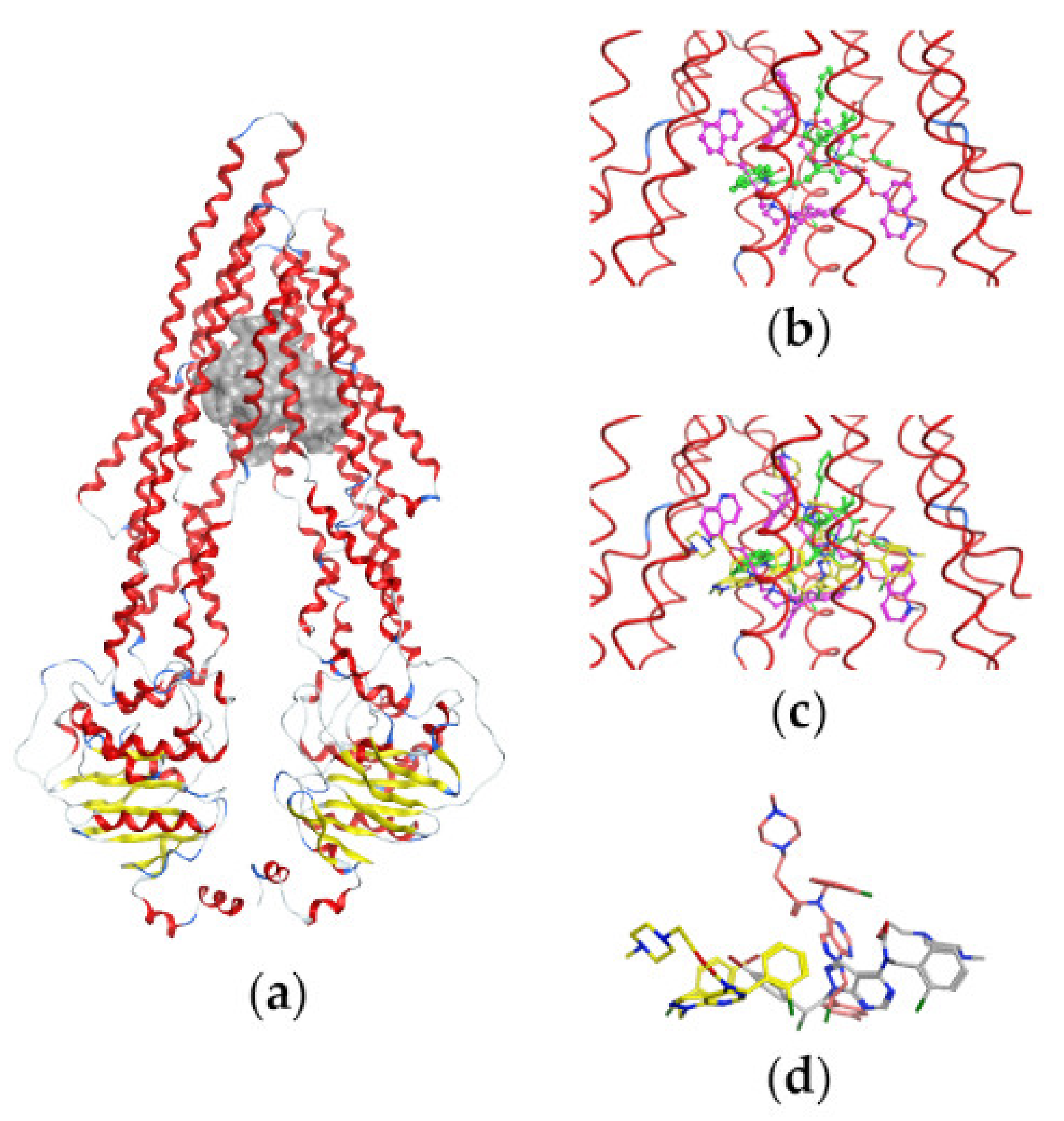
2.4. Docking Studies
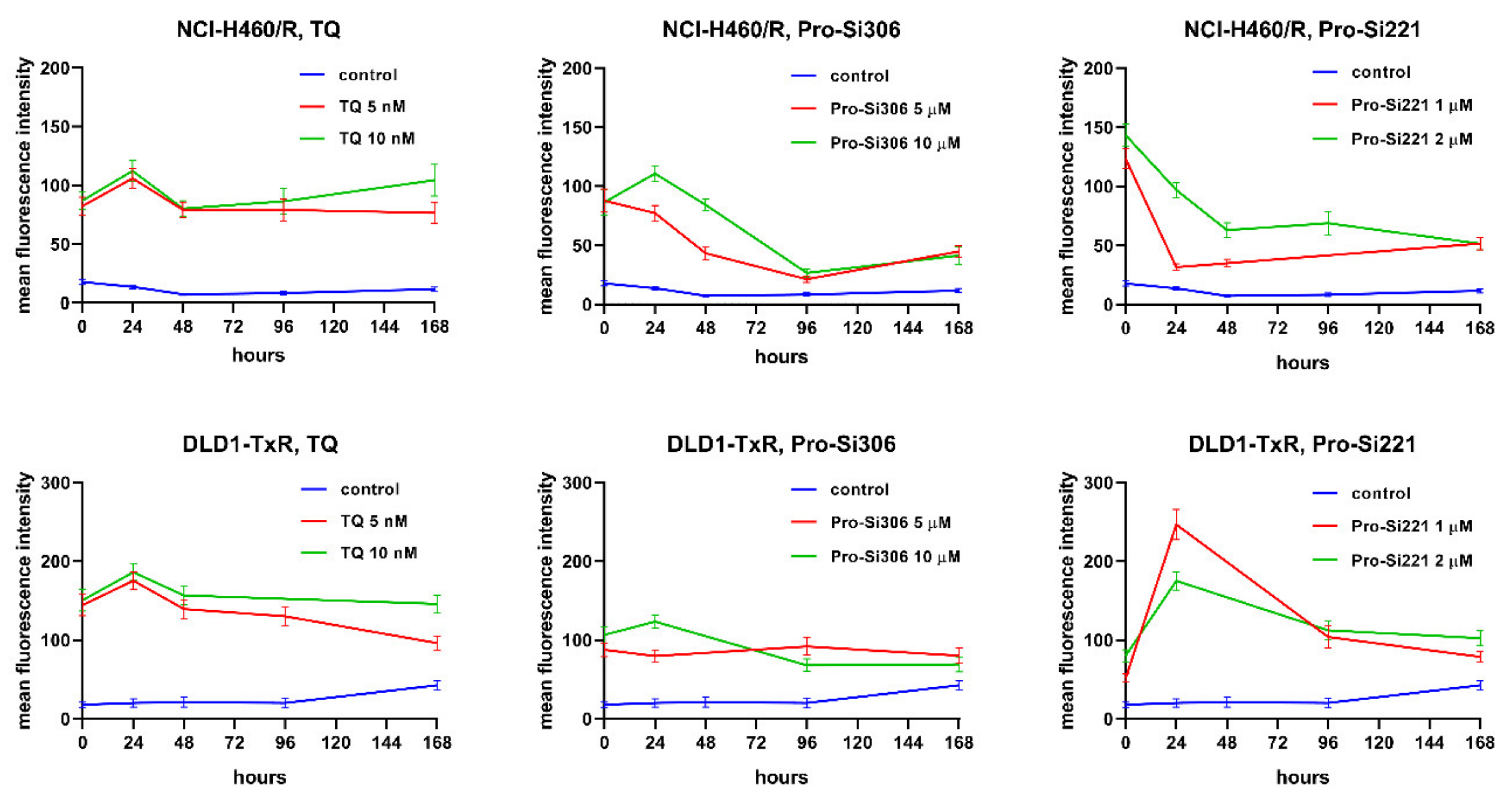
2.5. Kinetics of P-gp Inhibition
2.6. SFK Inhibitors Sensitize MDR Cancer Cells to DOX and PTX
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Chemicals
4.3. Cell Culture
4.4. MTT Assay
4.5. Rho 123 Accumulation Assay
4.6. P-gp ATPase Activity Assay
4.7. Docking Studies
4.8. Flow Cytometric Analysis of P-gp Expression
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, T.; Mizoi, K.; Kamioka, H.; Yano, K. Physiological Roles of ERM Proteins and Transcriptional Regulators in Supporting Membrane Expression of Efflux Transporters as Factors of Drug Resistance in Cancer. Cancers 2020, 12, 3352. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Matsuoka, K.; Kimura, Y.; Ueda, K.; Kato, H. Inward- and outward-facing X-ray crystal structures of homodimeric P-glycoprotein CmABCB1. Nat. Commun. 2019, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Ashby, C.R., Jr.; Assaraf, Y.G.; Chen, Z.S.; Liu, H.M. Chemical molecular-based approach to overcome multidrug resistance in cancer by targeting P-glycoprotein (P-gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef]
- Hall, M.D.; Handley, M.D.; Gottesman, M.M. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 2009, 30, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Krchniakova, M.; Skoda, J.; Neradil, J.; Chlapek, P.; Veselska, R. Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration. Int. J. Mol. Sci. 2020, 21, 3157. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Brullo, C.; Musumeci, F.; Botta, M. Novel dual Src/Abl inhibitors for hematologic and solid malignancies. Expert Opin. Investig. Drugs 2010, 19, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Radi, M.; Musumeci, F.; Brullo, C.; Botta, M. Biologically driven synthesis of pyrazolo[3,4-d]pyrimidines as protein kinase inhibitors: An old scaffold as a new tool for medicinal chemistry and chemical biology studies. Chem. Rev. 2014, 114, 7189–7238. [Google Scholar] [CrossRef] [PubMed]
- Navarra, M.; Celano, M.; Maiuolo, J.; Schenone, S.; Botta, M.; Angelucci, A.; Bramanti, P.; Russo, D. Antiproliferative and pro-apoptotic effects afforded by novel Src-kinase inhibitors in human neuroblastoma cells. BMC Cancer 2010, 10, 602. [Google Scholar] [CrossRef]
- Cozzi, M.; Giorgi, F.; Marcelli, E.; Pentimalli, F.; Forte, I.M.; Schenone, S.; D’Urso, V.; De Falco, G.; Botta, M.; Giordano, A.; et al. Antitumor activity of new pyrazolo[3,4-d]pyrimidine SRC kinase inhibitors in Burkitt lymphoma cell lines and its enhancement by WEE1 inhibition. Cell Cycle 2012, 11, 1029–1039. [Google Scholar] [CrossRef][Green Version]
- Kostic, A.; Jovanovic Stojanov, S.; Podolski-Renic, A.; Nesovic, M.; Dragoj, M.; Nikolic, I.; Tasic, G.; Schenone, S.; Pesic, M.; Dinic, J. Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells. Brain Sci. 2021, 11, 884. [Google Scholar] [CrossRef]
- Casini, N.; Forte, I.M.; Mastrogiovanni, G.; Pentimalli, F.; Angelucci, A.; Festuccia, C.; Tomei, V.; Ceccherini, E.; Di Marzo, D.; Schenone, S.; et al. SRC family kinase (SFK) inhibition reduces rhabdomyosarcoma cell growth In Vitro and In Vivo and triggers p38 MAP kinase-mediated differentiation. Oncotarget 2015, 6, 12421–12435. [Google Scholar] [CrossRef][Green Version]
- Vignaroli, G.; Iovenitti, G.; Zamperini, C.; Coniglio, F.; Calandro, P.; Molinari, A.; Fallacara, A.L.; Sartucci, A.; Calgani, A.; Colecchia, D.; et al. Prodrugs of Pyrazolo[3,4-d]pyrimidines: From Library Synthesis to Evaluation as Potential Anticancer Agents in an Orthotopic Glioblastoma Model. J. Med. Chem. 2017, 60, 6305–6320. [Google Scholar] [CrossRef]
- Nesovic, M.; Divac Rankov, A.; Podolski-Renic, A.; Nikolic, I.; Tasic, G.; Mancini, A.; Schenone, S.; Pesic, M.; Dinic, J. Src Inhibitors Pyrazolo[3,4-d]pyrimidines, Si306 and Pro-Si306, Inhibit Focal Adhesion Kinase and Suppress Human Glioblastoma Invasion In Vitro and In Vivo. Cancers 2020, 12, 1570. [Google Scholar] [CrossRef]
- Fallacara, A.L.; Zamperini, C.; Podolski-Renic, A.; Dinic, J.; Stankovic, T.; Stepanovic, M.; Mancini, A.; Rango, E.; Iovenitti, G.; Molinari, A.; et al. A New Strategy for Glioblastoma Treatment: In Vitro and In Vivo Preclinical Characterization of Si306, a Pyrazolo[3,4-d]Pyrimidine Dual Src/P-Glycoprotein Inhibitor. Cancers 2019, 11, 848. [Google Scholar] [CrossRef]
- Podolski-Renic, A.; Jadranin, M.; Stankovic, T.; Bankovic, J.; Stojkovic, S.; Chiourea, M.; Aljancic, I.; Vajs, V.; Tesevic, V.; Ruzdijic, S.; et al. Molecular and cytogenetic changes in multi-drug resistant cancer cells and their influence on new compounds testing. Cancer Chemother. Pharmacol. 2013, 72, 683–697. [Google Scholar] [CrossRef]
- Efferth, T.; Saeed, M.E.M.; Kadioglu, O.; Seo, E.J.; Shirooie, S.; Mbaveng, A.T.; Nabavi, S.M.; Kuete, V. Collateral sensitivity of natural products in drug-resistant cancer cells. Biotechnol. Adv. 2020, 38, 107342. [Google Scholar] [CrossRef]
- Kitazaki, T.; Oka, M.; Nakamura, Y.; Tsurutani, J.; Doi, S.; Yasunaga, M.; Takemura, M.; Yabuuchi, H.; Soda, H.; Kohno, S. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 2005, 49, 337–343. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, Y.; Xu, M.; Chen, L.; Zhang, X.; To, K.K.; Zhao, H.; Wang, F.; Xia, Z.; Chen, X.; et al. Osimertinib (AZD9291) Enhanced the Efficacy of Chemotherapeutic Agents in ABCB1- and ABCG2-Overexpressing Cells In Vitro, In Vivo, and Ex Vivo. Mol. Cancer Ther. 2016, 15, 1845–1858. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, X.; Wang, F.; Wang, X.; Yang, K.; Xu, M.; To, K.K.; Li, Q.; Fu, L. Effect of ceritinib (LDK378) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Oncotarget 2015, 6, 44643–44659. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Ozvegy-Laczka, C.; Apati, A.; Magocsi, M.; Nemet, K.; Orfi, L.; Keri, G.; Katona, M.; Takats, Z.; Varadi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kathawala, R.J.; Wang, Y.J.; Zhang, Y.K.; Patel, A.; Shukla, S.; Robey, R.W.; Talele, T.T.; Ashby, C.R., Jr.; Ambudkar, S.V.; et al. Linsitinib (OSI-906) antagonizes ATP-binding cassette subfamily G member 2 and subfamily C member 10-mediated drug resistance. Int. J. Biochem. Cell Biol. 2014, 51, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.N.; Zhang, Y.K.; Wang, Y.J.; Barbuti, A.M.; Zhu, X.J.; Yu, X.Y.; Wen, A.W.; Wurpel, J.N.D.; Chen, Z.S. Modulating the function of ATP-binding cassette subfamily G member 2 (ABCG2) with inhibitor cabozantinib. Pharmacol. Res. 2017, 119, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Laiolo, J.; Lanza, P.A.; Parravicini, O.; Barbieri, C.; Insuasty, D.; Cobo, J.; Vera, D.M.A.; Enriz, R.D.; Carpinella, M.C. Structure activity relationships and the binding mode of quinolinone-pyrimidine hybrids as reversal agents of multidrug resistance mediated by P-gp. Sci. Rep. 2021, 11, 16856. [Google Scholar] [CrossRef]
- Zawilska, J.B.; Wojcieszak, J.; Olejniczak, A.B. Prodrugs: A challenge for the drug development. Pharmacol. Rep. 2013, 65, 1–14. [Google Scholar] [CrossRef]
- Nakanishi, T.; Shiozawa, K.; Hassel, B.A.; Ross, D.D. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood 2006, 108, 678–684. [Google Scholar] [CrossRef]
- Sen, R.; Natarajan, K.; Bhullar, J.; Shukla, S.; Fang, H.B.; Cai, L.; Chen, Z.S.; Ambudkar, S.V.; Baer, M.R. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol. Cancer Ther. 2012, 11, 2033–2044. [Google Scholar] [CrossRef]
- Huang, W.C.; Chen, Y.J.; Li, L.Y.; Wei, Y.L.; Hsu, S.C.; Tsai, S.L.; Chiu, P.C.; Huang, W.P.; Wang, Y.N.; Chen, C.H.; et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Yoshioka, S.; Tsukahara, S.; Mitsuhashi, J.; Sugimoto, Y. Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein. Mol. Cancer Ther. 2007, 6, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Sodani, K.; Dai, C.L.; Abuznait, A.H.; Singh, S.; Xiao, Z.J.; Patel, A.; Talele, T.T.; Fu, L.; Kaddoumi, A.; et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013, 328, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Englinger, B.; Lotsch, D.; Pirker, C.; Mohr, T.; van Schoonhoven, S.; Boidol, B.; Lardeau, C.H.; Spitzwieser, M.; Szabo, P.; Heffeter, P.; et al. Acquired nintedanib resistance in FGFR1-driven small cell lung cancer: Role of endothelin—A receptor-activated ABCB1 expression. Oncotarget 2016, 7, 50161–50179. [Google Scholar] [CrossRef]
- Ellegaard, A.M.; Groth-Pedersen, L.; Oorschot, V.; Klumperman, J.; Kirkegaard, T.; Nylandsted, J.; Jaattela, M. Sunitinib and SU11652 inhibit acid sphingomyelinase, destabilize lysosomes, and inhibit multidrug resistance. Mol. Cancer Ther. 2013, 12, 2018–2030. [Google Scholar] [CrossRef]
- Pesic, M.; Markovic, J.Z.; Jankovic, D.; Kanazir, S.; Markovic, I.D.; Rakic, L.; Ruzdijic, S. Induced resistance in the human non small cell lung carcinoma (NCI-H460) cell line in vitro by anticancer drugs. J. Chemother. 2006, 18, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Podolski-Renic, A.; Andelkovic, T.; Bankovic, J.; Tanic, N.; Ruzdijic, S.; Pesic, M. The role of paclitaxel in the development and treatment of multidrug resistant cancer cell lines. Biomed. Pharmacother. 2011, 65, 345–353. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | NCI-H460 | NCI-H460/R | DLD1 | DLD1-TxR |
|---|---|---|---|---|
| Dasatinib | 3.931 ± 0.295 | 6.885 ± 0.605 | 3.244 ± 0.411 | 2.900 ± 0.223 |
| Si306 | 6.235 ± 0.685 | 1.893 ± 0.184 | 3.927 ± 0.679 | 2.564 ± 0.183 |
| Pro-Si306 | 2.405 ± 0.376 | 0.897 ± 0.102 | 2.451 ± 0.346 | 1.867 ± 0.128 |
| Si221 | 14.569 ± 1.477 | 18.136 ± 2.079 | 34.134 ± 2.322 | 11.642 ± 1.287 |
| Pro-Si221 | 4.269 ± 0.509 | 5.144 ± 0.573 | 9.798 ± 1.152 | 5.704 ± 0.543 |
| IC50 (µM) for P-gp Inhibition | ||
|---|---|---|
| Compounds | NCI-H460/R | DLD1-TxR |
| TQ a | 1.073 ± 0.174 | 0.925 ± 0.155 |
| Si306 | 13.267 ± 0.908 | 6.443 ± 0.667 |
| Pro-Si306 | 6.167 ± 0.479 | 2.952 ± 0.300 |
| Pro-Si221 | 0.612 ± 0.072 | 0.669 ± 0.061 |
| Compounds | Strongest Base pKa а ± sd b | %C c at pH = 7.4 | Prevalent Form at pH = 7.4 (%) |
|---|---|---|---|
| Si306 | 6.2 ± 0.1 | 38 | neutral (58%) |
 | |||
| Pro-Si306 | 7.5 ± 0.1 | 61 | protonated (99.5%) |
 | |||
| Si221 | 3.2 ± 0.1 | n.c. b | neutral (98%) |
 | |||
| Pro-Si221 | 7.5 ± 0.1 | 96 | protonated (99%) |
 | |||
| Compounds | Docking Scores S, kcal/mol | |||
|---|---|---|---|---|
| Neutral | Protonated a | |||
| 1st Pose | 10th Pose | 1st Pose | 10th Pose | |
| pro-Si221 | −13.75 b | −10.72 | −12.56 | −11.13 |
| pro-Si306 | −12.87 | −11.76 | −13.64 | −11.57 |
| Si306 | −11.91 | −10.17 | −12.41 | −10.56 |
| Si221 | −11.02 | −9.46 | - | - |
| TQ | −17.28 | −14.18 | −17.71 b | −14.21 |
| Compounds/Cell Lines | IC50 (nM) | Relative Reversal |
|---|---|---|
| DOX/NCI-H460/R | 905.4 ± 44.4 | |
| Pro-Si306 0.2 µM | 280.7 ± 23.3 | 3.2 |
| Pro-Si306 0.5 µM | 89.0 ± 5.1 | 10.2 |
| Pro-Si221 0.2 µM | 509.3 ± 45.3 | 1.8 |
| Pro-Si221 0.5 µM | 127.2 ± 5.2 | 7.1 |
| PTX/DLD1-TxR | 1052.0 ± 114.5 | |
| Pro-Si306 0.2 µM | 707.8 ± 49.9 | 1.5 |
| Pro-Si306 0.5 µM | 418.4 ± 23.6 | 2.5 |
| Pro-Si221 0.2 µM | 537.7 ± 24.4 | 2.0 |
| Pro-Si221 0.5 µM | 331.6 ± 15.7 | 3.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podolski-Renić, A.; Dinić, J.; Stanković, T.; Tsakovska, I.; Pajeva, I.; Tuccinardi, T.; Botta, L.; Schenone, S.; Pešić, M. New Therapeutic Strategy for Overcoming Multidrug Resistance in Cancer Cells with Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors. Cancers 2021, 13, 5308. https://doi.org/10.3390/cancers13215308
Podolski-Renić A, Dinić J, Stanković T, Tsakovska I, Pajeva I, Tuccinardi T, Botta L, Schenone S, Pešić M. New Therapeutic Strategy for Overcoming Multidrug Resistance in Cancer Cells with Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors. Cancers. 2021; 13(21):5308. https://doi.org/10.3390/cancers13215308
Chicago/Turabian StylePodolski-Renić, Ana, Jelena Dinić, Tijana Stanković, Ivanka Tsakovska, Ilza Pajeva, Tiziano Tuccinardi, Lorenzo Botta, Silvia Schenone, and Milica Pešić. 2021. "New Therapeutic Strategy for Overcoming Multidrug Resistance in Cancer Cells with Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors" Cancers 13, no. 21: 5308. https://doi.org/10.3390/cancers13215308
APA StylePodolski-Renić, A., Dinić, J., Stanković, T., Tsakovska, I., Pajeva, I., Tuccinardi, T., Botta, L., Schenone, S., & Pešić, M. (2021). New Therapeutic Strategy for Overcoming Multidrug Resistance in Cancer Cells with Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors. Cancers, 13(21), 5308. https://doi.org/10.3390/cancers13215308