
No Need to Stick Together to Be Connected: Multiple Types of Enhancers’ Networking


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. A Journey at the Center of the Dark Genome: The Challenge of Enhancers’ Functional Characterization
- (1)
- (2)
- (3)
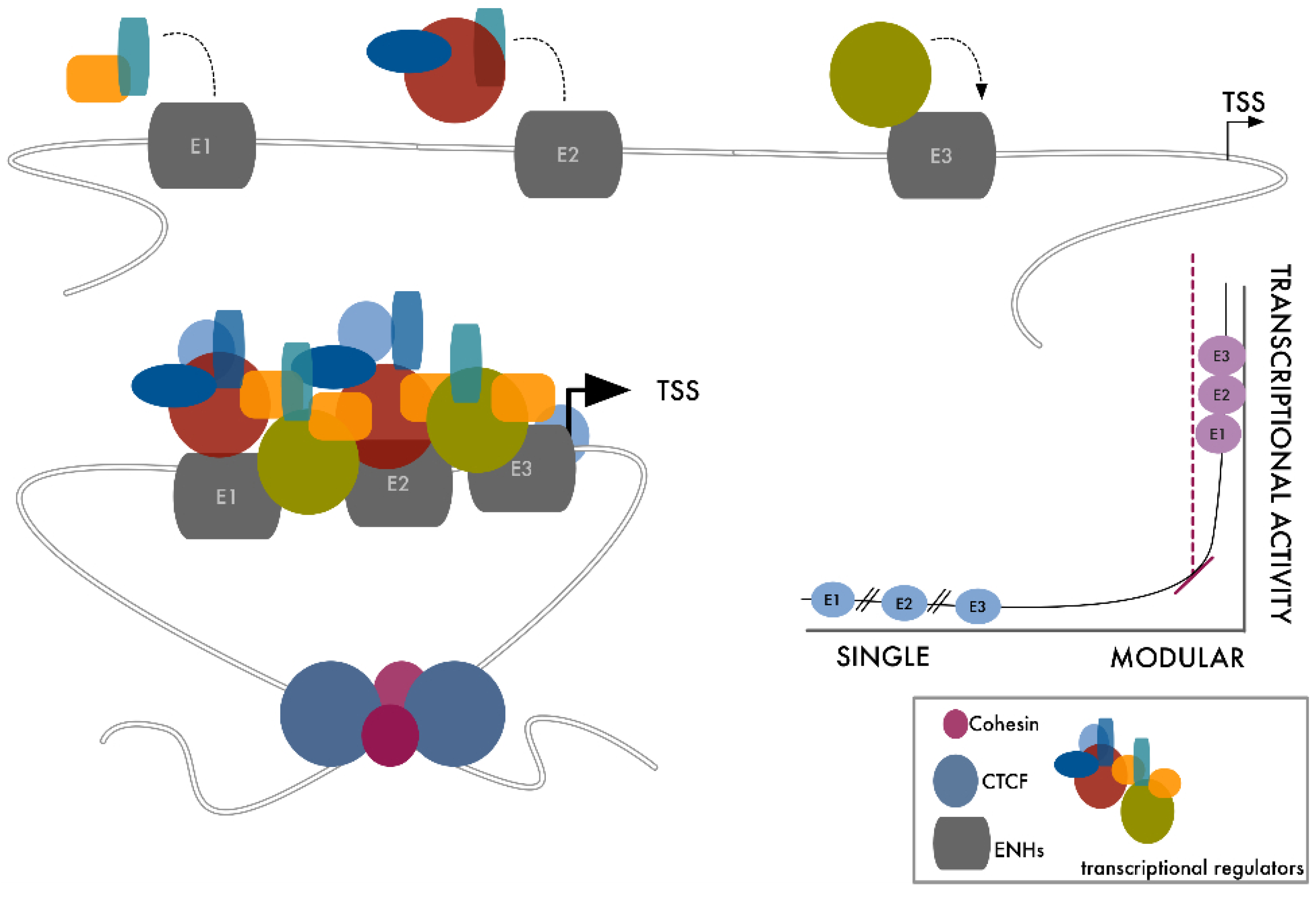
3. SUPER-Enhancers: Get Close to Maximize the Effect
3.1. SE Plasticity
3.2. Genetic Alterations and SEs
3.3. Pitfalls and Caveats of SEs
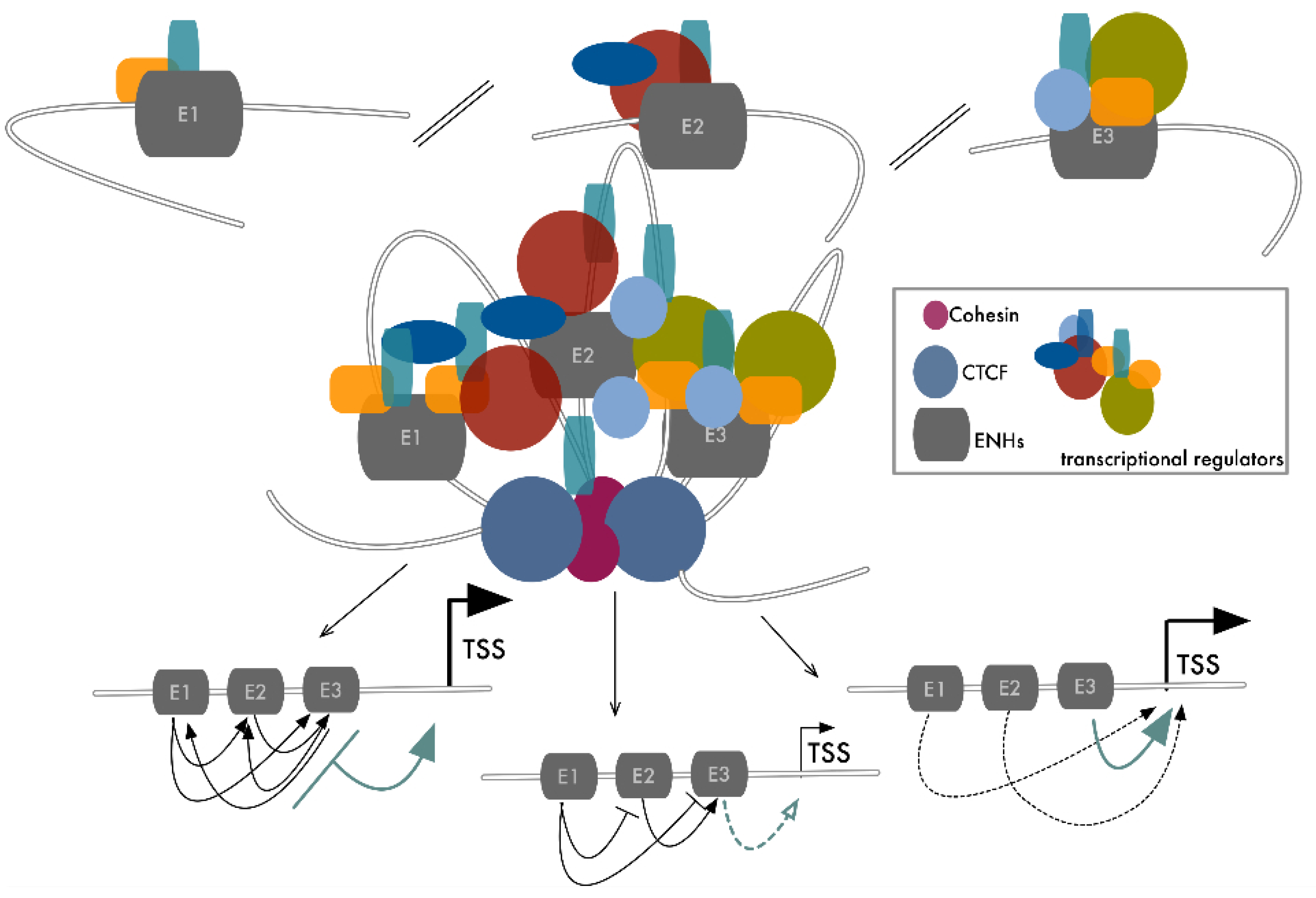
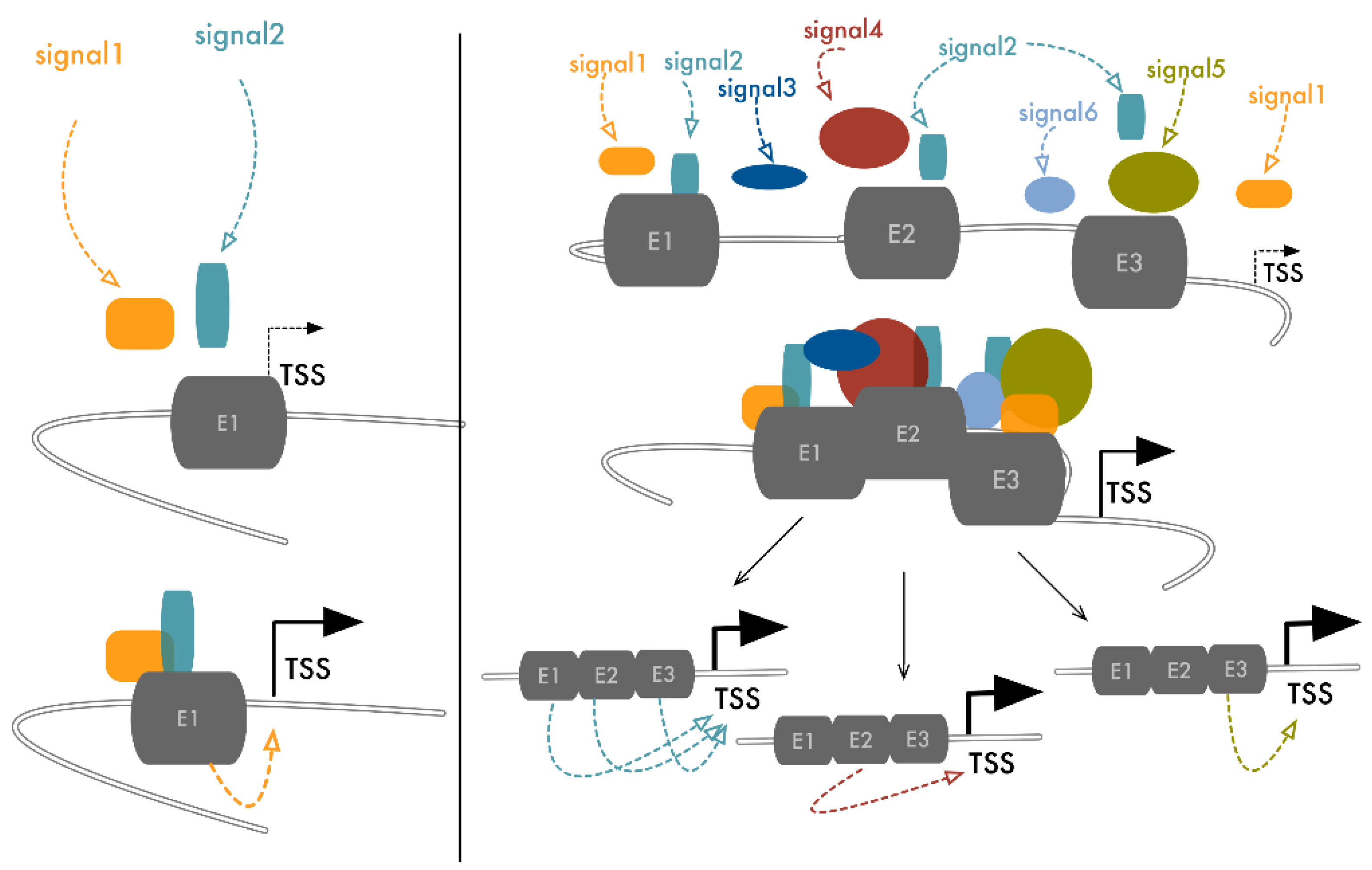
4. From Dependency to Redundancy: No Need to Stick Together to Be Connected
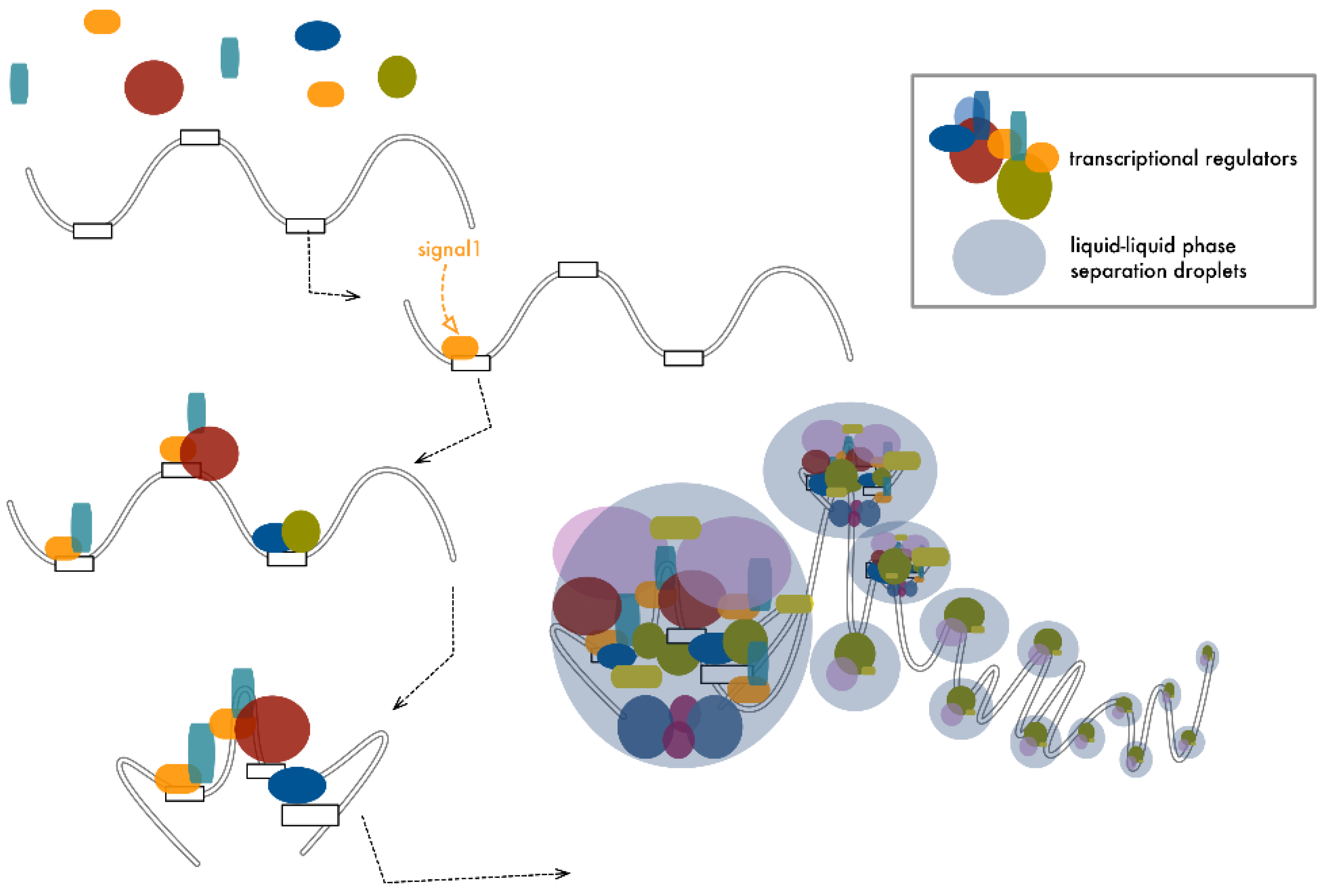
5. Transcriptional Control Is a Matter of Density
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Spitz, F.; Furlong, E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of Developmental and Onco-genic Signaling Pathways at Transcriptional Super-Enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [Green Version]
- The Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Rev. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef]
- Banerji, J.; Rusconi, S.; Schaffner, W. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell 1981, 27, 299–308. [Google Scholar] [CrossRef]
- Mercola, M.; Wang, X.F.; Olsen, J.; Calame, K. Transcriptional enhancer elements in the mouse immunoglobulin heavy chain locus. Science 1983, 221, 663–665. [Google Scholar] [CrossRef]
- Zhang, Y.; Wong, C.-H.; Birnbaum, R.; Li, G.; Favaro, R.; Ngan, C.Y.; Lim, J.; Tai, E.; Poh, H.M.; Wong, E.; et al. Chromatin connectivity maps reveal dynamic promoter–enhancer long-range associations. Nature 2013, 504, 306–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, C.-T.; Corces, V.G. Enhancer function: New insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Bulger, M.; Groudine, M. Functional and Mechanistic Diversity of Distal Transcription Enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Blum, R.; Vethantham, V.; Bowman, C.; Rudnicki, M.; Dynlacht, B.D. Genome-wide identification of enhancers in skeletal muscle: The role of MyoD1. Genes Dev. 2012, 26, 2763–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudkovsky, N.; Logie, C.; Hahn, S.; Peterson, C.L. Recruitment of the SWI/SNF chromatin remodeling complex by transcrip-tional activators. Genes Dev. 1999, 13, 2369–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Merkurjev, D.; Yang, F.; Li, W.; Oh, S.; Friedman, M.J.; Song, X.; Zhang, F.; Ma, Q.; Ohgi, K.A.; et al. Enhancer Activation Requires trans-Recruitment of a Mega Transcription Factor Complex. Cell 2014, 159, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cairns, M.J.; Yan, J. Super-enhancers in transcriptional regulation and genome organization. Nucleic Acids Res. 2019, 47, 11481–11496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, M. The Enhanceosome and Transcriptional Synergy. Cell 1998, 92, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Falvo, J.V.; Thanos, D.; Maniatis, T. Reversal of intrinsic DNA bends in the IFNβ gene enhancer by transcription factors and the architectural protein HMG I(Y). Cell 1995, 83, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Merika, M.; Williams, A.J.; Chen, G.; Collins, T.; Thanos, D. Recruitment of CBP/p300 by the IFNβ Enhanceosome Is Required for Synergistic Activation of Transcription. Mol. Cell 1998, 1, 277–287. [Google Scholar] [CrossRef]
- Robson, M.; Ringel, A.R.; Mundlos, S. Regulatory Landscaping: How Enhancer-Promoter Communication Is Sculpted in 3D. Mol. Cell 2019, 74, 1110–1122. [Google Scholar] [CrossRef]
- The Fantom Consortium; Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; et al. An atlas of active en-hancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Fulco, C.P.; Munschauer, M.; Anyoha, R.; Munson, G.; Grossman, S.; Perez, E.; Kane, M.; Cleary, B.; Lander, E.S.; Engreitz, J.M. Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 2016, 354, 769–773. [Google Scholar] [CrossRef] [Green Version]
- Carleton, J.B.; Berrett, K.C.; Gertz, J. Multiplex Enhancer Interference Reveals Collaborative Control of Gene Regulation by Estrogen Receptor α-Bound Enhancers. Cell Syst. 2017, 5, 333–344.e5. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Liu, Y.; Cao, H.; Zhang, Y.; Gu, Z.; Liu, X.; Yu, A.; Kaphle, P.; Dickerson, K.E.; Ni, M.; et al. Interrogation of enhancer function by enhancer-targeting CRISPR ep-igenetic editing. Nat. Commun. 2020, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Frankel, N.; Davis, G.K.; Vargas, D.; Wang, S.; Payre, F.; Stern, D.L. Phenotypic robustness conferred by apparently redun-dant transcriptional enhancers. Nature 2010, 466, 490–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterwalder, M.; Barozzi, I.; Tissières, V.; Fukuda-Yuzawa, Y.; Mannion, B.J.; Afzal, S.Y.; Lee, E.A.; Zhu, Y.; Plajzer-Frick, I.; Pickle, C.S.; et al. Enhancer redundancy pro-vides phenotypic robustness in mammalian development. Nature 2018, 554, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Me-diator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Iden-tity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Dis-ruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.E.; Golan, T.; Sheinboim, D.; Malcov, H.; Amar, D.; Salamon, A.; Liron, T.; Gelfman, S.; Gabet, Y.; Shamir, R.; et al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res. 2016, 26, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryń, M.; Rath, M.; Stark, A. Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef]
- Murtha, M.; Tokcaer-Keskin, Z.; Tang, Z.; Strino, F.; Chen, X.; Wang, Y.; Xi, X.; Basilico, C.; Brown, S.; Bonneau, R.; et al. FIREWACh: High-throughput functional de-tection of transcriptional regulatory modules in mammalian cells. Nat. Methods 2014, 11, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Ashuach, T.; Fischer, D.S.; Kreimer, A.; Ahituv, N.; Theis, F.J.; Yosef, N. MPRAnalyze: Statistical framework for massively parallel reporter assays. Genome Biol. 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinney, J.B.; McCandlish, D.M. Massively Parallel Assays and Quantitative Sequence–Function Relationships. Annu. Rev. Genom. Hum. Genet. 2019, 20, 99–127. [Google Scholar] [CrossRef] [Green Version]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signa-tures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Hallikas, O.; Palin, K.; Sinjushina, N.; Rautiainen, R.; Partanen, J.; Ukkonen, E.; Taipale, J. Genome-wide Prediction of Mammalian Enhancers Based on Analysis of Transcription-Factor Binding Affinity. Cell 2006, 124, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-Resolution Mapping and Characteri-zation of Open Chromatin across the Genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Pennacchio, L.A.; Ahituv, N.; Moses, A.M.; Prabhakar, S.; Nobrega, M.A.; Shoukry, M.; Minovitsky, S.; Dubchak, I.; Holt, A.; Lewis, K.D.; et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 2006, 444, 499–502. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensi-tive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Fullwood, M.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Bin Mohamed, Y.; Orlov, Y.; Velkov, S.; Thoreau, H.; Mei, P.H.; et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, G.; Lopes, R.; Ugalde, A.P.; Nevedomskaya, E.; Han, R.; Myacheva, K.; Zwart, W.; Elkon, R.; Agami, R. Functional genetic screens for en-hancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Li, B.; Meng, Z.; Jung, I.; Lee, A.Y.; Dixon, J.; Maliskova, L.; Guan, K.-L.; Shen, Y.; Ren, B. A new class of temporarily phenotypic enhancers identified by CRISPR/Cas9-mediated genetic screening. Genome Res. 2016, 26, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, N.; Srinivasan, S.; Kooshesh, K.; Guo, Y.; Edwards, M.D.; Banerjee, B.; Syed, T.; Emons, B.J.; Gifford, D.K.; Sherwood, R.I. High-throughput mapping of regu-latory DNA. Nat. Biotechnol. 2016, 34, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, N.J.; Savic, D.; A Nobrega, M. Transcriptional enhancers in development and disease. Genome Biol. 2012, 13, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, S.C.J.; Stitzel, M.L.; Taylor, L.; Orozco, J.M.; Erdos, M.R.; Akiyama, J.A.; van Bueren, K.L.; Chines, P.S.; Narisu, N.; Black, B.; et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. USA 2013, 110, 17921–17926. [Google Scholar] [CrossRef] [Green Version]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Semenkovich, N.P.; Planer, J.D.; Ahern, P.P.; Griffin, N.W.; Lin, C.Y.; Gordon, J.I. Impact of the gut microbiota on enhancer accessibility in gut intraepithelial lymphocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 14805–14810. [Google Scholar] [CrossRef] [Green Version]
- Ascensión, A.M.; Arrospide-Elgarresta, M.; Izeta, A.; Araúzo-Bravo, M.J. NaviSE: Superenhancer navigator integrating epigenomics signal algebra. BMC Bioinform. 2017, 18, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Siersbæk, R.; Rabiee, A.; Nielsen, R.; Sidoli, S.; Traynor, S.; Loft, A.; Poulsen, L.L.; Rogowska-Wrzesinska, A.; Jensen, O.N.; Mandrup, S. Transcription Factor Cooperativity in Early Adipo-genic Hotspots and Super-Enhancers. Cell Rep. 2014, 7, 1443–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, R.C.; Yang, H.; Rockowitz, S.; Larsen, S.; Nikolova, M.; Oristian, D.S.; Polak, L.; Kadaja, M.; Asare, A.; Zheng, D.; et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 2015, 521, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of Cell Identity Genes Occurs in Insu-lated Neighborhoods in Mammalian Chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ing-Simmons, E.; Seitan, V.; Faure, A.; Flicek, P.; Carroll, T.; Dekker, J.; Fisher, A.G.; Lenhard, B.; Merkenschlager, M. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 2015, 25, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Dadon, D.B.; Powell, B.E.; Fan, Z.P.; Borges-Rivera, D.; Shachar, S.; Weintraub, A.S.; Hnisz, D.; Pegoraro, G.; Lee, T.I.; et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 2015, 18, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.-K.; Jang, Y.J.; Kim, M.; Leblanc, L.; Rhee, C.; Lee, J.; Beck, S.; Shen, W.; Kim, J. Super-enhancer-guided mapping of regulatory networks controlling mouse trophoblast stem cells. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; George, R.E. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Bojcsuk, D.; Nagy, G.; Balint, B.L. Inducible super-enhancers are organized based on canonical signal-specific transcription factor binding elements. Nucleic Acids Res. 2016, 45, 3693–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, J.J.; Bayles, I.; Funnell, A.P.; Miller, T.E.; Saiakhova, A.; Lizardo, M.M.; Bartels, C.F.; Kapteijn, M.Y.; Hung, S.; Mendoza, A.; et al. Positively selected enhancer ele-ments endow osteosarcoma cells with metastatic competence. Nat. Med. 2018, 24, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xu, C.; Du, Y.; Junaid, M.; Kaushik, A.; Wei, D. Comprehensive epigenetic analyses reveal master regulators driving lung metastasis of breast cancer. J. Cell. Mol. Med. 2019, 23, 5415–5431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, W.F.; Xing, M.; Xu, C.; Yao, X.; Ramlee, M.K.; Lim, M.C.; Cao, F.; Lim, K.; Babu, D.; Poon, L.-F.; et al. Epigenomic profiling of primary gastric adenocarcinoma reveals super-enhancer heterogeneity. Nat. Commun. 2016, 7, 12983. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Erkek, S.; Tong, Y.; Yin, L.; Federation, A.J.; Zapatka, M.; Haldipur, P.; Kawauchi, D.; Risch, T.; Warnatz, H.J.; et al. Active medulloblastoma enhancers reveal sub-group-specific cellular origins. Nature 2016, 530, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; A Schnell, S.; Belver, L.; A Wendorff, A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.-S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [Green Version]
- Kubota, S.; Tokunaga, K.; Umezu, T.; Yokomizo-Nakano, T.; Sun, Y.; Oshima, M.; Tan, K.T.; Yang, H.; Kanai, A.; Iwanaga, E.; et al. Lineage-specific RUNX2 su-per-enhancer activates MYC and promotes the development of blastic plasmacytoid dendritic cell neoplasm. Nat. Commun. 2019, 10, 1653. [Google Scholar] [CrossRef] [Green Version]
- Schuijers, J.; Manteiga, J.C.; Weintraub, A.S.; Day, D.S.; Zamudio, A.V.; Hnisz, D.; Lee, T.I.; Young, R.A. Transcriptional Dysregulation of MYC Reveals Common Enhancer-Docking Mechanism. Cell Rep. 2018, 23, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, F.H.; Johnsen, S.A. DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc. Natl. Acad. Sci. USA 2018, 115, E12343–E12352. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Choi, P.; Francis, J.M.; Gao, G.; Campbell, J.D.; Ramachandran, A.; Mitsuishi, Y.; Ha, G.; Shih, J.; Vazquez, F.; et al. Somatic Superenhancer Duplications and Hotspot Mutations Lead to Oncogenic Activation of the KLF5 Transcription Factor. Cancer Discov. 2017, 8, 108–125. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. An oncogenic su-per-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef] [Green Version]
- Gröschel, S.; Sanders, M.A.; Hoogenboezem, R.; De Wit, E.; Bouwman, B.A.; Erpelinck, C.; Van Der Velden, V.H.; Havermans, M.; Avellino, R.; Van Lom, K.; et al. A Single Oncogenic En-hancer Rearrangement Causes Concomitant EVI1 and GATA2 Deregulation in Leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.; Molenaar, J.J.; et al. TERT rearrangements are fre-quent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Dubash, T.; Drainas, A.P.; Mardin, B.R.; Chen, Y.; Stütz, A.M.; Waszak, S.M.; Bosco, G.; Halvorsen, A.R.; Raeder, B.; et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 2016, 49, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stütz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.H.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef]
- Dukler, N.; Gulko, B.; Huang, Y.-F.; Siepel, A. Is a super-enhancer greater than the sum of its parts? Nat. Genet. 2017, 49, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, X.; Li, D.; Shao, Z.; Cao, H.; Zhang, Y.; Trompouki, E.; Bowman, T.V.; Zon, L.I.; Yuan, G.-C.; et al. Dynamic Control of Enhancer Repertoires Drives Lineage and Stage-Specific Transcription during Hematopoiesis. Dev. Cell 2016, 36, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, D.; Hughes, J.R.; Babbs, C.; Davies, J.O.; Graham, B.J.; Hanssen, L.L.; Kassouf, M.T.; Oudelaar, A.M.; Sharpe, J.A.; Suciu, M.C.; et al. Genetic dissection of the α-globin su-per-enhancer in vivo. Nat. Genet. 2016, 48, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y.; Willi, M.; Yoo, K.H.; Zeng, X.; Wang, C.; Metser, G.; Hennighausen, L. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 2016, 48, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.C. Dissecting super-enhancer hierarchy based on chromatin in-teractions. Nat. Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [Green Version]
- Moorthy, S.D.; Davidson, S.; Shchuka, V.M.; Singh, G.; Malek-Gilani, N.; Langroudi, L.; Martchenko, A.; So, V.; Macpherson, N.N.; Mitchell, J.A. Enhancers and su-per-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res. 2017, 27, 246–258. [Google Scholar] [CrossRef]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive Promoter-Centered Chromatin Interac-tions Provide a Topological Basis for Transcription Regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzaskoma, P.; Ruszczycki, B.; Lee, B.; Pels, K.K.; Krawczyk, K.; Bokota, G.; Szczepankiewicz, A.A.; Aaron, J.; Walczak, A.; Śliwińska, M.A.; et al. Ultrastructural visualization of 3D chroma-tin folding using volume electron microscopy and DNA in situ hybridization. Nat. Commun. 2020, 11, 2120. [Google Scholar] [CrossRef]
- Sancisi, V.; Manzotti, G.; Gugnoni, M.; Rossi, T.; Gandolfi, G.; Gobbi, G.; Torricelli, F.; Catellani, F.; Faria do Valle, I.; Remondini, D.; et al. RUNX2 expression in thyroid and breast can-cer requires the cooperation of three non-redundant enhancers under the control of BRD4 and c-JUN. Nucleic Acids Res. 2017, 45, 11249–11267. [Google Scholar] [CrossRef]
- Gertz, J.; Reddy, T.E.; Varley, K.E.; Garabedian, M.J.; Myers, R.M. Genistein and bisphenol A exposure cause estrogen recep-tor 1 to bind thousands of sites in a cell type-specific manner. Genome Res. 2012, 22, 2153–2162. [Google Scholar] [CrossRef] [Green Version]
- Gertz, J.; Savic, D.; Varley, K.; Partridge, E.C.; Safi, A.; Jain, P.; Cooper, G.M.; Reddy, T.; Crawford, G.E.; Myers, R.M. Distinct Properties of Cell-Type-Specific and Shared Transcription Factor Binding Sites. Mol. Cell 2013, 52, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.; Doudna, J.A.; Weissman, J.S.; Arkin, A.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regula-tion of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Will, A.J.; Cova, G.; Osterwalder, M.; Chan, W.-L.; Wittler, L.; Brieske, N.; Heinrich, V.; de Villartay, J.; Vingron, M.; Klopocki, E.; et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat. Genet. 2017, 49, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Montavon, T.; Soshnikova, N.; Mascrez, B.; Joye, E.; Thevenet, L.; Splinter, E.; de Laat, W.; Spitz, F.; Duboule, D. A Regulatory Archipelago Controls Hox Genes Transcription in Digits. Cell 2011, 147, 1132–1145. [Google Scholar] [CrossRef] [Green Version]
- Choi, O.-R.B.; Engel, J.D. Developmental regulation of β-globin gene switching. Cell 1988, 55, 17–26. [Google Scholar] [CrossRef]
- Ohtsuki, S.; Levine, M.; Cai, H.N. Different core promoters possess distinct regulatory activities in the Drosophila embryo. Genes Dev. 1998, 12, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagha, M.; Bothma, J.P.; Levine, M. Mechanisms of transcriptional precision in animal development. Trends Genet. 2012, 28, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.-W.; Hendrix, D.A.; Levine, M.S. Shadow Enhancers as a Source of Evolutionary Novelty. Science 2008, 321, 1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, M.; Boettiger, A.; Bothma, J.P.; Levine, M. Shadow Enhancers Foster Robustness of Drosophila Gastrulation. Curr. Biol. 2010, 20, 1562–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobert, O. Gene Regulation: Enhancers Stepping Out of the Shadow. Curr. Biol. 2010, 20, R697–R699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, D.D.; de Souza, F.S.; Nasif, S.; Yamashita, M.; López-Leal, R.; Otero-Corchon, V.; Meece, K.; Sampath, H.; Mercer, A.J.; Wardlaw, S.L.; et al. Partially Redundant Enhanc-ers Cooperatively Maintain Mammalian Pomc Expression Above a Critical Functional Threshold. Barsh, G.S., editor. PLoS Genet. 2015, 11, e1004935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, Z.; Bragdon, M.D.J.; Vincent, B.J.; White, J.A.; Estrada, J.; DePace, A.H. Krüppel Expression Levels Are Main-tained through Compensatory Evolution of Shadow Enhancers. Cell Rep. 2015, 12, 1740–1747. [Google Scholar] [CrossRef] [Green Version]
- Waymack, R.; Fletcher, A.; Enciso, G.; Wunderlich, Z. Shadow enhancers can suppress input transcription factor noise through distinct regulatory logic. eLife 2020, 9. [Google Scholar] [CrossRef]
- McGreal-Estrada, R.S.; Wolf, L.V.; Cvekl, A. Promoter-enhancer looping and shadow enhancers of the mouse αA-crystallin locus. Biol. Open 2018, 7, bio036897. [Google Scholar] [CrossRef] [Green Version]
- Ghiasvand, N.M.; Rudolph, D.D.; Mashayekhi, M.; Iv, J.A.B.; Goldman, D.; Glaser, T. Deletion of a remote enhancer near ATOH7 disrupts retinal neurogenesis, causing NCRNA disease. Nat. Neurosci. 2011, 14, 578–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte, C.; Jinks, T.; Wang, X.; Martinez Pastor, M.T.; Krumlauf, R. Shadow enhancers flanking the HoxB cluster direct dy-namic Hox expression in early heart and endoderm development. Dev. Biol. 2013, 383, 158–173. [Google Scholar] [CrossRef] [Green Version]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline Mutations inHOXB13and Prostate-Cancer Risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, H.; Lee, J.-Y.; Yun, H.J.; Park, B.-W.; Kim, M.H. Analysis of HOX Gene Expression Patterns in Human Breast Cancer. Mol. Biotechnol. 2013, 56, 64–71. [Google Scholar] [CrossRef]
- Ooki, A.; Dinalankara, W.; Marchionni, L.; Tsay, J.-C.; Goparaju, C.; Maleki, Z.; Rom, W.; Pass, H.; Hoque, M.O. Epigenetically regulated PAX6 drives cancer cells toward a stem-like state via GLI-SOX2 signaling axis in lung adenocarcinoma. Oncogene 2018, 37, 5967–5981. [Google Scholar] [CrossRef]
- Antosova, B.; Smolikova, J.; Klimova, L.; Lachova, J.; Bendova, M.; Kozmikova, I.; Machon, O.; Kozmik, Z. The Gene Regulatory Network of Lens Induction Is Wired through Meis-Dependent Shadow Enhancers of Pax6. PLoS Genet. 2016, 12, e1006441. [Google Scholar] [CrossRef] [Green Version]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bothma, J.P.; Garcia, H.G.; Ng, S.; Perry, M.W.; Gregor, T.; Levine, M. Enhancer additivity and non-additivity are deter-mined by enhancer strength in the Drosophila embryo. ELife 2015, 4, e07956. [Google Scholar] [CrossRef]
- El-Sherif, E.; Levine, M. Shadow Enhancers Mediate Dynamic Shifts of Gap Gene Expression in the Drosophila Embryo. Curr. Biol. 2016, 26, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Scholes, C.; Biette, K.M.; Harden, T.T.; DePace, A.H. Signal Integration by Shadow Enhancers and Enhancer Duplications Varies across the Drosophila Embryo. Cell Rep. 2019, 26, 2407–2418.e5. [Google Scholar] [CrossRef] [Green Version]
- Plank, J.L.; Dean, A. Enhancer Function: Mechanistic and Genome-Wide Insights Come Together. Mol. Cell 2014, 55, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.J.; Yang, L.; Meluzzi, D.; Oh, S.; Yang, F.; Friedman, M.J.; Wang, S.; Suter, T.; Alshareedah, I.; Gamliel, A.; et al. Phase separation of ligand-activated enhancers li-censes cooperative chromosomal enhancer assembly. Nat. Struct. Mol. Biol. 2019, 26, 193–203. [Google Scholar] [CrossRef]
- Cramer, P. Organization and regulation of gene transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at su-per-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.-K.; Spille, J.-H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitale, E.; Gugnoni, M.; Ciarrocchi, A. No Need to Stick Together to Be Connected: Multiple Types of Enhancers’ Networking. Cancers 2021, 13, 5201. https://doi.org/10.3390/cancers13205201
Vitale E, Gugnoni M, Ciarrocchi A. No Need to Stick Together to Be Connected: Multiple Types of Enhancers’ Networking. Cancers. 2021; 13(20):5201. https://doi.org/10.3390/cancers13205201
Chicago/Turabian StyleVitale, Emanuele, Mila Gugnoni, and Alessia Ciarrocchi. 2021. "No Need to Stick Together to Be Connected: Multiple Types of Enhancers’ Networking" Cancers 13, no. 20: 5201. https://doi.org/10.3390/cancers13205201
APA StyleVitale, E., Gugnoni, M., & Ciarrocchi, A. (2021). No Need to Stick Together to Be Connected: Multiple Types of Enhancers’ Networking. Cancers, 13(20), 5201. https://doi.org/10.3390/cancers13205201