
The Roles of Diacylglycerol Kinase α in Cancer Cell Proliferation and Apoptosis


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Structure, Enzymatic Properties, and Activation Mechanisms of DGKα
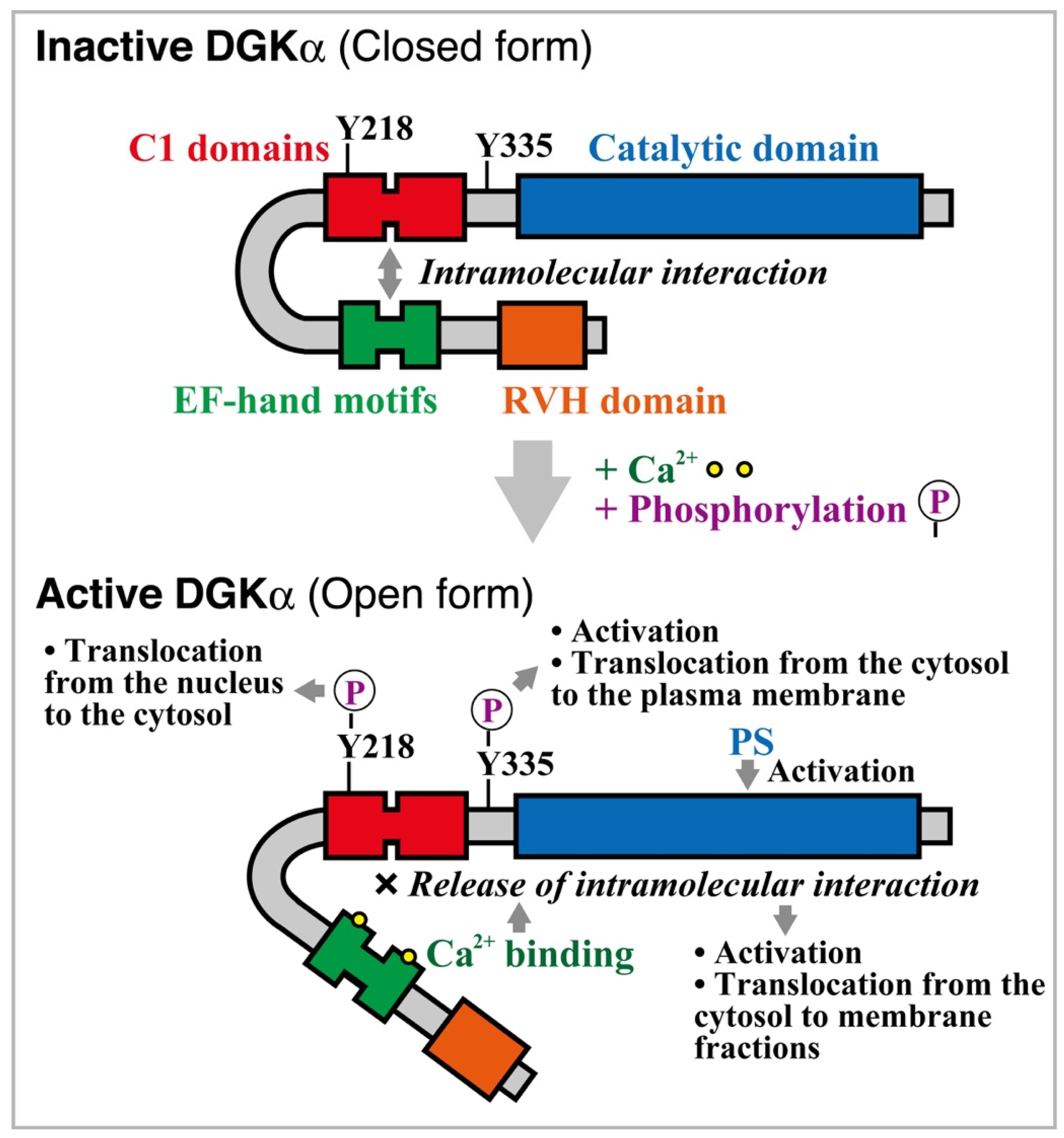
2.1. Structure
2.2. Enzymological Properties
2.3. Regulation of Activity and Subcellular Localization
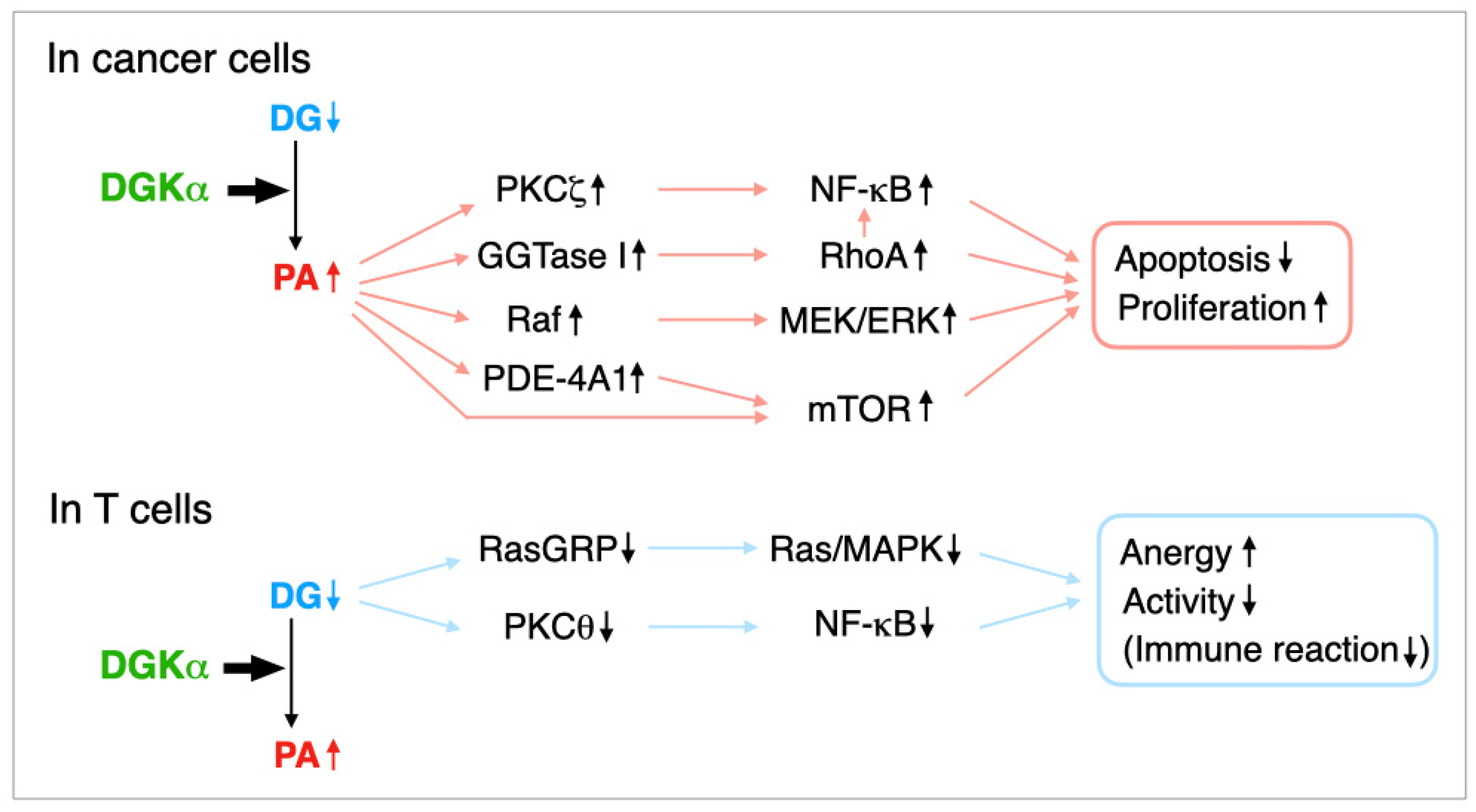
3. Regulation of Cancer Cell Proliferation and Apoptosis by DGKα
4. Regulation of T Cell Receptor Signaling by DGKα
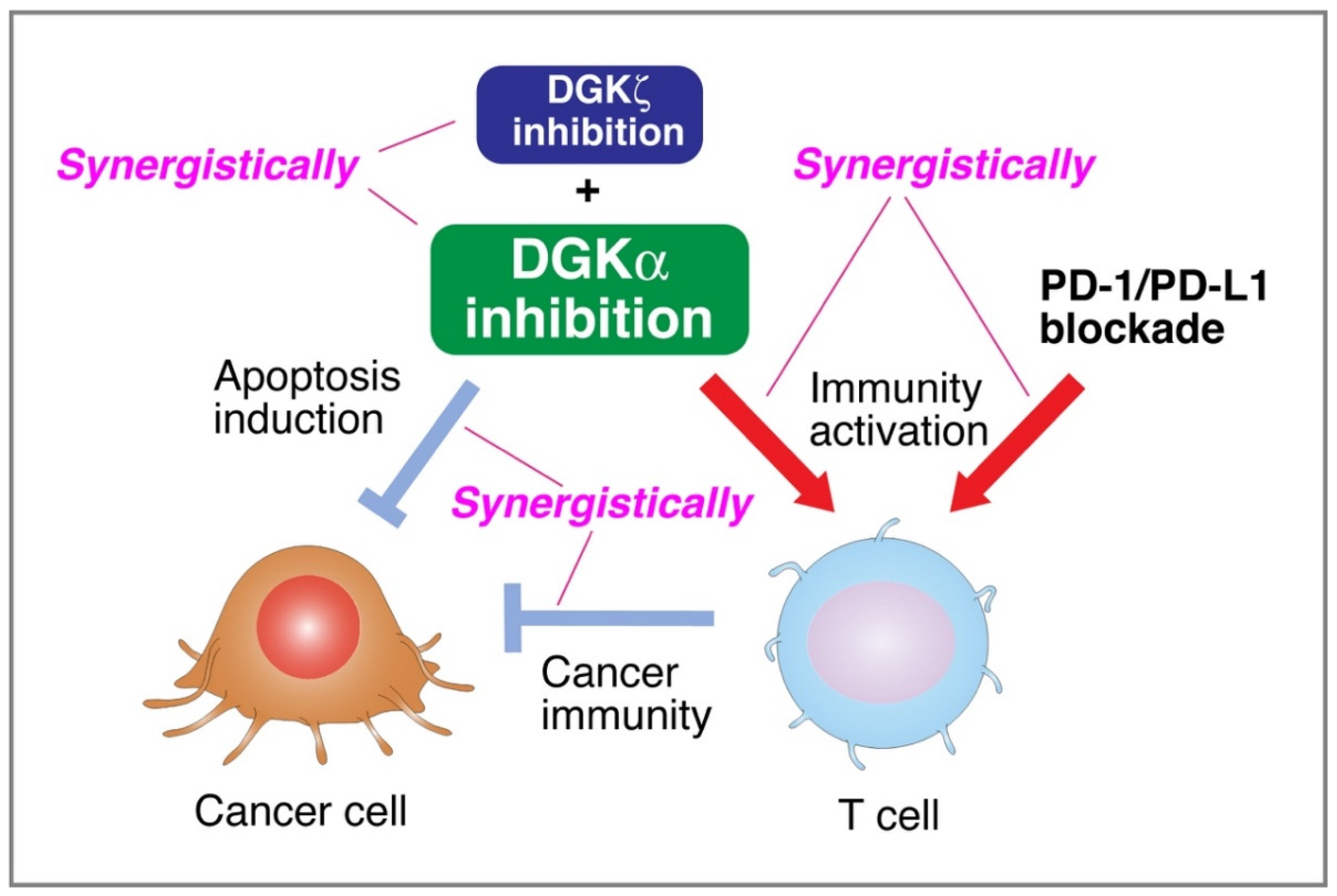
5. DGKα Inhibitors Simultaneously Attenuate Cancer Cell Proliferation and Activate T Cell Function
5.1. DGKα Inhibitors Simultaneously Attenuate Cancer Cell Proliferation and Activate T Cell Function
5.2. Synergistic Effect of PD-1/PD-L1 Blockade
5.3. Synergistic Effects of DGKα- and DGKζ-Inhibitions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Goto, K.; Hozumi, Y.; Kondo, H. Diacylglycerol, phosphatidic acid, and the converting enzyme, diacylglycerol kinase, in the nucleus. Biochim. Biophys. Acta 2006, 1761, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Merida, I.; Avila-Flores, A.; Merino, E. Diacylglycerol kinases: At the hub of cell signalling. Biochem. J. 2008, 409, 1–18. [Google Scholar] [CrossRef]
- Sakane, F.; Imai, S.; Kai, M.; Yasuda, S.; Kanoh, H. Diacylglycerol kinases: Why so many of them? Biochim. Biophys. Acta 2007, 1771, 793–806. [Google Scholar] [CrossRef]
- Sakane, F.; Mizuno, S.; Takahashi, D.; Sakai, H. Where do substrates of diacylglycerol kinases come from? Diacylglycerol kinases utilize diacylglycerol species supplied from phosphatidylinositol turnover-independent pathways. Adv. Biol. Regul. 2018, 67, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Topham, M.K.; Epand, R.M. Mammalian diacylglycerol kinases: Molecular interactions and biological functions of selected isoforms. Biochim. Biophys. Acta 2009, 1790, 416–424. [Google Scholar] [CrossRef]
- Sakane, F.; Yamada, K.; Kanoh, H.; Yokoyama, C.; Tanabe, T. Porcine diacylglycerol kinase sequence has zinc finger and E-F hand motifs. Nature 1990, 344, 345–348. [Google Scholar] [CrossRef]
- Schaap, D.; de Widt, J.; van der Wal, J.; Vandekerckhove, J.; van Damme, J.; Gussow, D.; Ploegh, H.L.; van Blitterswijk, W.J.; van der Bend, R.L. Purification, cDNA-cloning and expression of human diacylglycerol kinase. FEBS Lett. 1990, 275, 151–158. [Google Scholar] [CrossRef]
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; Yacur, M.N.; Abounader, R.; Lee, J.K.; Wilson, G.M.; et al. Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013, 3, 782–797. [Google Scholar] [CrossRef]
- Takeishi, K.; Taketomi, A.; Shirabe, K.; Toshima, T.; Motomura, T.; Ikegami, T.; Yoshizumi, T.; Sakane, F.; Maehara, Y. Diacylglycerol kinase alpha enhances hepatocellular carcinoma progression by activation of Ras-Raf-MEK-ERK pathway. J. Hepatol. 2012, 57, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Torres-Ayuso, P.; Daza-Martin, M.; Martin-Perez, J.; Avila-Flores, A.; Merida, I. Diacylglycerol kinase α promotes 3D cancer cell growth and limits drug sensitivity through functional interaction with Src. Oncotarget 2014, 5, 9710–9726. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Yasuda, S.; Kai, M.; Imai, S.; Yamada, K.; Yamashita, T.; Jimbow, K.; Kanoh, H.; Sakane, F. Diacylglycerol kinase α suppresses tumor necrosis factor-α-induced apoptosis of human melanoma cells through NF-κB activation. Biochim. Biophys. Acta 2007, 1771, 462–474. [Google Scholar] [CrossRef]
- Olenchock, B.A.; Guo, R.; Carpenter, J.H.; Jordan, M.; Topham, M.K.; Koretzky, G.A.; Zhong, X.P. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol. 2006, 7, 1174–1181. [Google Scholar] [CrossRef]
- Zha, Y.; Marks, R.; Ho, A.W.; Peterson, A.C.; Janardhan, S.; Brown, I.; Praveen, K.; Stang, S.; Stone, J.C.; Gajewski, T.F. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-α. Nat. Immunol. 2006, 7, 1166–1173. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, W.; Hawes, J.W.; Walsh, J.P. A domain with homology to neuronal calcium sensors is required for calcium-dependent activation of diacylglycerol kinase alpha. J. Biol. Chem. 2000, 275, 34092–34099. [Google Scholar] [CrossRef]
- Sakane, F.; Kai, M.; Wada, I.; Imai, S.; Kanoh, H. The C-terminal part of diacylglycerol kinase α lacking zinc fingers serves as a catalytic domain. Biochem. J. 1996, 318, 583–590. [Google Scholar] [CrossRef]
- Sakane, F.; Yamada, K.; Imai, S.; Kanoh, H. Porcine 80-kDa diacylglycerol kinase is a calcium-binding and calcium/phospholipid-dependent enzyme and undergoes calcium-dependent translocation. J. Biol. Chem. 1991, 266, 7096–7100. [Google Scholar] [CrossRef]
- Yamada, K.; Sakane, F.; Matsushima, N.; Kanoh, H. EF-hand motifs of α, β and γ isoforms of diacylglycerol kinase bind calcium with different affinities and conformational changes. Biochem. J. 1997, 321, 59–64. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Simons, T.J. Calcium and neuronal function. Neurosurg. Rev. 1988, 11, 119–129. [Google Scholar] [CrossRef]
- Kazanietz, M.G. Novel “nonkinase” phorbol ester receptors: The C1 domain connection. Mol. Pharmacol. 2002, 61, 759–767. [Google Scholar] [CrossRef]
- Nishizuka, Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984, 308, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Lu, X.; Jiang, Y.; Boccone, C.E.; Qian, S.; Vattem, K.M.; Wek, R.C.; Walsh, J.P. Site-directed mutagenesis of the active site of diacylglycerol kinase alpha: Calcium and phosphatidylserine stimulate enzyme activity via distinct mechanisms. Biochem. J. 2003, 375, 673–680. [Google Scholar] [CrossRef]
- Miller, D.J.; Jerga, A.; Rock, C.O.; White, S.W. Analysis of the Staphylococcus aureus DgkB structure reveals a common catalytic mechanism for the soluble diacylglycerol kinases. Structure 2008, 16, 1036–1046. [Google Scholar] [CrossRef]
- Sugiura, M.; Kono, K.; Liu, H.; Shimizugawa, T.; Minekura, H.; Spiegel, S.; Kohama, T. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J. Biol. Chem. 2002, 277, 23294–23300. [Google Scholar] [CrossRef] [PubMed]
- Komenoi, S.; Takemura, F.; Sakai, H.; Sakane, F. Diacylglycerol kinase eta1 is a high affinity isozyme for diacylglycerol. FEBS Lett. 2015, 589, 1272–1277. [Google Scholar] [CrossRef]
- Takahashi, D.; Sakane, F. Expression and purification of human diacylglycerol kinase alpha from baculovirus-infected insect cells for structural studies. PeerJ 2018, 6, e5449. [Google Scholar] [CrossRef]
- Sato, Y.; Murakami, C.; Yamaki, A.; Mizuno, S.; Sakai, H.; Sakane, F. Distinct 1-monoacylglycerol and 2-monoacylglycerol kinase activities of diacylglycerol kinase isozymes. Biochim. Biophys. Acta 2016, 1864, 1170–1176. [Google Scholar] [CrossRef]
- Marchan, R.; Buttner, B.; Lambert, J.; Edlund, K.; Glaeser, I.; Blaszkewicz, M.; Leonhardt, G.; Marienhoff, L.; Kaszta, D.; Anft, M.; et al. Glycerol-3-phosphate Acyltransferase 1 Promotes Tumor Cell Migration and Poor Survival in Ovarian Carcinoma. Cancer Res. 2017, 77, 4589–4601. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kunii, N.; Sakuma, M.; Yamaki, A.; Mizuno, S.; Sato, M.; Sakai, H.; Kado, S.; Kumagai, K.; Kojima, H.; et al. A novel diacylglycerol kinase α-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J. Lipid Res. 2016, 57, 368–379. [Google Scholar] [CrossRef]
- Murakami, Y.; Murakami, C.; Hoshino, F.; Lu, Q.; Akiyama, R.; Yamaki, A.; Takahashi, D.; Sakane, F. Palmitic acid- and/or palmitoleic acid-containing phosphatidic acids are generated by diacylglycerol kinase α in starved Jurkat T cells. Biochem. Biophys. Res. Commun. 2020, 525, 1054–1060. [Google Scholar] [CrossRef]
- Yamaki, A.; Akiyama, R.; Murakami, C.; Takao, S.; Murakami, Y.; Mizuno, S.; Takahashi, D.; Kado, S.; Taketomi, A.; Shirai, Y.; et al. Diacylglycerol kinase alpha-selective inhibitors induce apoptosis and reduce viability of melanoma and several other cancer cell lines. J. Cell Biochem. 2019, 120, 10043–10056. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef]
- Macara, I.G. Oncogenes, ions, and phospholipids. Am. J. Physiol. 1985, 248, C3–C11. [Google Scholar] [CrossRef]
- Komenoi, S.; Suzuki, Y.; Asami, M.; Murakami, C.; Hoshino, F.; Chiba, S.; Takahashi, D.; Kado, S.; Sakane, F. Microarray analysis of gene expression in the diacylglycerol kinase η knockout mouse brain. Biochem. Biophys. Rep. 2019, 19, 100660. [Google Scholar] [CrossRef]
- Lu, Q.; Murakami, C.; Murakami, Y.; Hoshino, F.; Asami, M.; Usuki, T.; Sakai, H.; Sakane, F. 1-Stearoyl-2-docosahexaenoyl-phosphatidic acid interacts with and activates Praja-1, the E3 ubiquitin ligase acting on the serotonin transporter in the brain. FEBS Lett. 2020, 594, 1787–1796. [Google Scholar] [CrossRef]
- Mizuno, S.; Kado, S.; Goto, K.; Takahashi, D.; Sakane, F. Diacylglycerol kinase ζ generates dipalmitoyl-phosphatidic acid species during neuroblastoma cell differentiation. Biochem. Biophys. Rep. 2016, 8, 352–359. [Google Scholar]
- Murakami, C.; Hoshino, F.; Sakai, H.; Hayashi, Y.; Yamashita, A.; Sakane, F. Diacylglycerol kinase delta and sphingomyelin synthase-related protein functionally interact via their sterile alpha motif domains. J. Biol. Chem. 2020, 295, 2932–2947. [Google Scholar] [CrossRef]
- Murakami, C.; Sakane, F. Sphingomyelin synthase-related protein generates diacylglycerol via the hydrolysis of glycerophospholipids in the absence of ceramide. J. Biol. Chem. 2021, 296, 100454. [Google Scholar] [CrossRef]
- Sakai, H.; Kado, S.; Taketomi, A.; Sakane, F. Diacylglycerol kinase δ phosphorylates phosphatidylcholine-specific phospholipase C-dependent, palmitic acid-containing diacylglycerol species in response to high glucose levels. J. Biol. Chem. 2014, 289, 26607–26617. [Google Scholar] [CrossRef]
- Sakane, F.; Hoshino, F.; Murakami, C. New Era of Diacylglycerol Kinase, Phosphatidic Acid and Phosphatidic Acid-Binding Protein. Int. J. Mol. Sci. 2020, 21, 6794. [Google Scholar] [CrossRef]
- Bozelli, J.C., Jr.; Yune, J.; Takahashi, D.; Sakane, F.; Epand, R.M. Membrane morphology determines diacylglycerol kinase alpha substrate acyl chain specificity. FASEB J. 2021, 35, e21602. [Google Scholar] [CrossRef]
- Ware, T.B.; Franks, C.E.; Granade, M.E.; Zhang, M.; Kim, K.B.; Park, K.S.; Gahlmann, A.; Harris, T.E.; Hsu, K.L. Reprogramming fatty acyl specificity of lipid kinases via C1 domain engineering. Nat. Chem. Biol. 2020, 16, 170–178. [Google Scholar] [CrossRef]
- Sakane, F.; Imai, S.; Yamada, K.; Kanoh, H. The regulatory role of EF-hand motifs of pig 80K diacylglycerol kinase as assessed using truncation and deletion mutants. Biochem. Biophys. Res. Commun. 1991, 181, 1015–1021. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamamoto, T.; Sakai, H.; Sakane, F. Calcium negatively regulates an intramolecular interaction between the N-terminal recoverin homology and EF-hand motif domains and the C-terminal C1 and catalytic domains of diacylglycerol kinase α. Biochem. Biophys. Res. Commun. 2012, 423, 571–576. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sakai, H.; Sakane, F. EF-hand motifs of diacylglycerol kinase α interact intra-molecularly with its C1 domains. FEBS Open Bio. 2014, 4, 387–392. [Google Scholar] [CrossRef]
- Takahashi, D.; Suzuki, K.; Sakamoto, T.; Iwamoto, T.; Murata, T.; Sakane, F. Crystal structure and calcium-induced conformational changes of diacylglycerol kinase alpha EF-hand domains. Protein Sci. A Publ. Protein Soc. 2019, 28, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.; Parker, P.J. The extended protein kinase C superfamily. Biochem. J. 1998, 332 Pt 2, 281–292. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [PubMed]
- Cipres, A.; Carrasco, S.; Merino, E.; Diaz, E.; Krishna, U.M.; Falck, J.R.; Martinez, A.C.; Merida, I. Regulation of diacylglycerol kinase α by phosphoinositide 3-kinase lipid products. J. Biol. Chem. 2003, 278, 35629–35635. [Google Scholar] [CrossRef] [PubMed]
- Sakane, F.; Yamada, K.; Kanoh, H. Different effects of sphingosine, R59022 and anionic amphiphiles on two diacylglycerol kinase isozymes purified from porcine thymus cytosol. FEBS Lett. 1989, 255, 409–413. [Google Scholar] [CrossRef]
- Yamada, K.; Sakane, F. The different effect of sphingosine on diacylglycerol kinase iszymes in Jurkat cells, a human T-cell line. Biochim. Biophys. Acta 1993, 1169, 211–216. [Google Scholar]
- Yamada, K.; Sakane, F.; Imai, S.; Takamura, H. Sphingosine activates cellular diacylglycerol kinase in intact Jurkat cells, a human T-cell line. Biochim. Biophys. Acta 1993, 1169, 217–224. [Google Scholar] [PubMed]
- Sanjuan, M.A.; Pradet-Balade, B.; Jones, D.R.; Martinez, A.C.; Stone, J.C.; Garcia-Sanz, J.A.; Merida, I. T cell activation in vivo targets diacylglycerol kinase alpha to the membrane: A novel mechanism for Ras attenuation. J. Immunol. 2003, 170, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Merino, E.; Sanjuan, M.A.; Moraga, I.; Cipres, A.; Merida, I. Role of the diacylglycerol kinase alpha-conserved domains in membrane targeting in intact T cells. J. Biol. Chem. 2007, 282, 35396–35404. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga-Takenaka, R.; Shirai, Y.; Yagi, K.; Adachi, N.; Sakai, N.; Merino, E.; Merida, I.; Saito, N. Importance of chroman ring and tyrosine phosphorylation in the subtype-specific translocation and activation of diacylglycerol kinase alpha by D-alpha-tocopherol. Genes Cells 2005, 10, 311–319. [Google Scholar] [CrossRef]
- Hayashi, D.; Wang, L.Q.; Ueda, S.; Yamanoue, M.; Ashida, H.; Shirai, Y. The mechanisms of ameliorating effect of a green tea polyphenol on diabetic nephropathy based on diacylglycerol kinase alpha. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Hayashi, D.; Ueda, S.; Yamanoue, M.; Saito, N.; Ashida, H.; Shirai, Y. Epigallocatechin-3-gallate activates diacylglycerol kinase alpha via a 67 kDa laminin receptor: A possibility of galloylated catechins as functional food to prevent and/or improve diabetic renal dysfunctions. J. Funct. Foods 2015, 15, 561–569. [Google Scholar] [CrossRef]
- Matsubara, T.; Ikeda, M.; Kiso, Y.; Sakuma, M.; Yoshino, K.; Sakane, F.; Merida, I.; Saito, N.; Shirai, Y. c-Abl Tyrosine Kinase Regulates Serum-induced Nuclear Export of Diacylglycerol Kinase α by Phosphorylation at Tyr-218. J. Biol. Chem. 2012, 287, 5507–5517. [Google Scholar] [CrossRef]
- Kai, M.; Yasuda, S.; Imai, S.; Toyota, M.; Kanoh, H.; Sakane, F. Diacylglycerol kinase α enhances protein kinase Cζ-dependent phosphorylation at Ser311 of p65/RelA subunit of nuclear factor-κB. FEBS Lett. 2009, 583, 3265–3268. [Google Scholar] [CrossRef]
- Olmez, I.; Love, S.; Xiao, A.; Manigat, L.; Randolph, P.; McKenna, B.D.; Neal, B.P.; Boroda, S.; Li, M.; Brenneman, B.; et al. Targeting the mesenchymal subtype in glioblastoma and other cancers via inhibition of diacylglycerol kinase alpha. Neuro Oncol. 2018, 20, 192–202. [Google Scholar] [CrossRef]
- Perez, Y.; Maffei, M.; Igea, A.; Amata, I.; Gairi, M.; Nebreda, A.R.; Bernado, P.; Pons, M. Lipid binding by the Unique and SH3 domains of c-Src suggests a new regulatory mechanism. Sci. Rep. 2013, 3, 1295. [Google Scholar] [CrossRef]
- Rainero, E.; Cianflone, C.; Porporato, P.E.; Chianale, F.; Malacarne, V.; Bettio, V.; Ruffo, E.; Ferrara, M.; Benecchia, F.; Capello, D.; et al. The diacylglycerol kinase alpha/atypical PKC/beta1 integrin pathway in SDF-1alpha mammary carcinoma invasiveness. PLoS ONE 2014, 9, e97144. [Google Scholar] [CrossRef]
- Poli, A.; Fiume, R.; Baldanzi, G.; Capello, D.; Ratti, S.; Gesi, M.; Manzoli, L.; Graziani, A.; Suh, P.G.; Cocco, L.; et al. Nuclear Localization of Diacylglycerol Kinase Alpha in K562 Cells Is Involved in Cell Cycle Progression. J. Cell. Physiol. 2017, 232, 2550–2557. [Google Scholar] [CrossRef]
- Kefas, B.; Floyd, D.H.; Comeau, L.; Frisbee, A.; Dominguez, C.; Dipierro, C.G.; Guessous, F.; Abounader, R.; Purow, B. A miR-297/hypoxia/DGK-alpha axis regulating glioblastoma survival. Neuro. Oncol. 2013, 15, 1652–1663. [Google Scholar] [CrossRef]
- Limatola, C.; Schaap, D.; Moolenaar, W.H.; van Blitterswijk, W.J. Phosphatidic acid activation of protein kinase C zeta overexpressed in COS cells: Comparison with other protein kinase C isotypes and other acidic lipids. Biochem. J. 1994, 304, 1001–1008. [Google Scholar] [CrossRef]
- Ghosh, S.; Moore, S.; Bell, R.M.; Dush, M. Functional analysis of a phosphatidic acid binding domain in human Raf-1 kinase: Mutations in the phosphatidate binding domain lead to tail and trunk abnormalities in developing zebrafish embryos. J. Biol. Chem. 2003, 278, 45690–45696. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Strum, J.C.; Sciorra, V.A.; Daniel, L.; Bell, R.M. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem. 1996, 271, 8472–8480. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.A.; Shome, K.; Watkins, S.C.; Romero, G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J. Biol. Chem. 2000, 275, 23911–23918. [Google Scholar] [CrossRef]
- Baillie, G.S.; Huston, E.; Scotland, G.; Hodgkin, M.; Gall, I.; Peden, A.H.; MacKenzie, C.; Houslay, E.S.; Currie, R.; Pettitt, T.R.; et al. TAPAS-1, a novel microdomain within the unique N-terminal region of the PDE4A1 cAMP-specific phosphodiesterase that allows rapid, Ca2+-triggered membrane association with selectivity for interaction with phosphatidic acid. J. Biol. Chem. 2002, 277, 28298–28309. [Google Scholar] [CrossRef]
- Kassas, N.; Tanguy, E.; Thahouly, T.; Fouillen, L.; Heintz, D.; Chasserot-Golaz, S.; Bader, M.F.; Grant, N.J.; Vitale, N. Comparative Characterization of Phosphatidic Acid Sensors and Their Localization during Frustrated Phagocytosis. J. Biol. Chem. 2017, 292, 4266–4279. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Sakane, F.; Kanoh, H. Immunoquantitation of 80 kDa diacylglycerol kinase in pig and human lymphocytes and several other cells. FEBS Lett. 1989, 244, 402–406. [Google Scholar] [CrossRef]
- Foell, J.; Hewes, B.; Mittler, R.S. T cell costimulatory and inhibitory receptors as therapeutic targets for inducing anti-tumor immunity. Curr. Cancer Drug Targets 2007, 7, 55–70. [Google Scholar] [CrossRef]
- Prinz, P.U.; Mendler, A.N.; Masouris, I.; Durner, L.; Oberneder, R.; Noessner, E. High DGK-alpha and disabled MAPK pathways cause dysfunction of human tumor-infiltrating CD8+ T cells that is reversible by pharmacologic intervention. J. Immunol. 2012, 188, 5990–6000. [Google Scholar] [CrossRef]
- Baldanzi, G.; Ragnoli, B.; Malerba, M. Potential role of diacylglycerol kinases in immune-mediated diseases. Clin. Sci. 2020, 134, 1637–1658. [Google Scholar] [CrossRef]
- Merida, I.; Andrada, E.; Gharbi, S.I.; Avila-Flores, A. Redundant and specialized roles for diacylglycerol kinases alpha and zeta in the control of T cell functions. Sci. Signal. 2015, 8, re6. [Google Scholar] [CrossRef]
- Noessner, E. DGK-alpha: A Checkpoint in Cancer-Mediated Immuno-Inhibition and Target for Immunotherapy. Front. Cell. Dev. Biol. 2017, 5, 16. [Google Scholar] [CrossRef]
- Riese, M.J.; Moon, E.K.; Johnson, B.D.; Albelda, S.M. Diacylglycerol Kinases (DGKs): Novel Targets for Improving T Cell Activity in Cancer. Front. Cell. Dev. Biol. 2016, 4, 108. [Google Scholar] [CrossRef]
- Sakane, F.; Mizuno, S.; Komenoi, S. Diacylglycerol Kinases as Emerging Potential Drug Targets for a Variety of Diseases: An Update. Front. Cell. Dev. Biol. 2016, 4, 82. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Shaffer, D.R.; Alvarez Arias, D.A.; Nakazaki, Y.; Pos, W.; Torres, A.J.; Cremasco, V.; Dougan, S.K.; Cowley, G.S.; Elpek, K.; et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 2014, 506, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Ebinu, J.O.; Bottorff, D.A.; Chan, E.Y.; Stang, S.L.; Dunn, R.J.; Stone, J.C. RasGRP, a Ras guanyl nucleotide- releasing protein with calcium- and diacylglycerol-binding motifs. Science 1998, 280, 1082–1086. [Google Scholar] [CrossRef]
- Jones, D.R.; Sanjuan, M.A.; Stone, J.C.; Merida, I. Expression of a catalytically inactive form of diacylglycerol kinase alpha induces sustained signaling through RasGRP. FASEB J. 2002, 16, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Flores, E.; Siliceo, M.; Martinez, A.C.; Merida, I. Membrane translocation of protein kinase Ctheta during T lymphocyte activation requires phospholipase C-gamma-generated diacylglycerol. J. Biol. Chem. 2003, 278, 29208–29215. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; Imai, S.; Sakane, F.; Kanoh, H. Isolation and characterization of the human diacylglycerol kinase gene. Biochem. J. 1993, 294, 443–449. [Google Scholar] [CrossRef][Green Version]
- Zheng, Y.; Zha, Y.; Driessens, G.; Locke, F.; Gajewski, T.F. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J. Exp. Med. 2012, 209, 2157–21563. [Google Scholar] [CrossRef] [PubMed]
- de Chaffoy de Courcelles, D.C.; Roevens, P.; Van Belle, H. R 59 022, a diacylglycerol kinase inhibitor. Its effect on diacylglycerol and thrombin-induced C kinase activation in the intact platelet. J. Biol. Chem. 1985, 260, 15762–15770. [Google Scholar] [CrossRef]
- de Chaffoy de Courcelles, D.; Roevens, P.; Van Belle, H.; Kennis, L.; Somers, Y.; De Clerck, F. The role of endogenously formed diacylglycerol in the propagation and termination of platelet activation. A biochemical and functional analysis using the novel diacylglycerol kinase inhibitor, R 59 949. J. Biol. Chem. 1989, 264, 3274–3285. [Google Scholar] [CrossRef]
- Jiang, Y.; Sakane, F.; Kanoh, H.; Walsh, J.P. Selectivity of the diacylglycerol kinase inhibitor 3-2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl-2, 3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem. Pharmacol. 2000, 59, 763–772. [Google Scholar] [CrossRef]
- Sato, M.; Liu, K.; Sasaki, S.; Kunii, N.; Sakai, H.; Mizuno, H.; Saga, H.; Sakane, F. Evaluations of the selectivities of the diacylglycerol kinase inhibitors R59022 and R59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology 2013, 92, 99–107. [Google Scholar] [CrossRef]
- Okada, N.; Sugiyama, K.; Shichi, S.; Shirai, Y.; Goto, K.; Sakane, F.; Kitamura, H.; Taketomi, A. Combination therapy for hepatocellular carcinoma with diacylglycerol kinase alpha inhibition and anti-programmed cell death-1 ligand blockade. Cancer Immunol. Immunother. 2021, 1–15. [Google Scholar] [CrossRef]
- Boroda, S.; Niccum, M.; Raje, V.; Purow, B.W.; Harris, T.E. Dual activities of ritanserin and R59022 as DGKalpha inhibitors and serotonin receptor antagonists. Biochem. Pharmacol. 2017, 123, 29–39. [Google Scholar] [CrossRef]
- Velnati, S.; Massarotti, A.; Antona, A.; Talmon, M.; Fresu, L.G.; Galetto, A.S.; Capello, D.; Bertoni, A.; Mercalli, V.; Graziani, A.; et al. Structure activity relationship studies on Amb639752: Toward the identification of a common pharmacophoric structure for DGKalpha inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 96–108. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Cancer Communications. The 150 most important questions in cancer research and clinical oncology series: Questions 94-101: Edited by Cancer Communications. Cancer Commun. 2018, 38, 1–9. [Google Scholar]
- Fu, L.; Li, S.; Xiao, W.; Yu, K.; Li, S.; Yuan, S.; Shen, J.; Dong, X.; Fang, Z.; Zhang, J.; et al. DGKA Mediates Resistance to PD-1 Blockade. Cancer Immunol. Res. 2021, 9, 371–385. [Google Scholar] [CrossRef]
- Arranz-Nicolas, J.; Martin-Salgado, M.; Adan-Barrientos, I.; Liebana, R.; Del Carmen Moreno-Ortiz, M.; Leitner, J.; Steinberger, P.; Avila-Flores, A.; Merida, I. Diacylglycerol kinase alpha inhibition cooperates with PD-1-targeted therapies to restore the T cell activation program. Cancer Immunol. Immunother. 2021, 70, 3277–3289. [Google Scholar] [CrossRef]
- Arranz-Nicolas, J.; Ogando, J.; Soutar, D.; Arcos-Perez, R.; Meraviglia-Crivelli, D.; Manes, S.; Merida, I.; Avila-Flores, A. Diacylglycerol kinase alpha inactivation is an integral component of the costimulatory pathway that amplifies TCR signals. Cancer Immunol. Immunother. 2018, 67, 965–980. [Google Scholar] [CrossRef]
- Avila-Flores, A.; Santos, T.; Rincon, E.; Merida, I. Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid. J. Biol. Chem. 2005, 280, 10091–10099. [Google Scholar] [CrossRef]
- Takao, S.; Akiyama, R.; Sakane, F. Combined inhibition/silencing of diacylglycerol kinase alpha and zeta simultaneously and synergistically enhances interleukin-2 production in T cells and induces cell death of melanoma cells. J. Cell Biochem. 2021, 122, 494–506. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Cancer Immunotherapy through the Inhibition of Diacylglycerol Kinases Alpha and Zeta. ACS Med. Chem. Lett. 2020, 11, 1083–1085. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakane, F.; Hoshino, F.; Ebina, M.; Sakai, H.; Takahashi, D. The Roles of Diacylglycerol Kinase α in Cancer Cell Proliferation and Apoptosis. Cancers 2021, 13, 5190. https://doi.org/10.3390/cancers13205190
Sakane F, Hoshino F, Ebina M, Sakai H, Takahashi D. The Roles of Diacylglycerol Kinase α in Cancer Cell Proliferation and Apoptosis. Cancers. 2021; 13(20):5190. https://doi.org/10.3390/cancers13205190
Chicago/Turabian StyleSakane, Fumio, Fumi Hoshino, Masayuki Ebina, Hiromichi Sakai, and Daisuke Takahashi. 2021. "The Roles of Diacylglycerol Kinase α in Cancer Cell Proliferation and Apoptosis" Cancers 13, no. 20: 5190. https://doi.org/10.3390/cancers13205190
APA StyleSakane, F., Hoshino, F., Ebina, M., Sakai, H., & Takahashi, D. (2021). The Roles of Diacylglycerol Kinase α in Cancer Cell Proliferation and Apoptosis. Cancers, 13(20), 5190. https://doi.org/10.3390/cancers13205190