
Crosstalk between KRAS, SRC and YAP Signaling in Pancreatic Cancer: Interactions Leading to Aggressive Disease and Drug Resistance


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. KRAS and PDAC
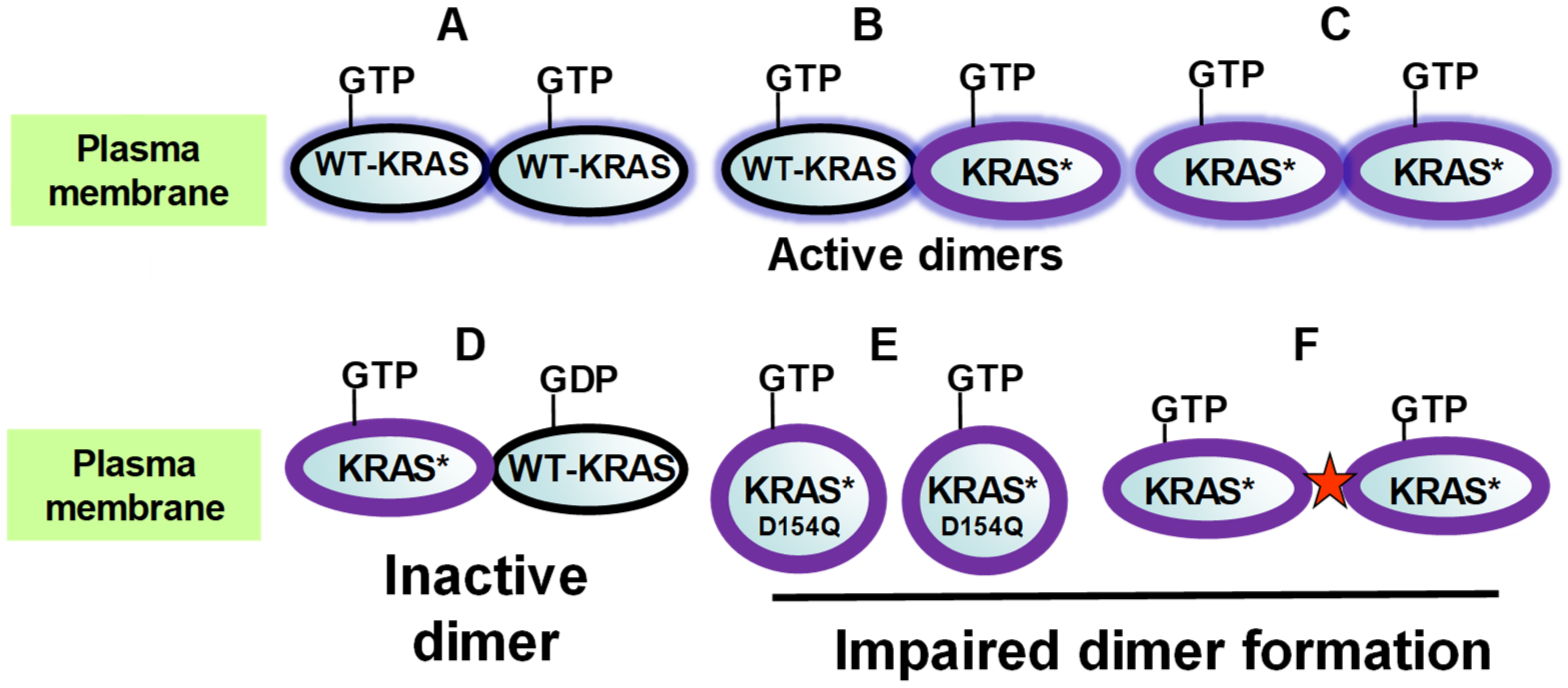
2.1. KRAS Dimerization in Signal Transduction
2.2. Regulation of KRAS Function by Tyrosine Phosphorylation: Contrasting Roles of SFK and SHP2
3. PDAC Subtypes and KRAS Dependency
4. Mechanisms That Circumvent KRAS in PDAC: The Role of YAP/TAZ
4.1. YAP Regulation: Succinct Description
4.2. Role of YAP in PanIN and PDAC Development and Maintenance
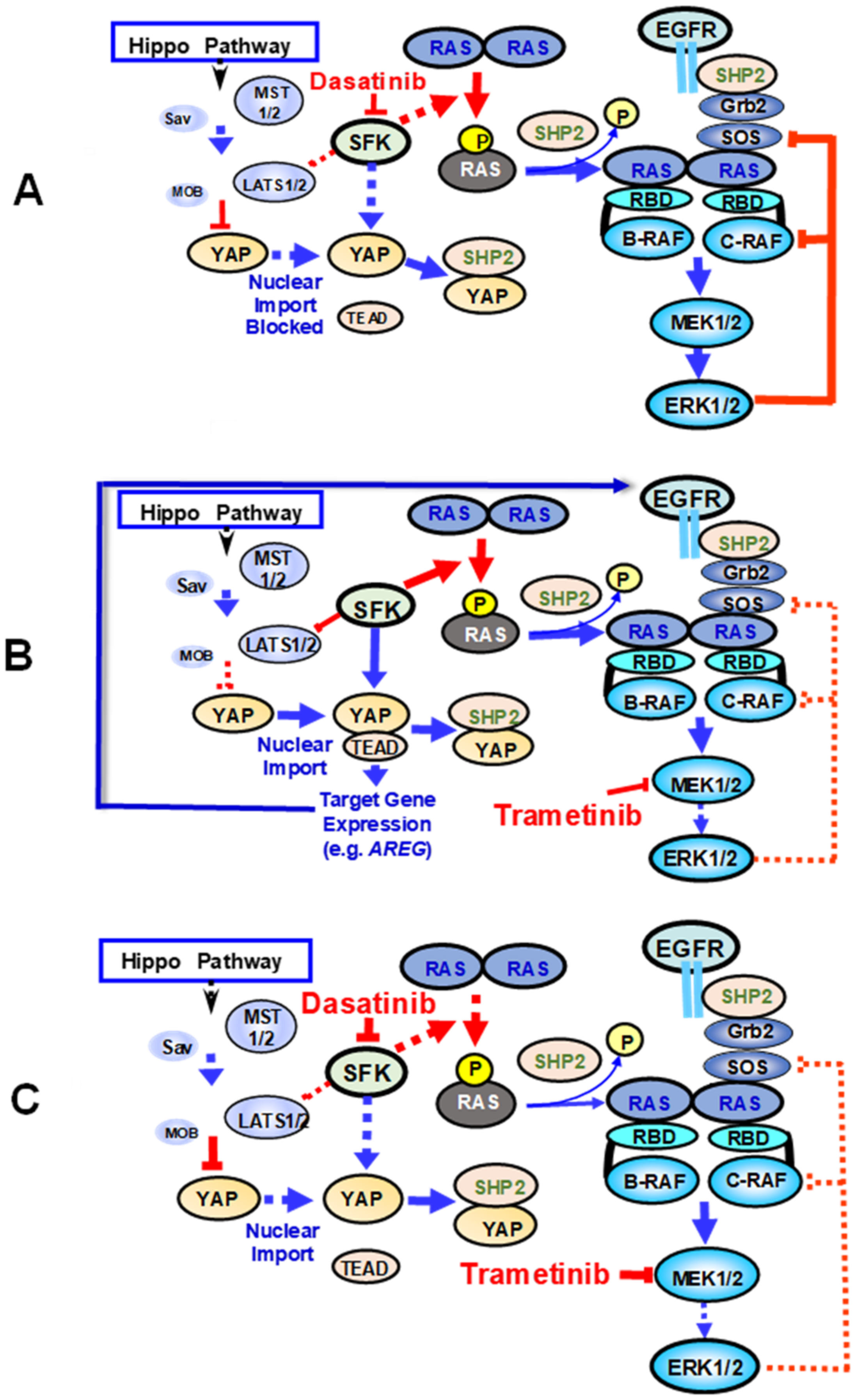
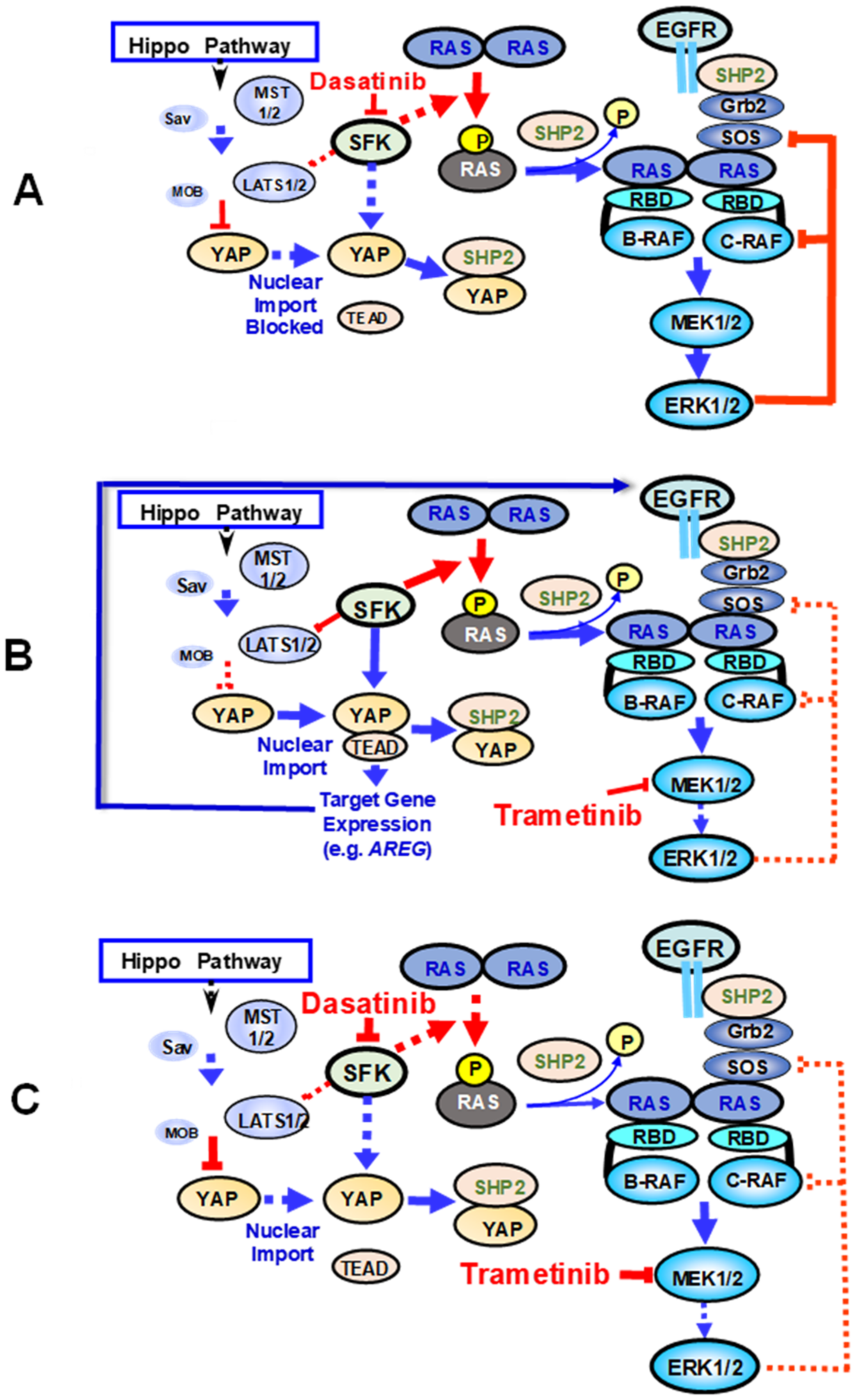
5. Regulation of YAP by SFK-Mediated Tyrosine Phosphorylation
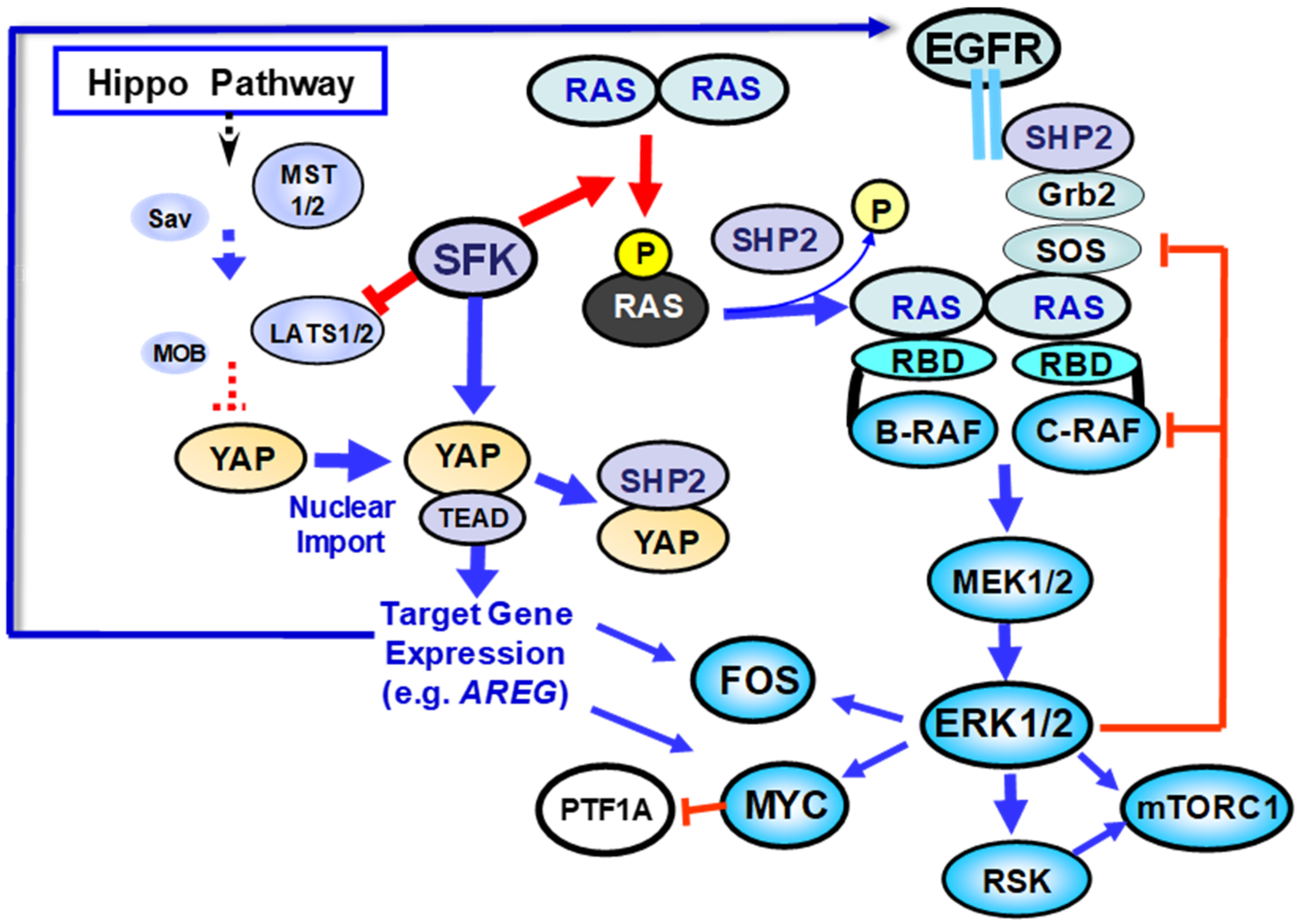
6. Implications
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. Folfirinox or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Garcia-Sampedro, A.; Gaggia, G.; Ney, A.; Mahamed, I.; Acedo, P. The state-of-the-art of phase ii/iii clinical trials for targeted pancreatic cancer therapies. J. Clin. Med. 2021, 10, 566. [Google Scholar] [CrossRef]
- Al-Share, B.; Hammad, N.; Diab, M. Pancreatic adenocarcinoma: Molecular drivers and the role of targeted therapy. Cancer Metastasis Rev. 2021, 40, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Eyres, M.; Lanfredini, S.; Xu, H.; Burns, A.; Blake, A.; Willenbrock, F.; Goldin, R.; Hughes, D.; Hughes, S.; Thapa, A.; et al. Tet2 drives 5hmc marking of gata6 and epigenetically defines pancreatic ductal adenocarcinoma transcriptional subtypes. Gastroenterology 2021, 161, 653–668.e16. [Google Scholar] [CrossRef] [PubMed]
- Koikawa, K.; Kibe, S.; Suizu, F.; Sekino, N.; Kim, N.; Manz, T.D.; Pinch, B.J.; Akshinthala, D.; Verma, A.; Gaglia, G.; et al. Targeting pin1 renders pancreatic cancer eradicable by synergizing with immunochemotherapy. Cell 2021, 184, 4753–4771.e27. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. Ras isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eibl, G.; Rozengurt, E. Kras, yap, and obesity in pancreatic cancer: A signaling network with multiple loops. Semin. Cancer Biol. 2019, 54, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. Kras: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Rhett, J.M.; Khan, I.; O’Bryan, J.P. Biology, pathology, and therapeutic targeting of ras. Adv. Cancer Res. 2020, 148, 69–146. [Google Scholar] [PubMed]
- Dhanaraman, T.; Singh, S.; Killoran, R.C.; Singh, A.; Xu, X.; Shifman, J.M.; Smith, M.J. Rassf effectors couple diverse ras subfamily gtpases to the hippo pathway. Sci. Signal. 2020, 13, eabb4778. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. Ras oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting kras mutant cancers with a covalent g12c-specific inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [Green Version]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; DelGiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. Egf receptor is required for kras-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. Egf receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Maitra, A.; Fukushima, N.; Takaori, K.; Hruban, R.H. Precursors to invasive pancreatic cancer. Adv. Anat. Pathol. 2005, 12, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733.e9. [Google Scholar] [CrossRef] [Green Version]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53r172h and krasg12d cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.M.; Hendley, A.M.; Lafaro, K.J.; Pruski, M.A.; Jones, N.C.; Alsina, J.; Younes, M.; Maitra, A.; McAllister, F.; Iacobuzio-Donahue, C.A.; et al. P53 mutations cooperate with oncogenic kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 2016, 35, 4282–4288. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.M.M.; Sancho, R.; Messal, H.A.; Nye, E.; Spencer-Dene, B.; Stone, R.K.; Stamp, G.; Rosewell, I.; Quaglia, A.; Behrens, A. Duct- and acinar-derived pancreatic ductal adenocarcinomas show distinct tumor progression and marker expression. Cell Rep. 2017, 21, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.Y.L.; Dubois, C.L.; Sarai, K.; Zarei, S.; Schaeffer, D.F.; Sander, M.; Kopp, J.L. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut 2019, 68, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Flowers, B.M.; Xu, H.; Mulligan, A.S.; Hanson, K.J.; Seoane, J.A.; Vogel, H.; Curtis, C.; Wood, L.D.; Attardi, L.D. Cell of origin influences pancreatic cancer subtype. Cancer Discov. 2021, 11, 660–677. [Google Scholar] [CrossRef]
- Rozengurt, E.; Eibl, G. Central role of yes-associated protein and ww-domain-containing transcriptional co-activator with pdz-binding motif in pancreatic cancer development. World J. Gastroenterol. 2019, 25, 1797–1816. [Google Scholar] [CrossRef]
- Makohon-Moore, A.P.; Matsukuma, K.; Zhang, M.; Reiter, J.G.; Gerold, J.M.; Jiao, Y.; Sikkema, L.; Attiyeh, M.A.; Yachida, S.; Sandone, C.; et al. Precancerous neoplastic cells can move through the pancreatic ductal system. Nature 2018, 561, 201–205. [Google Scholar] [CrossRef]
- Kim, M.P.; Li, X.; Deng, J.; Zhang, Y.; Dai, B.; Allton, K.L.; Hughes, T.G.; Siangco, C.; Augustine, J.J.; Kang, Y.; et al. Oncogenic kras recruits an expansive transcriptional network through mutant p53 to drive pancreatic cancer metastasis. Cancer Discov. 2021, 11, 2094–2111. [Google Scholar] [PubMed]
- Shain, A.H.; Giacomini, C.P.; Matsukuma, K.; Karikari, C.A.; Bashyam, M.D.; Hidalgo, M.; Maitra, A.; Pollack, J.R. Convergent structural alterations define switch/sucrose nonfermentable (swi/snf) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E252–E259. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, A.; Hong, J.; Iacobuzio-Donahue, C.A. The pancreatic cancer genome revisited. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. Ras proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, X.; Tamgüney, T.M.; Collisson, E.A.; Lin, L.-J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-gtp dimers activate the mitogen-activated protein kinase (mapk) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrogio, C.; Köhler, J.; Zhou, Z.-W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. Kras dimerization impacts mek inhibitor sensitivity and oncogenic activity of mutant kras. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef] [PubMed]
- Spencer-Smith, R.; Koide, A.; Zhou, Y.; Eguchi, R.R.; Sha, F.; Gajwani, P.; Santana, D.; Gupta, A.; Jacobs, M.; Herrero-Garcia, E.; et al. Inhibition of ras function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. [Google Scholar] [CrossRef]
- Khan, I.; Spencer-Smith, R.; O’Bryan, J.P. Targeting the α4-α5 dimerization interface of k-ras inhibits tumor formation in vivo. Oncogene 2019, 38, 2984–2993. [Google Scholar] [CrossRef]
- Rudack, T.; Teuber, C.; Scherlo, M.; Güldenhaupt, J.; Schartner, J.; Lübben, M.; Klare, J.; Gerwert, K.; Kötting, C. The ras dimer structure. Chem. Sci. 2021, 12, 8178–8189. [Google Scholar] [CrossRef]
- Rajakulendran, T.; Sahmi, M.; Lefrançois, M.; Sicheri, F.; Therrien, M. A dimerization-dependent mechanism drives raf catalytic activation. Nature 2009, 461, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.-J.; Jang, H. Is nanoclustering essential for all oncogenic kras pathways? Can it explain why wild-type kras can inhibit its oncogenic variant? Semin. Cancer Biol. 2019, 54, 114–120. [Google Scholar] [CrossRef]
- Terrell, E.M.; Morrison, D.K. Ras-mediated activation of the raf family kinases. Cold Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef]
- Lavoie, H.; Therrien, M. Regulation of raf protein kinases in erk signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Chan, A.H.; Young, L.C.; Bindu, L.; Neale, C.; Messing, S.; Dharmaiah, S.; Taylor, T.; Denson, J.P.; Esposito, D.; et al. Kras interaction with raf1 ras-binding domain and cysteine-rich domain provides insights into ras-mediated raf activation. Nat. Commun. 2021, 12, 1176. [Google Scholar] [CrossRef] [PubMed]
- Brummer, T.; McInnes, C. Raf kinase dimerization: Implications for drug discovery and clinical outcomes. Oncogene 2020, 39, 4155–4169. [Google Scholar] [CrossRef]
- Van, Q.N.; Prakash, P.; Shrestha, R.; Balius, T.E.; Turbyville, T.J.; Stephen, A.G. Ras nanoclusters: Dynamic signaling platforms amenable to therapeutic intervention. Biomolecules 2021, 11, 377. [Google Scholar] [CrossRef]
- Zhou, B.; Der, C.J.; Cox, A.D. The role of wild type ras isoforms in cancer. Semin. Cell Dev. Biol. 2016, 58, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Guha, S.; Lunn, J.A.; Santiskulvong, C.; Rozengurt, E. Neurotensin stimulates protein kinase c-dependent mitogenic signaling in human pancreatic carcinoma cell line panc-1. Cancer Res. 2003, 63, 2379–2387. [Google Scholar]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Öllinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and kras dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Qiu, W.; Sahin, F.; Iacobuzio-Donahue, C.A.; Garcia-Carracedo, D.; Wang, W.M.; Kuo, C.Y.; Chen, D.; Arking, D.E.; Lowy, A.M.; Hruban, R.H.; et al. Disruption of p16 and activation of kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget 2011, 2, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Sheffels, E.; Kortum, R.L. The role of wild-type ras in oncogenic ras transformation. Genes 2021, 12, 662. [Google Scholar] [CrossRef]
- Rozengurt, E.; Sinnett-Smith, J.; Eibl, G. Yes-associated protein (yap) in pancreatic cancer: At the epicenter of a targetable signaling network associated with patient survival. Signal Transduct. Target. Ther. 2018, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Je, D.W.; Moon, Y.; Ji, Y.G.; Cho, Y.; Lee, D.H. The inhibition of src family kinase suppresses pancreatic cancer cell proliferation, migration, and invasion. Pancreas 2014, 43, 768–776. [Google Scholar] [CrossRef]
- Ortiz, M.A.; Mikhailova, T.; Li, X.; Porter, B.A.; Bah, A.; Kotula, L. Src family kinases, adaptor proteins and the actin cytoskeleton in epithelial-to-mesenchymal transition. Cell Commun. Signal. 2021, 19, 67. [Google Scholar] [CrossRef]
- Kano, Y.; Gebregiworgis, T.; Marshall, C.B.; Radulovich, N.; Poon, B.P.K.; St-Germain, J.; Cook, J.D.; Valencia-Sama, I.; Grant, B.M.M.; Herrera, S.G.; et al. Tyrosyl phosphorylation of kras stalls gtpase cycle via alteration of switch i and ii conformation. Nat. Commun. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Buday, L.; Vas, V. Novel regulation of ras proteins by direct tyrosine phosphorylation and dephosphorylation. Cancer Metastasis Rev. 2020, 39, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Heir, P.; Srikumar, T.; Cook, J.D.; Burrell, K.; Kano, Y.; Lee, J.E.; Zadeh, G.; Raught, B.; Ohh, M. Src promotes gtpase activity of ras via tyrosine 32 phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, E3785–E3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Liu, M.; Li, D.; Tan, Y.; Zhang, R.; Xia, Z.; Wang, P.; Jiao, B.; Liu, P.; Ren, R. Ptpn2 regulates the activation of kras and plays a critical role in proliferation and survival of kras-driven cancer cells. J. Biol. Chem. 2020, 295, 18343–18354. [Google Scholar] [CrossRef]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of shp2-mediated dephosphorylation of ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef] [Green Version]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant kras-driven cancers depend on ptpn11/shp2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of shp2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Fodor, M.; Price, E.; Wang, P.; Lu, H.; Argintaru, A.; Chen, Z.; Glick, M.; Hao, H.-X.; Kato, M.; Koenig, R.; et al. Dual allosteric inhibition of shp2 phosphatase. ACS Chem. Biol. 2018, 13, 647–656. [Google Scholar] [CrossRef]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. Shp2 inhibition prevents adaptive resistance to mek inhibitors in multiple cancer models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, C.; Velazquez, R.; Wang, H.; Dunkl, L.M.; Kazic-Legueux, M.; Haberkorn, A.; Billy, E.; Manchado, E.; Brachmann, S.M.; et al. Shp2 inhibition overcomes rtk-mediated pathway reactivation in kras-mutant tumors treated with mek inhibitors. Mol. Cancer Ther. 2019, 18, 1323–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Gonçalves-Ribeiro, S.; et al. Shp2 is required for growth of kras-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Rozengurt, E.; Soares, H.P.; Sinnet-Smith, J. Suppression of feedback loops mediated by pi3k/mtor induces multiple overactivation of compensatory pathways: An unintended consequence leading to drug resistance. Mol. Cancer Ther. 2014, 13, 2477–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, T.A.; Adamopoulos, C.; Karoulia, Z.; Wu, X.; Sachidanandam, R.; Aaronson, S.A.; Poulikakos, P.I. Shp2 drives adaptive resistance to erk signaling inhibition in molecularly defined subsets of erk-dependent tumors. Cell Rep. 2019, 26, 65–78.e5. [Google Scholar] [CrossRef] [Green Version]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. Ras nucleotide cycling underlies the shp2 phosphatase dependence of mutant braf-, nf1- and ras-driven cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the mapk pathway in kras-driven tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Lefesvre, P.; Nicolle, R.; Biankin, A.V.; Puleo, F.; Van Laethem, J.L.; Rooman, I. Different shades of pancreatic ductal adenocarcinoma, different paths towards precision therapeutic applications. Ann. Oncol. 2019, 30, 1428–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. Gata6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. Gata6 regulates emt and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2017, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, K.; Jang, G.H.; Grant, R.C.; Wilson, J.M.; Notta, F.; O’Kane, G.M.; Knox, J.J.; Gallinger, S.; Fischer, S. The value of gata6 immunohistochemistry and computer-assisted diagnosis to predict clinical outcome in advanced pancreatic cancer. Sci. Rep. 2021, 11, 14951. [Google Scholar] [CrossRef]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Birnbaum, D.J.; Begg, S.K.S.; Finetti, P.; Vanderburg, C.; Kulkarni, A.S.; Neyaz, A.; Hank, T.; Tai, E.; Deshpande, V.; Bertucci, F.; et al. Transcriptomic analysis of laser capture microdissected tumors reveals cancer- and stromal-specific molecular subtypes of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2021, 27, 2314–2325. [Google Scholar] [CrossRef]
- Juiz, N.; Elkaoutari, A.; Bigonnet, M.; Gayet, O.; Roques, J.; Nicolle, R.; Iovanna, J.; Dusetti, N. Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. FASEB J. 2020, 34, 12214–12228. [Google Scholar] [CrossRef] [PubMed]
- Milan, M.; Diaferia, G.R.; Natoli, G. Tumor cell heterogeneity and its transcriptional bases in pancreatic cancer: A tale of two cell types and their many variants. EMBO J. 2021, 40, e107206. [Google Scholar] [CrossRef]
- Hayashi, A.; Fan, J.; Chen, R.; Ho, Y.-J.; Makohon-Moore, A.P.; Lecomte, N.; Zhong, Y.; Hong, J.; Huang, J.; Sakamoto, H.; et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nat. Cancer 2020, 1, 59–74. [Google Scholar] [CrossRef] [Green Version]
- Miyabayashi, K.; Baker, L.A.; Deschênes, A.; Traub, B.; Caligiuri, G.; Plenker, D.; Alagesan, B.; Belleau, P.; Li, S.; Kendall, J.; et al. Intraductal transplantation models of human pancreatic ductal adenocarcinoma reveal progressive transition of molecular subtypes. Cancer Discov. 2020, 10, 1566–1589. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “k-ras addiction” reveals regulators of emt and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzumdar, M.D.; Chen, P.Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking kras function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef] [Green Version]
- Tu, B.; Yao, J.; Ferri-Borgogno, S.; Zhao, J.; Chen, S.; Wang, Q.; Yan, L.; Zhou, X.; Zhu, C.; Bang, S.; et al. Yap1 oncogene is a context-specific driver for pancreatic ductal adenocarcinoma. JCI Insight 2019, 4, e130811. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest. 2012, 122, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target ras signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 116. [Google Scholar] [CrossRef]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; Ackroyd, J.J.; et al. Tumor microenvironment remodeling enables bypass of oncogenic kras dependency in pancreatic cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef] [PubMed]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic networks in mutant kras-driven tumours: Tissue specificities and the microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef]
- Sudol, M.; Bork, P.; Einbond, A.; Kastury, K.; Druck, T.; Negrini, M.; Huebner, K.; Lehman, D. Characterization of the mammalian yap (yes-associated protein) gene and its role in defining a novel protein module, the ww domain. J. Biol. Chem. 1995, 270, 14733–14741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, F.; Marignani, P.A.; Sarbassova, D.; Yagi, R.; Hall, R.A.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; et al. Taz: A novel transcriptional co-activator regulated by interactions with 14-3-3 and pdz domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of yap/taz activity by metabolic and nutrient-sensing pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. Yap/taz at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. Yap and taz: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J. Yap/taz: Drivers of tumor growth, metastasis, and resistance to therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kim, D.-H.; Shah, S.R.; Kim, H.-N.; Kshitiz; Kim, P.; Quiñones-Hinojosa, A.; Levchenko, A. Switch-like enhancement of epithelial-mesenchymal transition by yap through feedback regulation of wt1 and rho-family gtpases. Nat. Commun. 2019, 10, 2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhough, A.; Bagley, C.; Heesom, K.J.; Gurevich, D.B.; Gay, D.; Bond, M.; Collard, T.J.; Paraskeva, C.; Martin, P.; Sansom, O.J.; et al. Cancer cell adaptation to hypoxia involves a hif-gprc5a-yap axis. EMBO Mol. Med. 2018, 10, e8699. [Google Scholar] [CrossRef]
- Gundogdu, R.; Hergovich, A. Mob (mps one binder) proteins in the hippo pathway and cancer. Cells 2019, 8, 569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes yap/taz transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and hippo pathways regulates rhamm transcription via yap to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Wang, W.; Liu, B.; Deng, H.; Uster, E.; Pan, D. Identification of happyhour/map4k as alternative hpo/mst-like kinases in the hippo kinase cascade. Dev. Cell 2015, 34, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.-S.; Lu, W.; Lu, S.; et al. Map4k family kinases act in parallel to mst1/2 to activate lats1/2 in the hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef]
- Hergovich, A. The roles of ndr protein kinases in hippo signalling. Genes 2016, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Mana-Capelli, S.; McCollum, D. Angiomotins stimulate lats kinase autophosphorylation and act as scaffolds that promote hippo signaling. J. Biol. Chem. 2018, 293, 18230–18241. [Google Scholar] [CrossRef] [Green Version]
- Höffken, V.; Hermann, A.; Pavenstädt, H.; Kremerskothen, J. Wwc proteins: Important regulators of hippo signaling in cancer. Cancers 2021, 13, 306. [Google Scholar] [CrossRef]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L.; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.-L. The hippo pathway effector proteins yap and taz have both distinct and overlapping functions in the cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [Green Version]
- Totaro, A.; Panciera, T.; Piccolo, S. Yap/taz upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.-L. The emerging roles of yap and taz in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X. The hippo pathway effectors taz and yap in development, homeostasis and disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [Green Version]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of taz-tead complex reveals a distinct interaction mode from that of yap-tead complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef]
- Finch-Edmondson, M.L.; Strauss, R.P.; Passman, A.M.; Sudol, M.; Yeoh, G.C.; Callus, B.A. Taz protein accumulation is negatively regulated by yap abundance in mammalian cells. J. Biol. Chem. 2015, 290, 27928–27938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Guan, K.-L. The hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straßburger, K.; Tiebe, M.; Pinna, F.; Breuhahn, K.; Teleman, A.A. Insulin/igf signaling drives cell proliferation in part via yorkie/yap. Dev. Biol. 2012, 367, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sinnett-Smith, J.; Stevens, J.V.; Young, S.H.; Rozengurt, E. Biphasic regulation of yes-associated protein (yap) cellular localization, phosphorylation, and activity by g protein-coupled receptor agonists in intestinal epithelial cells: A novel role for protein kinase d (pkd). J. Biol. Chem. 2016, 291, 17988–18005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudol, M. Yap1 oncogene and its eight isoforms. Oncogene 2013, 32, 3922. [Google Scholar] [CrossRef]
- Ben, C.; Wu, X.; Takahashi-Kanemitsu, A.; Knight, C.T.; Hayashi, T.; Hatakeyama, M. Alternative splicing reverses the cell-intrinsic and cell-extrinsic pro-oncogenic potentials of yap1. J. Biol. Chem. 2020, 295, 13965–13980. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Jiang, J. Hippo-independent regulation of yki/yap/taz: A non-canonical view. Front. Cell Dev. Biol. 2021, 9, 658481. [Google Scholar] [CrossRef]
- Mo, J.-S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.-S.; Guan, K.-L. Cellular energy stress induces ampk-mediated regulation of yap and the hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.-D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. Ampk modulates hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, L.; Liu, M.; Chong, R.; Ding, S.J.; Chen, Y.; Dong, J. Cdk1 phosphorylation of yap promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 2013, 73, 6722–6733. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Li, S.; Wang, X.; Zhu, J.; Zhuo, S.; Han, Y.; Yue, T.; Yang, Y.; Jiang, J. Cdk7 regulates organ size and tumor growth by safeguarding the hippo pathway effector yki/yap/taz in the nucleus. Genes Dev. 2020, 34, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, M.H.; Hong, H.; Cho, H.; Park, S.; Kim, S.K.; Kim, J. Mk5 regulates yap stability and is a molecular target in yap-driven cancers. Cancer Res. 2019, 79, 6139–6152. [Google Scholar] [CrossRef] [Green Version]
- An, L.; Nie, P.; Chen, M.; Tang, Y.; Zhang, H.; Guan, J.; Cao, Z.; Hou, C.; Wang, W.; Zhao, Y.; et al. Mst4 kinase suppresses gastric tumorigenesis by limiting yap activation via a non-canonical pathway. J. Exp. Med. 2020, 217, e20191817. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.; Kim, W.; Kim, S.; Kim, Y.; Song, Y.; Bilousov, O.; Kim, J.; Lee, T.; Cha, B.; Kim, M.; et al. Phosphorylation by nlk inhibits yap-14-3-3-interactions and induces its nuclear localization. EMBO Rep. 2017, 18, 61–71. [Google Scholar] [CrossRef]
- Xie, D.; Cui, J.; Xia, T.; Jia, Z.; Wang, L.; Wei, W.; Zhu, A.; Gao, Y.; Xie, K.; Quan, M. Hippo transducer taz promotes epithelial mesenchymal transition and supports pancreatic cancer progression. Oncotarget 2015, 6, 35949–35963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, L.; Purohit, V.; Shukla, S.K.; Chen, X.; Yu, F.; Fu, K.; Chen, Y.; Solheim, J.; Singh, P.K.; et al. Active yap promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through lpar3. Oncotarget 2015, 6, 36019–36031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcedo Allende, M.T.; Zeron-Medina, J.; Hernandez, J.; Macarulla, T.; Balsells, J.; Merino, X.; Allende, H.; Tabernero, J.; Ramon y Cajal Agüeras, S. Overexpression of yes associated protein 1, an independent prognostic marker in patients with pancreatic ductal adenocarcinoma, correlated with liver metastasis and poor prognosis. Pancreas 2017, 46, 913–920. [Google Scholar] [CrossRef]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant kras, the transcription regulator yap is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. Yap1 and taz control pancreatic cancer initiation in mice by direct up-regulation of jak–stat3 signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Kodama, T.; Sato, K.; Murai, K.; Yoshioka, T.; Shigekawa, M.; Yamada, R.; Hikita, H.; Sakamori, R.; Akita, H.; et al. Dysregulation of pi3k and hippo signaling pathways synergistically induces chronic pancreatitis via ctgf upregulation. J. Clin. Invest. 2021, 131. [Google Scholar] [CrossRef]
- Murakami, S.; Nemazanyy, I.; White, S.M.; Chen, H.; Nguyen, C.D.K.; Graham, G.T.; Saur, D.; Pende, M.; Yi, C. A yap-myc-sox2-p53 regulatory network dictates metabolic homeostasis and differentiation in kras-driven pancreatic ductal adenocarcinomas. Dev. Cell 2019, 51, 113–128.e119. [Google Scholar] [CrossRef]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. Kras and yap1 converge to regulate emt and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [Green Version]
- King, B.; Araki, J.; Palm, W.; Thompson, C.B. Yap/taz promote the scavenging of extracellular nutrients through macropinocytosis. Genes Dev. 2020, 34, 1345–1358. [Google Scholar] [CrossRef]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The hippo effector yap promotes resistance to raf- and mek-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Eisenbarth, D.; Choi, W.; Kim, H.; Choi, C.; Lee, D.; Lim, D.S. Yap and ap-1 cooperate to initiate pancreatic cancer development from ductal cells in mice. Cancer Res. 2020, 80, 4768–4779. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, M.; Nipper, M.; Deng, J.; Sharkey, F.E.; Johnson, R.L.; Crawford, H.C.; Chen, Y.; Wang, P. Activation of the intrinsic fibroinflammatory program in adult pancreatic acinar cells triggered by hippo signaling disruption. PLoS Biol. 2019, 17, e3000418. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative wnt signaling activates yap/taz. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bo, H.; Gao, L.; Chen, Y.; Zhang, J.; Zhu, M. Upregulation of the expression of wnt5a promotes the proliferation of pancreatic cancer cells in vitro and in a nude mouse model. Mol. Med. Rep. 2016, 13, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, W.; Mossmann, D.; Kleemann, J.; Mock, K.; Meisinger, C.; Brummer, T.; Herr, R.; Brabletz, S.; Stemmler, M.P.; Brabletz, T. Zeb1 turns into a transcriptional activator by interacting with yap1 in aggressive cancer types. Nat. Commun. 2016, 7, 10498. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, Y.; Yang, J.; Zhan, H.; Zhou, Z.; Jiang, Y.; Shi, X.; Fan, X.; Zhang, J.; Luo, W.; et al. Zinc-dependent regulation of zeb1 and yap1 coactivation promotes epithelial-mesenchymal transition plasticity and metastasis in pancreatic cancer. Gastroenterology 2021, 160, 1771–1783.e1. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The emt-activator zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The swi/snf complex is a mechanoregulated inhibitor of yap and taz. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.; Bam, R. Reciprocal crosstalk between yap1/hippo pathway and the p53 family proteins: Mechanisms and outcomes in cancer. Front. Cell Dev. Biol. 2019, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. Actl6a is co-amplified with p63 in squamous cell carcinoma to drive yap activation, regenerative proliferation, and poor prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Pham, T.; Chang, M.T.; Barnes, D.; Cai, A.G.; Noubade, R.; Totpal, K.; Chen, X.; Tran, C.; Hagenbeek, T.; et al. The tumor suppressor bap1 regulates the hippo pathway in pancreatic ductal adenocarcinoma. Cancer Res. 2020, 80, 1656–1668. [Google Scholar] [CrossRef] [Green Version]
- Hasan, N.; Ahuja, N. The emerging roles of atp-dependent chromatin remodeling complexes in pancreatic cancer. Cancers 2019, 11, 1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, F.; Xu, Q.; Zhao, Y.; Stevens, J.V.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Insulin receptor and gpcr crosstalk stimulates yap via pi3k and pkd in pancreatic cancer cells. Mol. Cancer Res. 2017, 15, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Kisfalvi, K.; Eibl, G.; Sinnett-Smith, J.; Rozengurt, E. Metformin disrupts crosstalk between g protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009, 69, 6539–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozengurt, E.; Sinnett-Smith, J.; Kisfalvi, K. Crosstalk between insulin/insulin-like growth factor-1 receptors and g protein-coupled receptor signaling systems: A novel target for the antidiabetic drug metformin in pancreatic cancer. Clin. Cancer Res. 2010, 16, 2505–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, F.; Xu, Q.; Wang, J.; Yu, S.; Chang, H.-H.; Sinnett-Smith, J.; Eibl, G.; Rozengurt, E. Lipophilic statins inhibit yap nuclear localization, co-activator activity and colony formation in pancreatic cancer cells and prevent the initial stages of pancreatic ductal adenocarcinoma in krasg12d mice. PLoS ONE 2019, 14, e0216603. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.L.; Krop, I.E. Advances in targeting src in the treatment of breast cancer and other solid malignancies. Clin. Cancer Res. 2010, 16, 3526–3532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Moro, L.; Simoneschi, D.; Kurz, E.; Arbini, A.A.; Jang, S.; Guaragnella, N.; Giannattasio, S.; Wang, W.; Chen, Y.-A.; Pires, G.; et al. Epigenetic silencing of the ubiquitin ligase subunit fbxl7 impairs c-src degradation and promotes epithelial-to-mesenchymal transition and metastasis. Nat. Cell Biol. 2020, 22, 1130–1142. [Google Scholar] [CrossRef]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src family kinases as therapeutic targets in advanced solid tumors: What we have learned so far. Cancers 2020, 12, 1448. [Google Scholar] [CrossRef]
- Parkin, A.; Man, J.; Timpson, P.; Pajic, M. Targeting the complexity of src signalling in the tumour microenvironment of pancreatic cancer: From mechanism to therapy. FEBS J. 2019, 286, 3510–3539. [Google Scholar] [CrossRef] [Green Version]
- Morton, J.P.; Karim, S.A.; Graham, K.; Timpson, P.; Jamieson, N.; Athineos, D.; Doyle, B.; McKay, C.; Heung, M.Y.; Oien, K.A.; et al. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 2010, 139, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Shields, D.J.; Murphy, E.A.; Desgrosellier, J.S.; Mielgo, A.; Lau, S.K.; Barnes, L.A.; Lesperance, J.; Huang, M.; Schmedt, C.; Tarin, D.; et al. Oncogenic ras/src cooperativity in pancreatic neoplasia. Oncogene 2011, 30, 2123–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosch, A.R.; Dai, X.; Gaidarski Iii, A.A.; Shi, C.; Castellanos, J.A.; VanSaun, M.N.; Merchant, N.B.; Nagathihalli, N.S. Src kinase inhibition restores e-cadherin expression in dasatinib-sensitive pancreatic cancer cells. Oncotarget 2019, 10, 1056–1069. [Google Scholar] [CrossRef]
- Ye, X.; Weinberg, R.A. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, Y.; Ji, X.; Cao, X.; Dai, X.; Xu, L.; Zhao, H.; Guo, X.; Yan, H.; Zhang, H.; Zhu, C.; et al. Src inhibits the hippo tumor suppressor pathway through tyrosine phosphorylation of lats1. Cancer Res. 2017, 77, 4868–4880. [Google Scholar] [CrossRef] [Green Version]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.-G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. Src tyrosine kinase activates the yap/taz axis and thereby drives tumor growth and metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef] [Green Version]
- Sugihara, T.; Werneburg, N.W.; Hernandez, M.C.; Yang, L.; Kabashima, A.; Hirsova, P.; Yohanathan, L.; Sosa, C.; Truty, M.J.; Vasmatzis, G.; et al. Yap tyrosine phosphorylation and nuclear localization in cholangiocarcinoma cells are regulated by lck and independent of lats activity. Mol. Cancer Res. 2018, 16, 1556–1567. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Wu, L.-W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.-X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-src-yap module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. Β-catenin driven cancers require a yap1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Silvis, M.R.; Honaker, Y.; Lien, W.-H.; Arron, S.T.; Vasioukhin, V. Ae-catenin inhibits a src-yap1 oncogenic module that couples tyrosine kinases and the effector of hippo signaling pathway. Genes Dev. 2016, 30, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Buckarma, E.H.; Werneburg, N.W.; Conboy, C.B.; Kabashima, A.; Brien, D.R.; Wang, C.; Rizvi, S.; Smoot, R.L. The yap-interacting phosphatase shp2 can regulate transcriptional coactivity and modulate sensitivity to chemotherapy in cholangiocarcinoma. Mol. Cancer Res. 2020, 18, 1574–1588. [Google Scholar] [CrossRef]
- Mettu, N.B.; Niedzwiecki, D.; Rushing, C.; Nixon, A.B.; Jia, J.; Haley, S.; Honeycutt, W.; Hurwitz, H.; Bendell, J.C.; Uronis, H. A phase i study of gemcitabine + dasatinib (gd) or gemcitabine + dasatinib + cetuximab (gdc) in refractory solid tumors. Cancer Chemother. Pharmacol. 2019, 83, 1025–1035. [Google Scholar] [CrossRef]
- Evans, T.R.J.; Van Cutsem, E.; Moore, M.J.; Bazin, I.S.; Rosemurgy, A.; Bodoky, G.; Deplanque, G.; Harrison, M.; Melichar, B.; Pezet, D.; et al. Phase 2 placebo-controlled, double-blind trial of dasatinib added to gemcitabine for patients with locally-advanced pancreatic cancer. Ann. Oncol. 2017, 28, 354–361. [Google Scholar] [CrossRef]
- George, T.J.; Ali, A.; Wang, Y.; Lee, J.H.; Ivey, A.M.; DeRemer, D.; Daily, K.C.; Allegra, C.J.; Hughes, S.J.; Fan, Z.H.; et al. Phase ii study of 5-fluorouracil, oxaliplatin plus dasatinib (folfox-d) in first-line metastatic pancreatic adenocarcinoma. Oncologist 2021, 26, 825.e1674. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral mek inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Balaji, U.; Eslinger, C.; McMillan, E.; Conway, W.; Posner, B.; Mills, G.B.; O’Reilly, E.M.; Knudsen, E.S. Integrated patient-derived models delineate individualized therapeutic vulnerabilities of pancreatic cancer. Cell Rep. 2016, 16, 2017–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, G.; Kim, I.-K.; Conforti, F.; Liu, J.; Zhang, Y.-W.; Giaccone, G. Dasatinib sensitises kras-mutant cancer cells to mitogen-activated protein kinase kinase inhibitor via inhibition of taz activity. Eur. J. Cancer 2018, 99, 37–48. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozengurt, E.; Eibl, G. Crosstalk between KRAS, SRC and YAP Signaling in Pancreatic Cancer: Interactions Leading to Aggressive Disease and Drug Resistance. Cancers 2021, 13, 5126. https://doi.org/10.3390/cancers13205126
Rozengurt E, Eibl G. Crosstalk between KRAS, SRC and YAP Signaling in Pancreatic Cancer: Interactions Leading to Aggressive Disease and Drug Resistance. Cancers. 2021; 13(20):5126. https://doi.org/10.3390/cancers13205126
Chicago/Turabian StyleRozengurt, Enrique, and Guido Eibl. 2021. "Crosstalk between KRAS, SRC and YAP Signaling in Pancreatic Cancer: Interactions Leading to Aggressive Disease and Drug Resistance" Cancers 13, no. 20: 5126. https://doi.org/10.3390/cancers13205126
APA StyleRozengurt, E., & Eibl, G. (2021). Crosstalk between KRAS, SRC and YAP Signaling in Pancreatic Cancer: Interactions Leading to Aggressive Disease and Drug Resistance. Cancers, 13(20), 5126. https://doi.org/10.3390/cancers13205126