
PPARγ Targets-Derived Diagnostic and Prognostic Index for Papillary Thyroid Cancer

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Targeted DNA Sequencing and Analysis
2.3. Mapping of Protein Mutation Data
2.4. MSKCC-ATC and TCGA-THCA Data
2.5. MMD-THCA Data
2.6. ScRNA-Seq Data
2.7. DE, GSEA, and GO Analysis
2.8. CIBERSORT
2.9. ROC and Survival Analysis
2.10. PPARGi Derivation
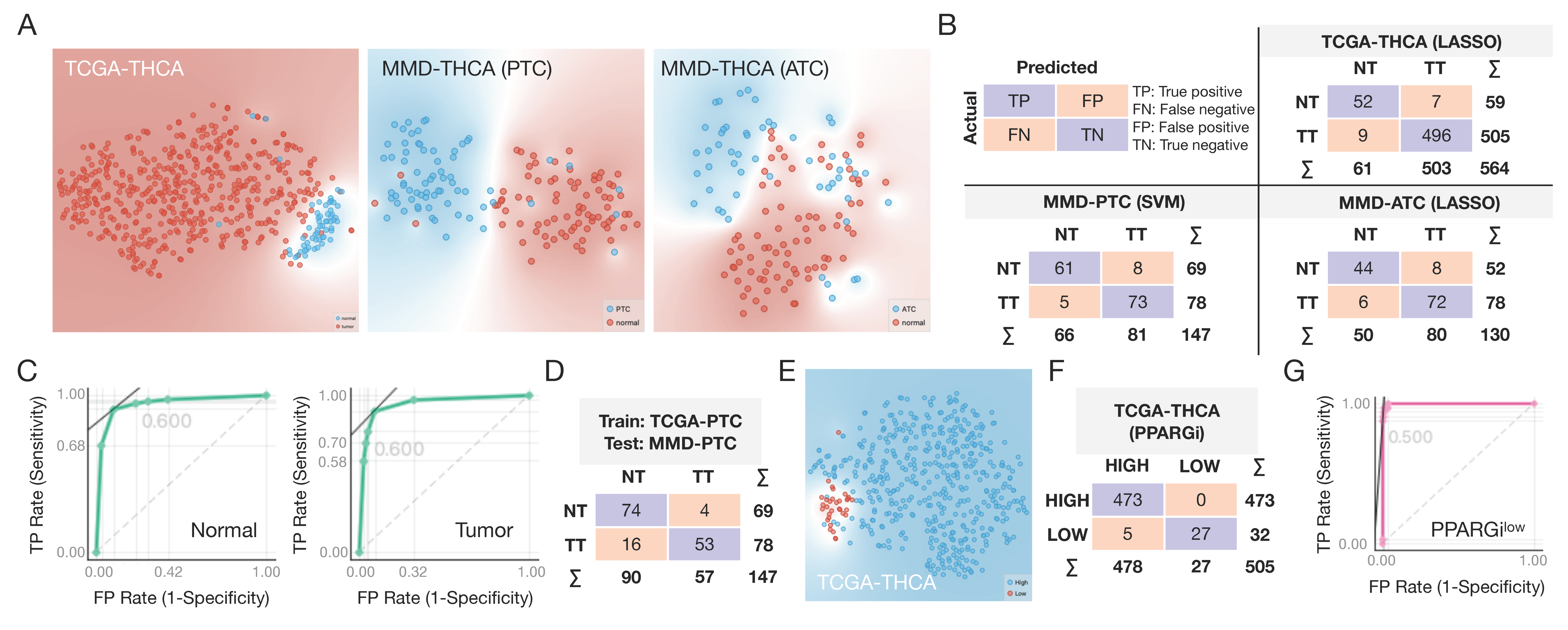
2.11. Machine Learning Algorithms for Disease Selection
3. Results
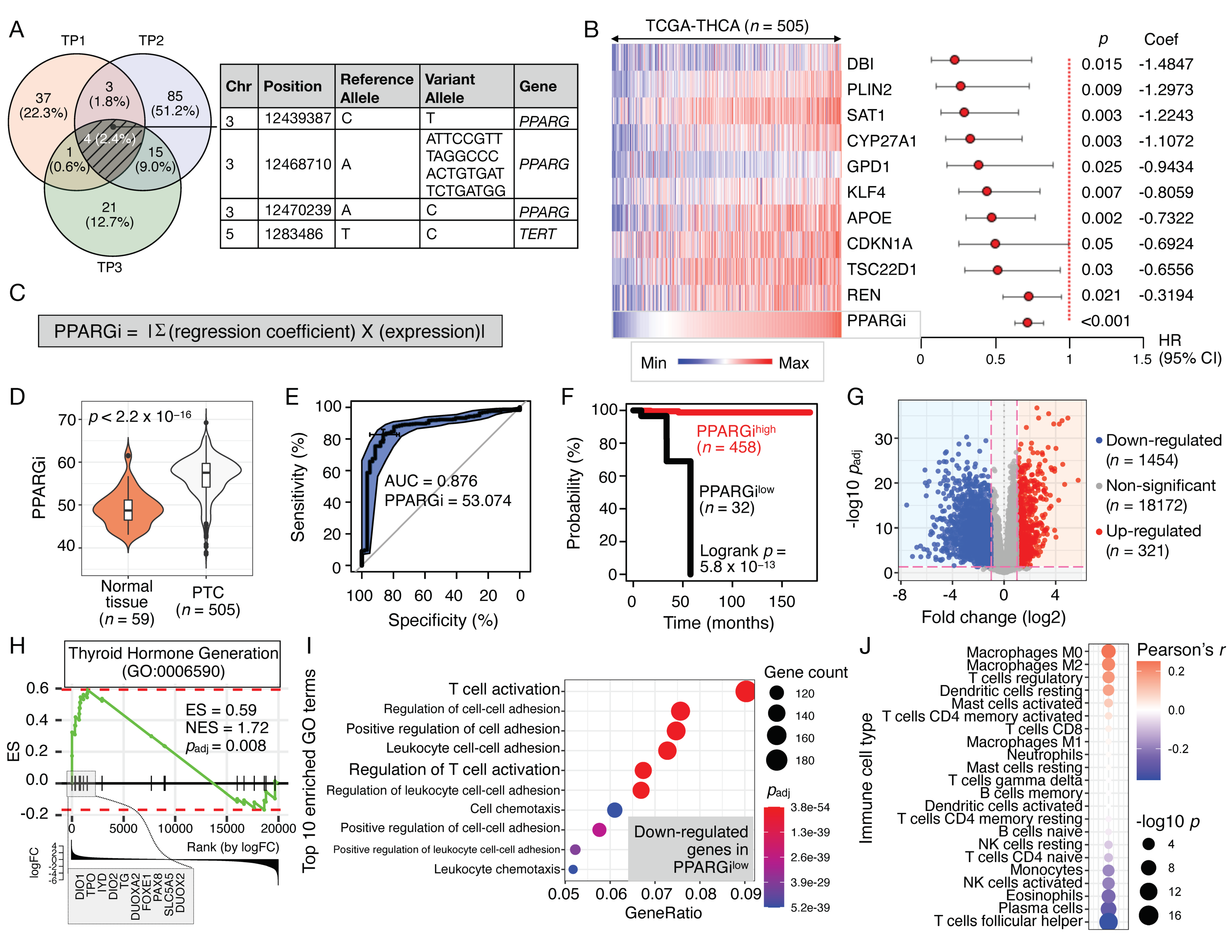
3.1. Targeted NGS of Thyroid Cancer-Related Genes in Monozygotic Twins with PTC
3.2. Clinical Significance of Pparγ Target Genes in Thyroid Cancer
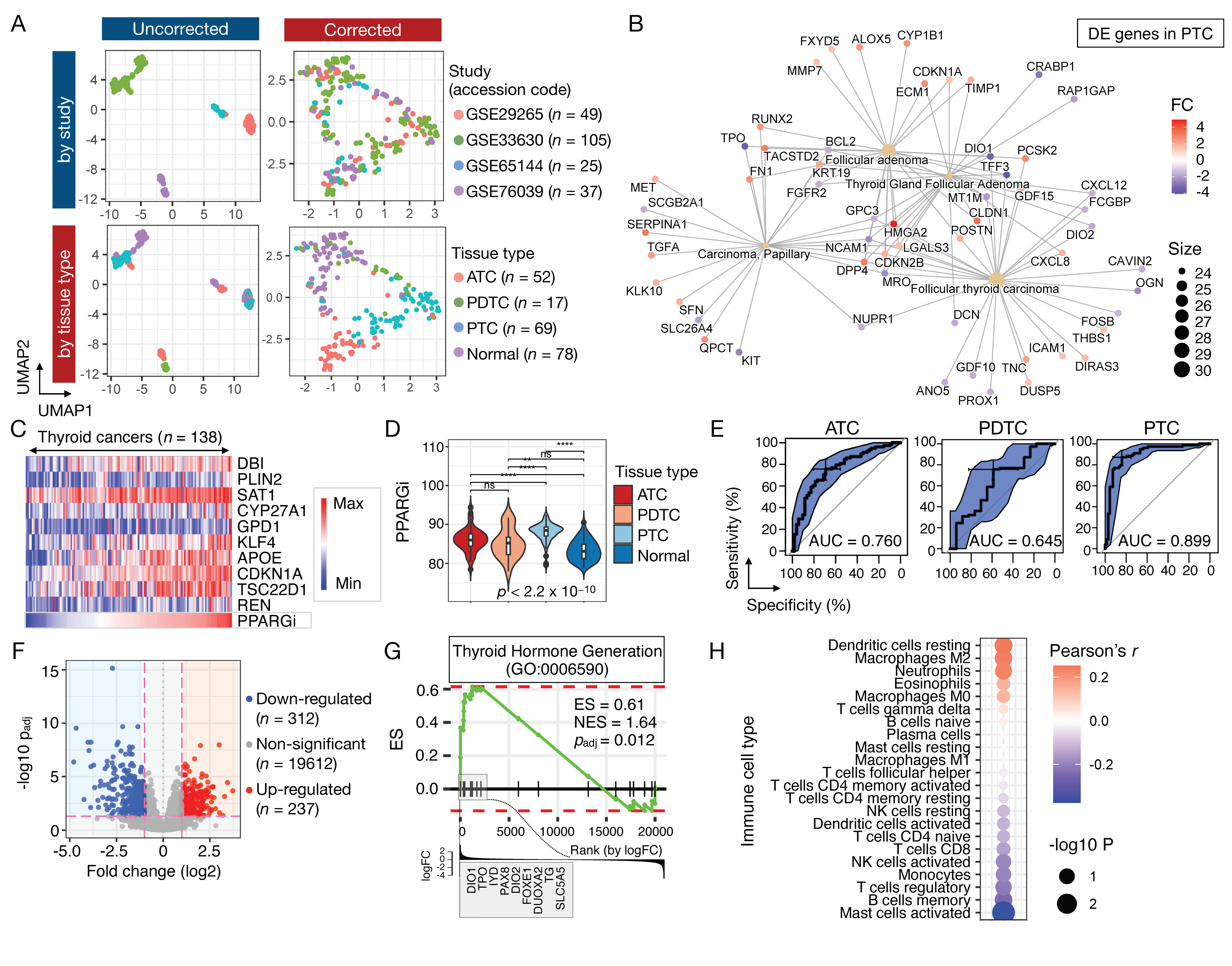
3.3. Validation of PPARGi in MMD-THCA
3.4. Single-Cell Analysis of Ppargi-Comprising Genes in PTC
3.5. Machine Learning for Disease Selection and Risk Stratification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miranda-Filho, A.; Lortet-Tieulent, J.; Bray, F.; Cao, B.; Franceschi, S.; Vaccarella, S.; Dal Maso, L. Thyroid cancer incidence trends by histology in 25 countries: A population-based study. Lancet Diabetes Endocrinol. 2021, 9, 225–234. [Google Scholar] [CrossRef]
- Park, S.; Oh, C.-M.; Cho, H.; Lee, J.Y.; Jung, K.-W.; Jun, J.K.; Won, Y.-J.; Kong, H.-J.; Choi, K.S.; Lee, Y.J.; et al. Association between screening and the thyroid cancer “epidemic” in South Korea: Evidence from a nationwide study. BMJ 2016, 355, i5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, I.J.; Kuk, D.; Wreesmann, V.; Morris, L.; Palmer, F.L.; Ganly, I.; Patel, S.G.; Singh, B.; Tuttle, R.M.; Shaha, A.R.; et al. Defining a valid age cutoff in staging of well-differentiated thyroid cancer. Ann. Surg. Oncol. 2016, 23, 410–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugazzola, L.; Muzza, M.; Pogliaghi, G.; Vitale, M. Intratumoral genetic heterogeneity in papillary thyroid cancer: Occurrence and clinical significance. Cancers 2020, 12, 383. [Google Scholar] [CrossRef] [Green Version]
- Muzza, M.; Pogliaghi, G.; Persani, L.; Fugazzola, L.; Colombo, C. Combined mutational and clonality analyses support the existence of intra-tumor heterogeneity in papillary thyroid cancer. J. Clin. Med. 2021, 10, 2645. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.A.; Hussein, M.H.; Zerfaoui, M.; Attia, A.S.; Marzouk Ellythy, A.; Mostafa, A.; Ruiz, E.M.L.; Shama, M.A.; Russell, J.O.; Randolph, G.W.; et al. Site-specific metastasis and survival in papillary thyroid cancer: The importance of brain and multi-organ disease. Cancers 2021, 13, 1625. [Google Scholar] [CrossRef]
- Khatami, F.; Tavangar, S.M. A review of driver genetic alterations in thyroid cancers. Iran. J. Pathol. 2018, 13, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Koenig, R.J. Pax-8–PPAR-γ fusion protein in thyroid carcinoma. Nat. Rev. Endocrinol. 2014, 10, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Vu-Phan, D.; Grachtchouk, V.; Yu, J.; Colby, L.A.; Wicha, M.S.; Koenig, R.J. The thyroid cancer PAX8-PPARG fusion protein activates Wnt/TCF-responsive cells that have a transformed phenotype. Endocr. Relat. Cancer 2013, 20, 725–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, N.L.; Grebe, S.K.; McIver, B.; Reddi, H.V. The role of the PAX8/PPARgamma fusion oncogene in the pathogenesis of follicular thyroid cancer. Mol. Cell. Endocrinol. 2010, 321, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in metabolism, immunity, and cancer: Unified and diverse mechanisms of action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef] [PubMed]
- Aprile, M.; Cataldi, S.; Ambrosio, M.R.; D’Esposito, V.; Lim, K.; Dietrich, A.; Bluher, M.; Savage, D.B.; Formisano, P.; Ciccodicola, A.; et al. PPARgammaDelta5, a naturally occurring dominant-negative splice isoform, impairs PPARgamma function and adipocyte differentiation. Cell Rep. 2018, 25, 1577–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARgamma and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Pedersen, T.A.; Hagenbeek, D.; Moulos, P.; Siersbaek, R.; Megens, E.; Denissov, S.; Borgesen, M.; Francoijs, K.J.; Mandrup, S.; et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008, 22, 2953–2967. [Google Scholar] [CrossRef] [Green Version]
- Sarhangi, N.; Sharifi, F.; Hashemian, L.; Hassani Doabsari, M.; Heshmatzad, K.; Rahbaran, M.; Jamaldini, S.H.; Aghaei Meybodi, H.R.; Hasanzad, M. PPARG (Pro12Ala) genetic variant and risk of T2DM: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 12764. [Google Scholar] [CrossRef]
- Le Menn, G.; Neels, J.G. Regulation of immune cell function by PPARs and the connection with metabolic and neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeles, L.; Torocsik, D.; Nagy, L. PPARgamma in immunity and inflammation: Cell types and diseases. Biochim. Biophys. Acta 2007, 1771, 1014–1030. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Rosen, E.D.; Fitzgerald, M.L.; Randow, F.; Andersson, L.P.; Altshuler, D.; Milstone, D.S.; Mortensen, R.M.; Spiegelman, B.M.; Freeman, M.W. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat. Med. 2001, 7, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Penas, F.; Mirkin, G.A.; Vera, M.; Cevey, A.; Gonzalez, C.D.; Gomez, M.I.; Sales, M.E.; Goren, N.B. Treatment in vitro with PPARalpha and PPARgamma ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim. Biophys. Acta 2015, 1852, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, A.; Barak, Y.; Nagy, L.; Liao, D.; Tontonoz, P.; Evans, R.M. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001, 7, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ricote, M.; Akiyama, T.E.; Gonzalez, F.J.; Glass, C.K. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 6712–6717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, V.; Gallo, M.A.; Letizia, F.; Aprile, M.; Casamassimi, A.; Ciccodicola, A. PPARG: Gene expression regulation and next-generation sequencing for unsolved issues. PPAR Res. 2010, 2010, 409168. [Google Scholar] [CrossRef] [PubMed]
- Vohra, S.; Biggin, P.C. Mutationmapper: A tool to aid the mapping of protein mutation data. PLoS ONE 2013, 8, e71711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, N.P.; Reznik, E.; Gao, J.; Sumer, S.O.; Schultz, N.; Sander, C.; Miller, M.L. MutationAligner: A resource of recurrent mutation hotspots in protein domains in cancer. Nucleic Acids Res. 2016, 44, D986–D991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahceci, I.; Dogrusoz, U.; La, K.C.; Babur, O.; Gao, J.; Schultz, N. PathwayMapper: A collaborative visual web editor for cancer pathways and genomic data. Bioinformatics 2017, 33, 2238–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.B.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.-T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. NPJ Precis. Oncol. 2019, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.B.; Tan, S.J.; Lim, W.T.; Lim, C.T. A merged lung cancer transcriptome dataset for clinical predictive modeling. Sci. Data 2018, 5, 180136. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.B.; Tan, S.J.; Lim, W.T.; Lim, C.T. An extracellular matrix-related prognostic and predictive indicator for early-stage non-small cell lung cancer. Nat. Commun. 2017, 8, 1734. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.B.; Tan, S.J.; Lim, W.T.; Lim, C.T. Compendiums of cancer transcriptomes for machine learning applications. Sci. Data 2019, 6, 194. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Wei, G.; Zhang, Y.; Li, H.; Lai, Y.; Guo, Y.; Chen, Y.; Liu, L.; Xiao, H.; Guan, H.; et al. Single-cell transcriptomic landscape reveals the differences in cell differentiation and immune microenvironment of papillary thyroid carcinoma between genders. Cell Biosci. 2021, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021, 060012. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.B.; Yeo, T.; Lee, W.D.; Bhagat, A.A.S.; Tan, S.J.; Tan, D.S.W.; Lim, W.T.; Lim, C.T. Addressing cellular heterogeneity in tumor and circulation for refined prognostication. Proc. Natl. Acad. Sci. USA 2019, 116, 17957–17962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Gyorffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Zhang, M.; Li, Y.; Liu, Y.; Cui, Q.; Wang, N. PPARgene: A database of experimentally verified and computationally predicted PPAR target genes. PPAR Res. 2016, 2016, 6042162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Seo, J.; Park, J.; Nam, J.Y.; Choi, A.; Ignatius, J.S.; Bjornson, R.D.; Chae, J.H.; Jang, I.J.; Lee, S.; et al. Korean Variant Archive (KOVA): A reference database of genetic variations in the Korean population. Sci. Rep. 2017, 7, 4287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, D.M.; Jang, J.Y.; Kim, H.J.; Han, B.W. Differential effects of cancer-associated mutations enriched in helix H3 of PPARgamma. Cancers 2020, 12, 3580. [Google Scholar] [CrossRef] [PubMed]
- Yousefnia, S.; Momenzadeh, S.; Seyed Forootan, F.; Ghaedi, K.; Nasr Esfahani, M.H. The influence of peroxisome proliferator-activated receptor gamma (PPARgamma) ligands on cancer cell tumorigenicity. Gene 2018, 649, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Puyang, X.; Jeremy Wu, Z.; Seiler, R.; Furman, C.; Oo, H.Z.; Seiler, M.; Irwin, S.; Subramanian, V.; Julie Joshi, J.; et al. Evasion of immunosurveillance by genomic alterations of PPARgamma/RXRalpha in bladder cancer. Nat. Commun. 2017, 8, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heming, M.; Gran, S.; Jauch, S.L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schafers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome proliferator-activated receptor-gamma modulates the response of macrophages to lipopolysaccharide and glucocorticoids. Front. Immunol. 2018, 9, 893. [Google Scholar] [CrossRef]
- Setoguchi, K.; Misaki, Y.; Terauchi, Y.; Yamauchi, T.; Kawahata, K.; Kadowaki, T.; Yamamoto, K. Peroxisome proliferator-activated receptor-gamma haploinsufficiency enhances B cell proliferative responses and exacerbates experimentally induced arthritis. J. Clin. Investig. 2001, 108, 1667–1675. [Google Scholar] [CrossRef]
- Lee, C.H.; Chawla, A.; Urbiztondo, N.; Liao, D.; Boisvert, W.A.; Evans, R.M.; Curtiss, L.K. Transcriptional repression of atherogenic inflammation: Modulation by PPARdelta. Science 2003, 302, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Kuenzli, S.; Saurat, J.H. Effect of topical PPARbeta/delta and PPARgamma agonists on plaque psoriasis. A pilot study. Dermatology 2003, 206, 252–256. [Google Scholar] [CrossRef]
- Kobawala, T.P.; Trivedi, T.I.; Gajjar, K.K.; Patel, D.H.; Patel, G.H.; Ghosh, N.R. Significance of interleukin-6 in papillary thyroid carcinoma. J. Thyroid Res. 2016, 2016, 6178921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Su, C.; Xu, J.; Zhou, D.; Yan, H.; Li, W.; Chen, G.; Zhang, N.; Xu, D.; Hu, H. Immunohistochemical analysis of matrix metalloproteinase-9 predicts papillary thyroid carcinoma prognosis. Oncol. Lett. 2019, 17, 2308–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Model | AUC | CA | F1 | Precision | Recall |
|---|---|---|---|---|---|---|
| TCGA-THCA | kNN | 0.980 | 0.961 | 0.962 | 0.963 | 0.961 |
| SVM | 0.989 | 0.968 | 0.968 | 0.967 | 0.968 | |
| Random forest | 0.975 | 0.956 | 0.954 | 0.954 | 0.956 | |
| Neural network | 0.986 | 0.975 | 0.976 | 0.976 | 0.975 | |
| Logistic regression | 0.990 | 0.972 | 0.972 | 0.972 | 0.972 | |
| MMD-THCA (PTC) | kNN | 0.925 | 0.884 | 0.884 | 0.885 | 0.884 |
| SVM | 0.937 | 0.912 | 0.911 | 0.912 | 0.912 | |
| Random forest | 0.914 | 0.871 | 0.871 | 0.871 | 0.871 | |
| Neural network | 0.928 | 0.912 | 0.911 | 0.913 | 0.912 | |
| Logistic regression | 0.929 | 0.912 | 0.912 | 0.912 | 0.912 | |
| MMD-THCA (ATC) | kNN | 0.944 | 0.838 | 0.835 | 0.841 | 0.838 |
| SVM | 0.929 | 0.862 | 0.861 | 0.861 | 0.862 | |
| Random forest | 0.897 | 0.808 | 0.807 | 0.807 | 0.808 | |
| Neural network | 0.938 | 0.862 | 0.862 | 0.862 | 0.862 | |
| Logistic regression | 0.945 | 0.892 | 0.892 | 0.892 | 0.892 |
| Dataset | Model | AUC | CA | F1 | Precision | Recall |
|---|---|---|---|---|---|---|
| Train-test data | kNN | 0.942 | 0.864 | 0.862 | 0.873 | 0.864 |
| SVM | 0.890 | 0.469 | 0.300 | 0.220 | 0.469 | |
| Random forest | 0.828 | 0.599 | 0.551 | 0.721 | 0.599 | |
| Neural network | 0.925 | 0.837 | 0.836 | 0.860 | 0.837 | |
| Logistic regression | 0.922 | 0.531 | 0.368 | 0.282 | 0.531 |
| Dataset | Model | AUC | CA | F1 | Precision | Recall |
|---|---|---|---|---|---|---|
| TCGA-THCA | kNN | 0.965 | 0.980 | 0.979 | 0.979 | 0.980 |
| SVM | 0.998 | 0.990 | 0.990 | 0.990 | 0.990 | |
| Random forest | 0.988 | 0.966 | 0.962 | 0.964 | 0.966 | |
| Neural network | 0.997 | 0.982 | 0.982 | 0.982 | 0.982 | |
| Logistic regression | 0.974 | 0.976 | 0.975 | 0.975 | 0.976 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Kim, S.Y.; Ma, S.-X.; Kim, S.-M.; Shin, S.-J.; Lee, Y.S.; Chang, H.; Chang, H.-S.; Park, C.S.; Lim, S.B. PPARγ Targets-Derived Diagnostic and Prognostic Index for Papillary Thyroid Cancer. Cancers 2021, 13, 5110. https://doi.org/10.3390/cancers13205110
Kim J, Kim SY, Ma S-X, Kim S-M, Shin S-J, Lee YS, Chang H, Chang H-S, Park CS, Lim SB. PPARγ Targets-Derived Diagnostic and Prognostic Index for Papillary Thyroid Cancer. Cancers. 2021; 13(20):5110. https://doi.org/10.3390/cancers13205110
Chicago/Turabian StyleKim, Jaehyung, Soo Young Kim, Shi-Xun Ma, Seok-Mo Kim, Su-Jin Shin, Yong Sang Lee, Hojin Chang, Hang-Seok Chang, Cheong Soo Park, and Su Bin Lim. 2021. "PPARγ Targets-Derived Diagnostic and Prognostic Index for Papillary Thyroid Cancer" Cancers 13, no. 20: 5110. https://doi.org/10.3390/cancers13205110
APA StyleKim, J., Kim, S. Y., Ma, S.-X., Kim, S.-M., Shin, S.-J., Lee, Y. S., Chang, H., Chang, H.-S., Park, C. S., & Lim, S. B. (2021). PPARγ Targets-Derived Diagnostic and Prognostic Index for Papillary Thyroid Cancer. Cancers, 13(20), 5110. https://doi.org/10.3390/cancers13205110