
The Evolution of Clinically Aggressive Triple-Negative Breast Cancer Shows a Large Mutational Diversity and Early Metastasis to Lymph Nodes

 ,
,  , , , ,
, , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Information
2.2. Sample Preparation and DNA Extraction
2.3. Library Preparation
2.4. SNV e Indels Identification
2.5. Tumor Mutational Burden Analysis
2.6. CNV Identification
2.7. Mutational Signature Analysis
2.8. Pathway Enrichment Analysis
2.9. Global Actionable Alterations
2.10. Evolutionary Tree Construction
3. Results
3.1. Patients with TNBC Exhibit a Variety of Clinical Scenarios
3.2. Patients with TNBC Display an Extensive Genetic Heterogeneity
3.3. Mutational Processes Act on Different Stages of TNBC
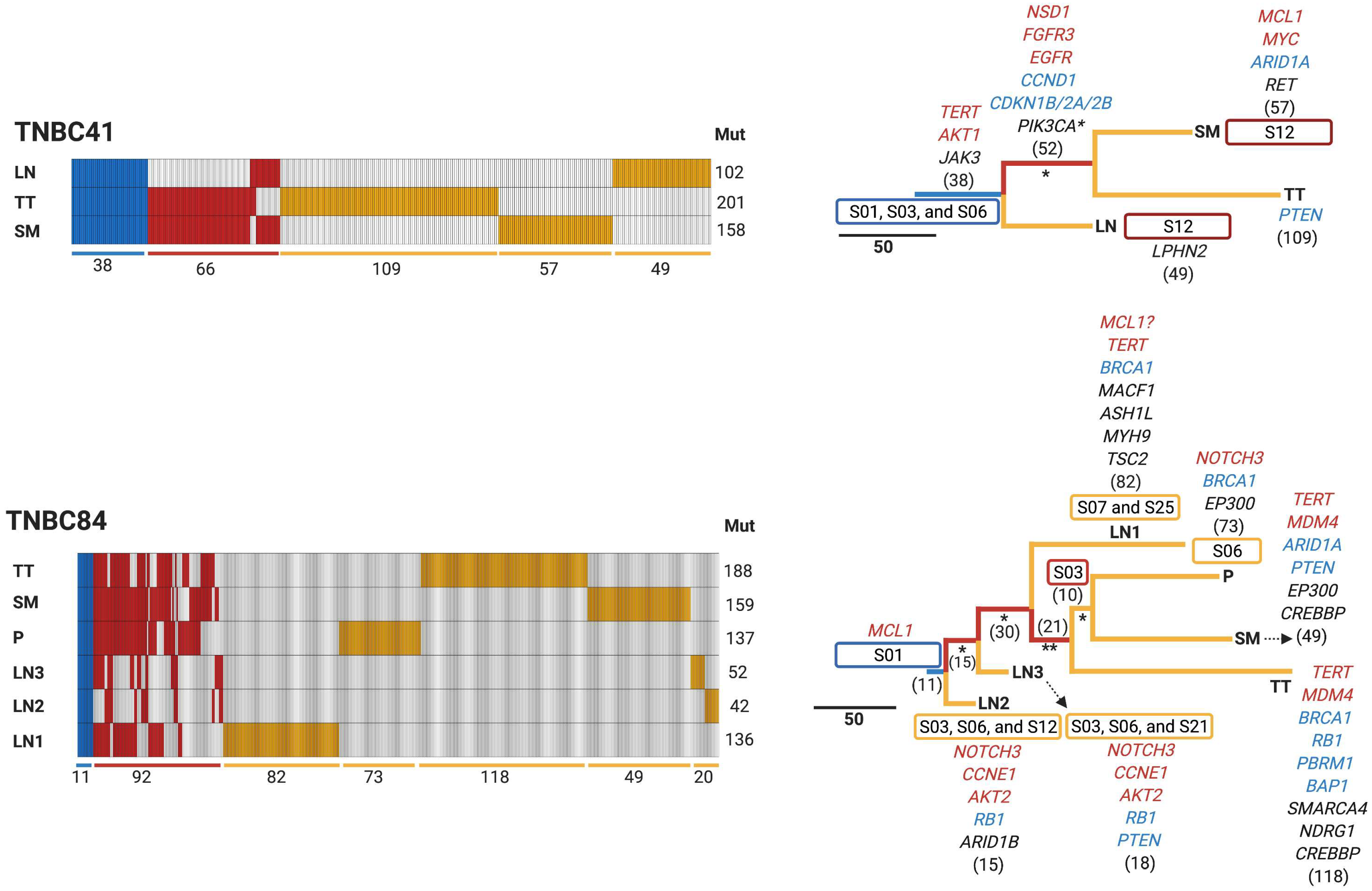
3.4. Early Divergence in TNBC
3.5. Potential Clinical Implications of Mutations Detected in TNBC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primer 2019, 5, 66. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Jones, S.; Chen, W.-D.; Parmigiani, G.; Diehl, F.; Beerenwinkel, N.; Antal, T.; Traulsen, A.; Nowak, M.A.; Siegel, C.; Velculescu, V.E.; et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 4283–4288. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Buonomo, O.C.; Caredda, E.; Portarena, I.; Vanni, G.; Orlandi, A.; Bagni, C.; Petrella, G.; Palombi, L.; Orsaria, P. New insights into the metastatic behavior after breast cancer surgery, according to well-established clinicopathological variables and molecular subtypes. PLoS ONE 2017, 12, e0184680. [Google Scholar] [CrossRef] [PubMed]
- Colleoni, M.; Zahrieh, D.; Gelber, R.D.; Holmberg, S.B.; Mattsson, J.E.; Rudenstam, C.-M.; Lindtner, J.; Erzen, D.; Snyder, R.; Collins, J.; et al. Site of primary tumor has a prognostic role in operable breast cancer: The international breast cancer study group experience. J. Clin. Oncol. 2005, 23, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.R.A. Skin metastasis: A pathologist’s perspective. J. Cutan. Pathol. 2010, 37, e1–e20. [Google Scholar] [CrossRef]
- Kalmykow, B.; Walker, S. Cutaneous metastases in breast cancer. Clin. J. Oncol. Nurs. 2011, 15, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef]
- Brown, D.; Smeets, D.; Székely, B.; Larsimont, D.; Szász, A.M.; Adnet, P.-Y.; Rothé, F.; Rouas, G.; Nagy, Z.I.; Faragó, Z.; et al. Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat. Commun. 2017, 8, 14944. [Google Scholar] [CrossRef]
- Ullah, I.; Karthik, G.-M.; Alkodsi, A.; Kjällquist, U.; Stålhammar, G.; Lövrot, J.; Martinez, N.-F.; Lagergren, J.; Hautaniemi, S.; Hartman, J.; et al. Evolutionary history of metastatic breast cancer reveals minimal seeding from axillary lymph nodes. J. Clin. Investig. 2018, 128, 1355–1370. [Google Scholar] [CrossRef]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinform. Oxf. Engl. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 1–11. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef]
- Fitch, W.M. Toward Defining the Course of Evolution: Minimum Change for a Specific Tree Topology. Syst. Biol. 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Efron, B.; Halloran, E.; Holmes, S. Bootstrap confidence levels for phylogenetic trees. Proc. Natl. Acad. Sci. USA 1996, 93, 13429–13434. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Peng, L.; Sahin, A.A.; Huo, L.; Ward, K.C.; O’Regan, R.; Torres, M.A.; Meisel, J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 161, 279–287. [Google Scholar] [CrossRef]
- Reynoso-Noverón, N.; Villarreal-Garza, C.; Soto-Perez-de-Celis, E.; Arce-Salinas, C.; Matus-Santos, J.; Ramírez-Ugalde, M.T.; Alvarado-Miranda, A.; Cabrera-Galeana, P.; Meneses-García, A.; Lara-Medina, F.; et al. Clinical and Epidemiological Profile of Breast Cancer in Mexico: Results of the Seguro Popular. J. Glob. Oncol. 2017, 3, 757–764. [Google Scholar] [CrossRef]
- Rojas-Jiménez, E.; Mejía-Gómez, J.C.; Díaz-Velásquez, C.; Quezada-Urban, R.; Martínez Gregorio, H.; Vallejo-Lecuona, F.; de la Cruz-Montoya, A.; Porras Reyes, F.I.; Pérez-Sánchez, V.M.; Maldonado-Martínez, H.A.; et al. Comprehensive Genomic Profile of Heterogeneous Long Follow-Up Triple-Negative Breast Cancer and Its Clinical Characteristics Shows DNA Repair Deficiency Has Better Prognostic. Genes 2020, 11, 1367. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.; Vatsiou, A.; Spiteri, I.; Nichol, D.; Cresswell, G.D.; Acar, A.; Trahearn, N.; Hrebien, S.; Garcia-Murillas, I.; Chkhaidze, K.; et al. The Spatiotemporal Evolution of Lymph Node Spread in Early Breast Cancer. Clin. Cancer Res. 2018, 24, 4763–4770. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Grönroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Pich, O.; Muiños, F.; Lolkema, M.P.; Steeghs, N.; Gonzalez-Perez, A.; Lopez-Bigas, N. The mutational footprints of cancer therapies. Nat. Genet. 2019, 51, 1732–1740. [Google Scholar] [CrossRef]
- Szikriszt, B.; Póti, Á.; Pipek, O.; Krzystanek, M.; Kanu, N.; Molnár, J.; Ribli, D.; Szeltner, Z.; Tusnády, G.E.; Csabai, I.; et al. A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 2016, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Morganella, S. Mutational Signatures in Breast Cancer: The Problem at the DNA Level. Clin. Cancer Res. 2017, 23, 2617–2629. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.-C.; Massard, C.; Lévy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.; Leung, S.C.Y.; Gao, D.; Burugu, S.; Anurag, M.; Ellis, M.J.; Nielsen, T.O. Mismatch repair protein loss in breast cancer: Clinicopathological associations in a large British Columbia cohort. Breast Cancer Res. Treat. 2020, 179, 3–10. [Google Scholar] [CrossRef]
- Chitty, J.L.; Filipe, E.C.; Lucas, M.C.; Herrmann, D.; Cox, T.R.; Timpson, P. Recent advances in understanding the complexities of metastasis. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Halsted, W.S.I. The Results of Operations for the Cure of Cancer of the Breast Performed at the Johns Hopkins Hospital from June, 1889, to January, 1894. Ann. Surg. 1894, 20, 497–555. [Google Scholar] [CrossRef]
- Patey, D.H.; Dyson, W.H. The prognosis of carcinoma of the breast in relation to the type of operation performed. Br. J. Cancer 1948, 2, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Venet, D.; Fimereli, D.; Rothé, F.; Boeckx, B.; Maetens, M.; Majjaj, S.; Rouas, G.; Capra, M.; Bonizzi, G.; Contaldo, F.; et al. Phylogenetic reconstruction of breast cancer reveals two routes of metastatic dissemination associated with distinct clinical outcome. EBioMedicine 2020, 56, 102793. [Google Scholar] [CrossRef]
- Chen, X.; Yu, X.; Chen, J.; Zhang, Z.; Tuan, J.; Shao, Z.; Guo, X.; Feng, Y. Analysis in early stage triple-negative breast cancer treated with mastectomy without adjuvant radiotherapy: Patterns of failure and prognostic factors. Cancer 2013, 119, 2366–2374. [Google Scholar] [CrossRef]
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 2020, 52, 701–708. [Google Scholar] [CrossRef]
- Wang, X.-X.; Jiang, Y.-Z.; Li, J.-J.; Song, C.-G.; Shao, Z.-M. Effect of nodal status on clinical outcomes of triple-negative breast cancer: A population-based study using the SEER 18 database. Oncotarget 2016, 7, 46636–46645. [Google Scholar] [CrossRef][Green Version]
- Hunter, K.W.; Amin, R.; Deasy, S.; Ha, N.-H.; Wakefield, L. Genetic insights into the morass of metastatic heterogeneity. Nat. Rev. Cancer 2018, 18, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Gregorio, H.; Rojas-Jiménez, E.; Mejía-Gómez, J.C.; Díaz-Velásquez, C.; Quezada-Urban, R.; Vallejo-Lecuona, F.; de la Cruz-Montoya, A.; Porras-Reyes, F.I.; Pérez-Sánchez, V.M.; Maldonado-Martínez, H.A.; et al. The Evolution of Clinically Aggressive Triple-Negative Breast Cancer Shows a Large Mutational Diversity and Early Metastasis to Lymph Nodes. Cancers 2021, 13, 5091. https://doi.org/10.3390/cancers13205091
Martínez-Gregorio H, Rojas-Jiménez E, Mejía-Gómez JC, Díaz-Velásquez C, Quezada-Urban R, Vallejo-Lecuona F, de la Cruz-Montoya A, Porras-Reyes FI, Pérez-Sánchez VM, Maldonado-Martínez HA, et al. The Evolution of Clinically Aggressive Triple-Negative Breast Cancer Shows a Large Mutational Diversity and Early Metastasis to Lymph Nodes. Cancers. 2021; 13(20):5091. https://doi.org/10.3390/cancers13205091
Chicago/Turabian StyleMartínez-Gregorio, Héctor, Ernesto Rojas-Jiménez, Javier César Mejía-Gómez, Clara Díaz-Velásquez, Rosalía Quezada-Urban, Fernando Vallejo-Lecuona, Aldo de la Cruz-Montoya, Fany Iris Porras-Reyes, Víctor Manuel Pérez-Sánchez, Héctor Aquiles Maldonado-Martínez, and et al. 2021. "The Evolution of Clinically Aggressive Triple-Negative Breast Cancer Shows a Large Mutational Diversity and Early Metastasis to Lymph Nodes" Cancers 13, no. 20: 5091. https://doi.org/10.3390/cancers13205091
APA StyleMartínez-Gregorio, H., Rojas-Jiménez, E., Mejía-Gómez, J. C., Díaz-Velásquez, C., Quezada-Urban, R., Vallejo-Lecuona, F., de la Cruz-Montoya, A., Porras-Reyes, F. I., Pérez-Sánchez, V. M., Maldonado-Martínez, H. A., Robles-Estrada, M., Bargalló-Rocha, E., Cabrera-Galeana, P., Ramos-Ramírez, M., Chirino, Y. I., Alonso Herrera, L., Terrazas, L. I., Frecha, C., Oliver, J., ... Vaca-Paniagua, F. (2021). The Evolution of Clinically Aggressive Triple-Negative Breast Cancer Shows a Large Mutational Diversity and Early Metastasis to Lymph Nodes. Cancers, 13(20), 5091. https://doi.org/10.3390/cancers13205091