The Taming of Nuclear Factor Erythroid-2-Related Factor-2 (Nrf2) Deglycation by Fructosamine-3-Kinase (FN3K)-Inhibitors-A Novel Strategy to Combat Cancers


 ,
,
Simple Summary
Abstract
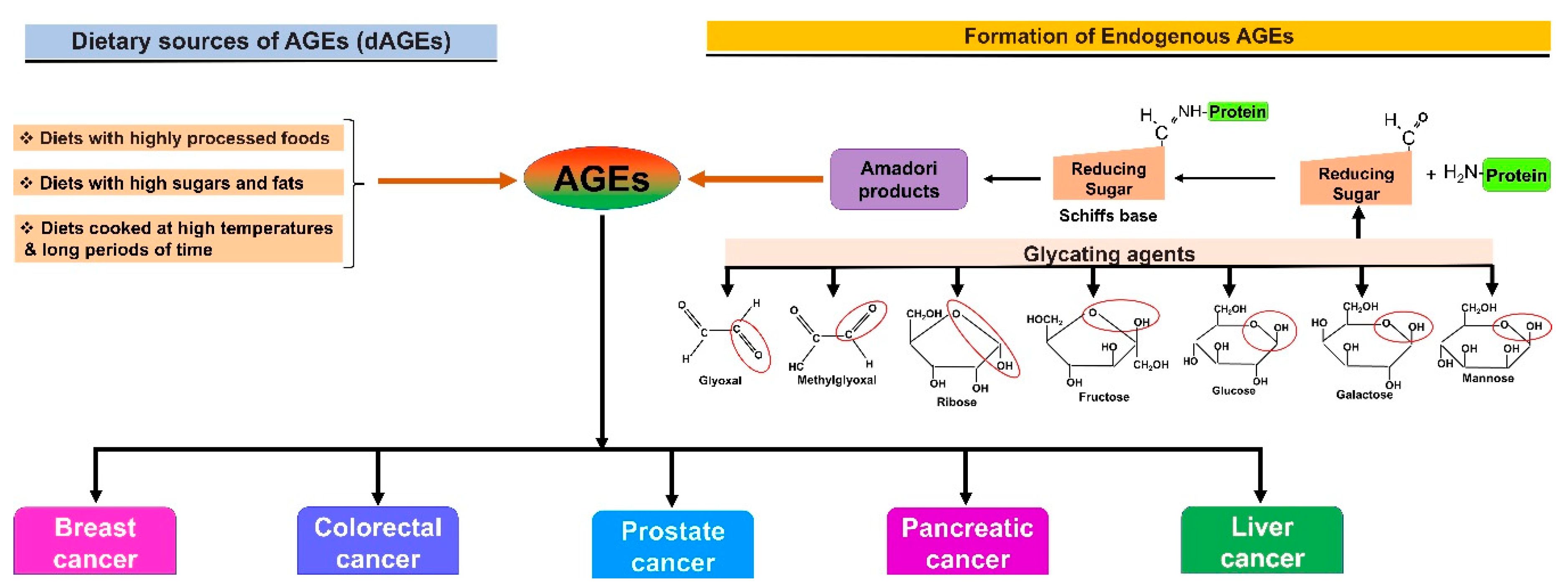
1. Introduction
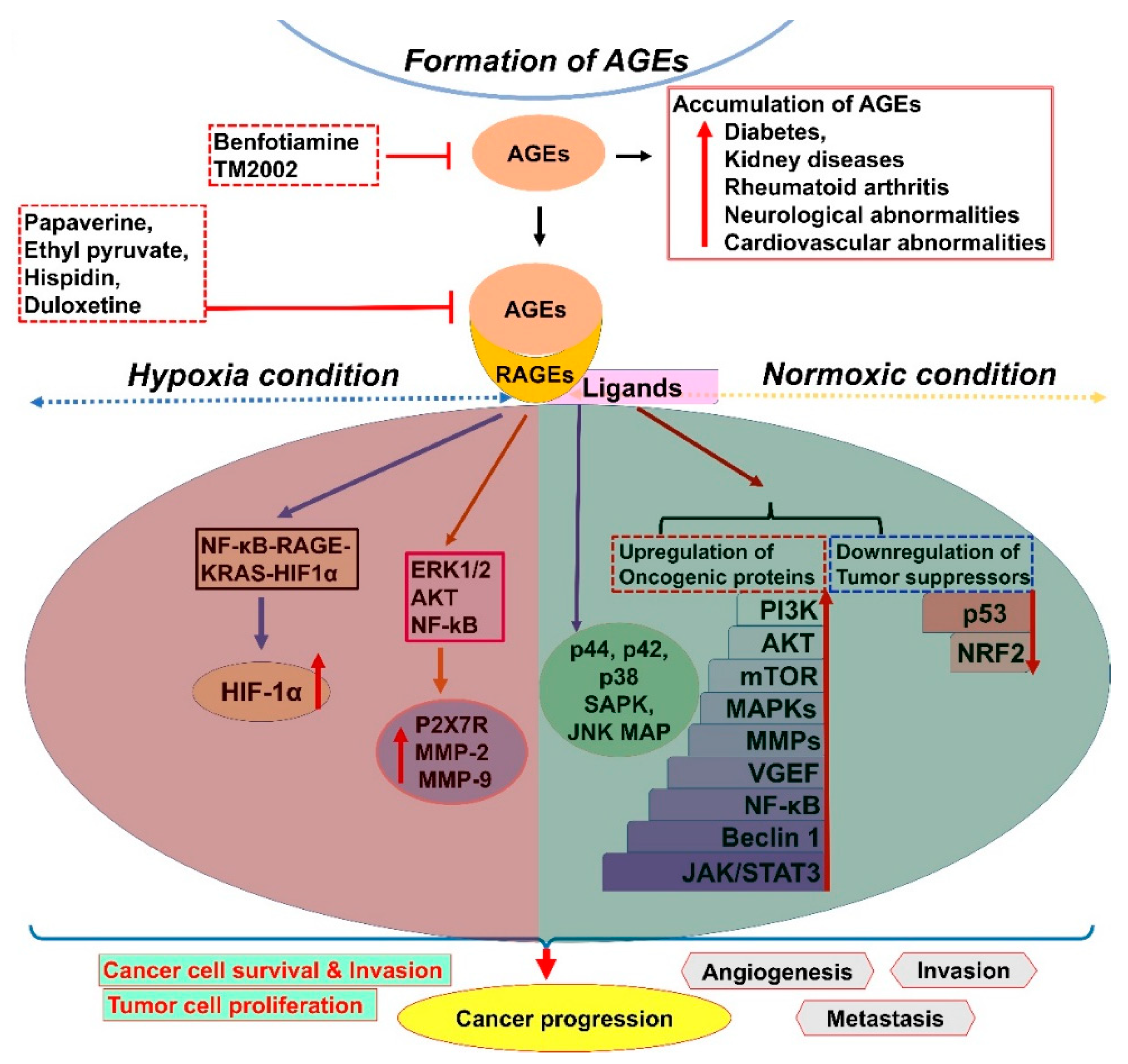
2. AGE–RAGE Signaling and Cancer Progression
3. Role of Glycation in the Modulation of Target Protein Expression and Activity
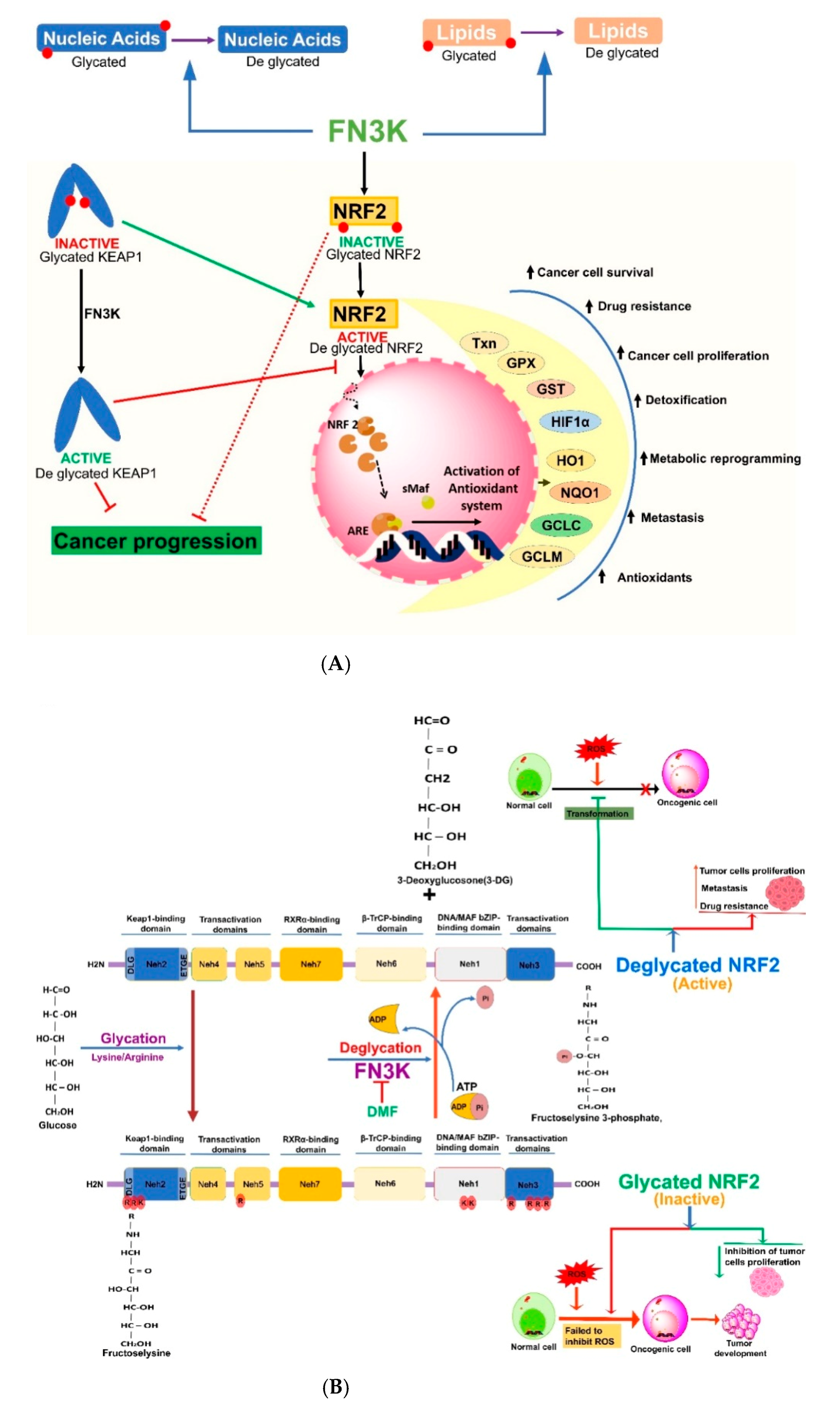
4. Nrf2 and Its Glycation and Deglycation Mechanisms in Regulation of Cancers
4.1. Nuclear Factor Erythroid-2-Related Factor 2 (Nrf2)
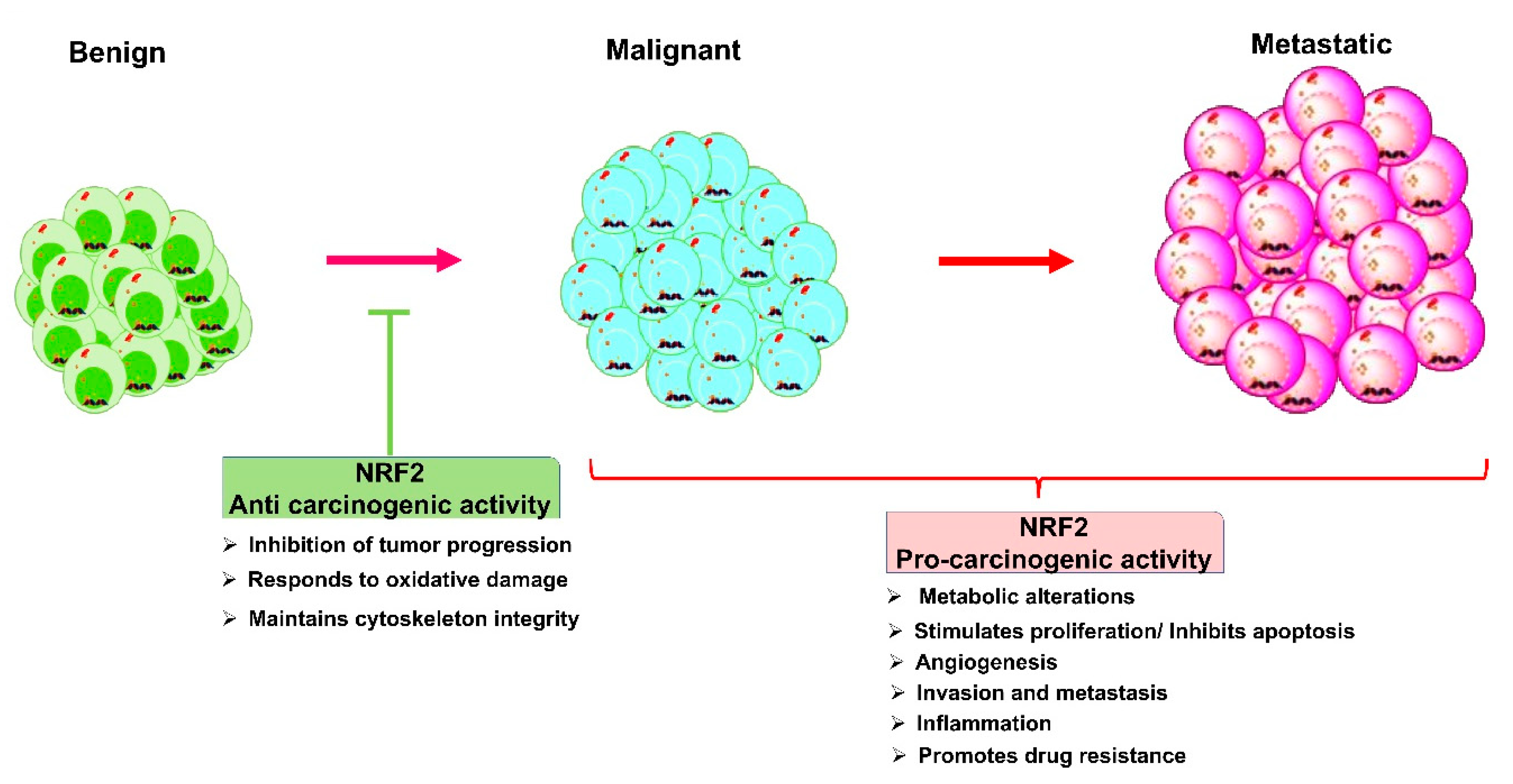
4.2. Nrf2 Role in Cancers: A Double-Edged Sword
4.3. Glycation induced AGE-RAGE Signaling and Nrf2
4.4. Global Inhibition of Deglycation and Nrf2: Limitations
5. Fructosamine Kinases (FN3K and FN3K-RP) in the Regulation of Cancers
6. Cross-Talk between Nrf2 and FN3K-Mediated Deglycation
7. Need for the Development of FN3K Inhibitors against Breast Cancer
8. Phytochemical and Drug Interventions to Modulate Glycation of Proteins
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-DG | 3-deoxyglucosone |
| 4-HNE | 4-hydroxynonenal |
| AGEs | Advanced glycation end products |
| AP-1 | Activator protein 1 |
| ARE | Antioxidant response element |
| BC | Breast cancer |
| BCRP | Breast cancer resistance protein |
| BSA | Bovine serum albumin |
| CAND1 | Cullin-associated NEDD8-dissociated protein 1 |
| ChREBP | Carbohydrate response element binding protein |
| CUL3 | Cullin3 |
| eIF1 | Eukaryotic translation initiation factor 1 |
| eIF3G | Eukaryotic translation initiation factor 3 subunit G |
| elF4A1 | Eukaryotic initiation factor 4A-I |
| eNOS | Endothelial nitric oxide synthase |
| EMT | Epithelial–mesenchymal transition |
| FGFR | Human fibroblast growth factor receptor |
| FL6PDG | Fructoselysine-6-phosphate deglycase |
| FN3K | Fructosamine-3-kinase |
| FN3K-RP | Fructosamine-3-kinase-related-protein |
| FN6K | Fructosamine-6-kinase |
| GCLC | Glutamate-cysteine ligase catalytic subunit |
| GCLM | Glutamate cysteine ligase modifier subunit |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| GST | Glutathione S-transferase |
| HbA1c | Glycated hemoglobin |
| HBXIP | Mammalian hepatitis B X-interacting protein |
| HCC | Hepatocellular carcinoma |
| HELB | DNA helicase B |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HMGB1 | High mobility group box-1 |
| HO1 | Heme oxygenase-1 |
| HSP | Heat shock protein |
| HSP90AA1 | Heat shock protein 90 alpha family class A member 1 |
| HSP90AA4 | Heat shock protein 90 alpha family class A member 4 |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin 6 |
| JAK | Janus kinase |
| Keap1 | Kelch-like ECH-associated protein 1 |
| KRAS | Kirsten rat sarcoma viral oncogene homologue |
| LDHA | Lactate dehydrogenase A |
| LDHC | Lactate dehydrogenase C |
| MAF | Musculoaponeurotic fibrosarcoma |
| Maf | Musculoaponeurotic fibrosarcoma |
| MAPK | Mitogen activated protein kinase |
| MCM3 | Minichromosome maintenance complex component 3 |
| MD | Malondialdehyde |
| MGO | Methylglyoxal |
| MMPs | Matrix metalloproteinases/Matrix metallopeptidases |
| mTOR | Mammalian target of rapamycin |
| NAD(P)H | Reduced nicotinamide adenine dinucleotide phosphate |
| NFE2L2 | Nuclear factor erythroid-derived 2-like 2 gene |
| NF-κB | Nuclear factor kappa—B |
| NOX | NADPH oxidases |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor erythroid 2 (NFE2)-related factor 2 |
| NSCLC | Non small cell lung carcinoma |
| NSMIs | Novel small molecule inhibitors |
| p53 | Tumor protein p53/phosphoprotein p53/tumor suppressor p53 |
| pERK | Protein kinase R-like endoplasmic reticulum kinase |
| PI3K | Phosphoinositide 3-kinase |
| PNG-1 | Peptide N-Glycanase 1 |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| PUF60 | Poly(U)-binding-splicing factor |
| RAGEs | Receptors for advanced glycation end products |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SCLCs | Squamous cell lung carcinomas |
| sMaf | small Maf proteins |
| SOD | Superoxide dismutase |
| SRSF7 | Serine/arginine-rich splicing factor 7 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAMs | Tumor-associated macrophages |
| TCGA | The cancer genome atlas |
| TNF | Tumor necrosis factor |
| TNFR-1 | Tumor necrosis factor receptor 1 |
| TNF-α | Tumor necrosis factor alpha |
| Txn | Thioredoxin |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
| YES1 | YES proto-oncogene 1 |
References
- Malik, P.; Chaudhry, N.; Mittal, R.; Mukherjee, T.K. Role of receptor for advanced glycation end products in the complication and progression of various types of cancers. Biochim. Biophys. Acta 2015, 1850, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Song, X.L.; Jia, L.Y.; Song, F.L.; Zhao, S.C.; Jiang, Y. Differential expressions of the receptor for advanced glycation end products in prostate cancer and normal prostate. Zhonghua Nan Ke Xue 2010, 16, 405–409. [Google Scholar] [PubMed]
- Khan, M.I.; Rath, S.; Adhami, V.M.; Mukhtar, H. Hypoxia driven glycation: Mechanisms and therapeutic opportunities. Semin. Cancer Biol. 2018, 49, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Gkogkolou, P.; Bohm, M. Advanced glycation end products: Key players in skin aging? Dermatoendocrinology 2012, 4, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Younus, H.; Anwar, S. Prevention of non-enzymatic glycosylation (glycation): Implication in the treatment of diabetic complication. Int. J. Health Sci. 2016, 10, 261–277. [Google Scholar] [CrossRef]
- Turner, D.P. Advanced glycation end-products: A biological consequence of lifestyle contributing to cancer disparity. Cancer Res. 2015, 75, 1925–1929. [Google Scholar] [CrossRef]
- Bansal, S.; Siddarth, M.; Chawla, D.; Banerjee, B.D.; Madhu, S.V.; Tripathi, A.K. Advanced glycation end products enhance reactive oxygen and nitrogen species generation in neutrophils in vitro. Mol. Cell. Biochem. 2012, 361, 289–296. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta 2000, 1498, 99–111. [Google Scholar] [CrossRef]
- Lindsey, J.B.; Cipollone, F.; Abdullah, S.M.; McGuire, D.K. Receptor for advanced glycation end-products (RAGE) and soluble RAGE (sRAGE): Cardiovascular implications. Diab. Vasc. Dis. Res. 2009, 6, 7–14. [Google Scholar] [CrossRef]
- Lalla, E.; Lamster, I.B.; Stern, D.M.; Schmidt, A.M. Receptor for advanced glycation end products, inflammation, and accelerated periodontal disease in diabetes: Mechanisms and insights into therapeutic modalities. Ann. Periodontol. 2001, 6, 113–118. [Google Scholar] [CrossRef]
- Takada, M.; Hirata, K.; Ajiki, T.; Suzuki, Y.; Kuroda, Y. Expression of receptor for advanced glycation end products (RAGE) and MMP-9 in human pancreatic cancer cells. Hepatogastroenterology 2004, 51, 928–930. [Google Scholar] [PubMed]
- Hirata, K.; Takada, M.; Suzuki, Y.; Kuroda, Y. Expression of receptor for advanced glycation end products (RAGE) in human biliary cancer cells. Hepatogastroenterology 2003, 50, 1205–1207. [Google Scholar] [PubMed]
- El-Far, A.H.; Sroga, G.; Jaouni, S.K.A.; Mousa, S.A. Role and Mechanisms of RAGE-Ligand Complexes and RAGE-Inhibitors in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 3613. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Cui, M.; Wang, J.; Wang, H. Receptor for advanced glycation end products (RAGE) soluble form (sRAGE): A new biomarker for lung cancer. Neoplasma 2010, 57, 55–61. [Google Scholar] [CrossRef]
- Tesarova, P.; Kalousova, M.; Jachymova, M.; Mestek, O.; Petruzelka, L.; Zima, T. Receptor for advanced glycation end products (RAGE)-soluble form (sRAGE) and gene polymorphisms in patients with breast cancer. Cancer Investig. 2007, 25, 720–725. [Google Scholar] [CrossRef]
- Ishiguro, H.; Nakaigawa, N.; Miyoshi, Y.; Fujinami, K.; Kubota, Y.; Uemura, H. Receptor for advanced glycation end products (RAGE) and its ligand, amphoterin are overexpressed and associated with prostate cancer development. Prostate 2005, 64, 92–100. [Google Scholar] [CrossRef]
- Jiao, L.; Taylor, P.R.; Weinstein, S.J.; Graubard, B.I.; Virtamo, J.; Albanes, D.; Stolzenberg-Solomon, R.Z. Advanced glycation end products, soluble receptor for advanced glycation end products, and risk of colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1430–1438. [Google Scholar] [CrossRef]
- Zhang, S.; Hou, X.; Zi, S.; Wang, Y.; Chen, L.; Kong, B. Polymorphisms of receptor for advanced glycation end products and risk of epithelial ovarian cancer in Chinese patients. Cell. Physiol. Biochem. 2013, 31, 525–531. [Google Scholar] [CrossRef]
- Li, M.L.; Wang, X.F.; Tan, Z.J.; Dong, P.; Gu, J.; Lu, J.H.; Wu, X.S.; Zhang, L.; Ding, Q.C.; Wu, W.G.; et al. Ethyl pyruvate administration suppresses growth and invasion of gallbladder cancer cells via downregulation of HMGB1-RAGE axis. Int. J. Immunopathol. Pharm. 2012, 25, 955–965. [Google Scholar] [CrossRef]
- El-Far, A.; Munesue, S.; Harashima, A.; Sato, A.; Shindo, M.; Nakajima, S.; Inada, M.; Tanaka, M.; Takeuchi, A.; Tsuchiya, H.; et al. In vitro anticancer effects of a RAGE inhibitor discovered using a structure-based drug design system. Oncol. Lett. 2018, 15, 4627–4634. [Google Scholar] [CrossRef]
- Ko, S.Y.; Ko, H.A.; Shieh, T.M.; Chi, T.C.; Chen, H.I.; Chen, Y.T.; Yu, Y.H.; Yang, S.H.; Chang, S.S. Advanced glycation end products influence oral cancer cell survival via Bcl-xl and Nrf2 regulation in vitro. Oncol. Lett. 2017, 13, 3328–3334. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, H.K.; Yoon, J.S.; Kim, S.J.; Kim, E.S.; Ahn, K.S.; Kim, D.S.; Yoon, S.S.; Kim, B.K.; Lee, Y.Y. Advanced glycation end product (AGE)-induced proliferation of HEL cells via receptor for AGE-related signal pathways. Int. J. Oncol. 2008, 33, 493–501. [Google Scholar] [CrossRef]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J. Gastroenterol. 2012, 18, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Chihara, Y.; Kondo, H. Differential effects between amphoterin and advanced glycation end products on colon cancer cells. Int. J. Cancer 2003, 104, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, H.; Matou-Nasri, S.; Wang, Q.; Rabhan, Z.; Al-Eidi, H.; Al Abdulrahman, A.; Ahmed, N. Advanced glycation endproducts increase proliferation, migration and invasion of the breast cancer cell line MDA-MB-231. Biochim. Biophys. Acta 2015, 1852, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Stolzenberg-Solomon, R.; Zimmerman, T.P.; Duan, Z.; Chen, L.; Kahle, L.; Risch, A.; Subar, A.F.; Cross, A.J.; Hollenbeck, A.; et al. Dietary consumption of advanced glycation end products and pancreatic cancer in the prospective NIH-AARP Diet and Health Study. Am. J. Clin. Nutr. 2015, 101, 126–134. [Google Scholar] [CrossRef]
- Shang, L.; Ananthakrishnan, R.; Li, Q.; Quadri, N.; Abdillahi, M.; Zhu, Z.; Qu, W.; Rosario, R.; Toure, F.; Yan, S.F.; et al. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS ONE 2010, 5, e10092. [Google Scholar] [CrossRef]
- Hiwatashi, K.; Ueno, S.; Abeyama, K.; Kubo, F.; Sakoda, M.; Maruyama, I.; Hamanoue, M.; Natsugoe, S.; Aikou, T. A novel function of the receptor for advanced glycation end-products (RAGE) in association with tumorigenesis and tumor differentiation of HCC. Ann. Surg. Oncol. 2008, 15, 923–933. [Google Scholar] [CrossRef]
- Tafani, M.; Schito, L.; Pellegrini, L.; Villanova, L.; Marfe, G.; Anwar, T.; Rosa, R.; Indelicato, M.; Fini, M.; Pucci, B.; et al. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-{kappa}B. Carcinogenesis 2011, 32, 1167–1175. [Google Scholar] [CrossRef]
- Kang, R.; Hou, W.; Zhang, Q.; Chen, R.; Lee, Y.J.; Bartlett, D.L.; Lotze, M.T.; Tang, D.; Zeh, H.J. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. 2014, 5, e1480. [Google Scholar] [CrossRef] [PubMed]
- Stirban, A.; Negrean, M.; Stratmann, B.; Gawlowski, T.; Horstmann, T.; Gotting, C.; Kleesiek, K.; Mueller-Roesel, M.; Koschinsky, T.; Uribarri, J.; et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 2006, 29, 2064–2071. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Izuhara, Y. Inhibition of advanced glycation end products: An implicit goal in clinical medicine for the treatment of diabetic nephropathy? Ann. N. Y. Acad. Sci. 2008, 1126, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Ryu, T.Y.; Park, J.; Scherer, P.E. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab. J. 2014, 38, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Moinuddin; Dixit, K.; Shahab, U.; Alam, K.; Ali, A. Genotoxicity and immunogenicity of DNA-advanced glycation end products formed by methylglyoxal and lysine in presence of Cu2+. Biochem. Biophys. Res. Commun. 2011, 407, 568–574. [Google Scholar] [CrossRef]
- Ashraf, J.M.; Ahmad, S.; Rabbani, G.; Jan, A.T.; Lee, E.J.; Khan, R.H.; Choi, I. Physicochemical analysis of structural alteration and advanced glycation end products generation during glycation of H2A histone by 3-deoxyglucosone. IUBMB Life 2014, 66, 686–693. [Google Scholar] [CrossRef]
- Shahab, U.; Tabrez, S.; Khan, M.S.; Akhter, F.; Khan, M.S.; Saeed, M.; Ahmad, K.; Srivastava, A.K.; Ahmad, S. Immunogenicity of DNA-advanced glycation end product fashioned through glyoxal and arginine in the presence of Fe(3)(+): Its potential role in prompt recognition of diabetes mellitus auto-antibodies. Chem. Biol. Interact. 2014, 219, 229–240. [Google Scholar] [CrossRef]
- Ashraf, J.M.; Ahmad, S.; Rabbani, G.; Hasan, Q.; Jan, A.T.; Lee, E.J.; Khan, R.H.; Alam, K.; Choi, I. 3-Deoxyglucosone: A potential glycating agent accountable for structural alteration in H3 histone protein through generation of different AGEs. PLoS ONE 2015, 10, e0116804. [Google Scholar] [CrossRef]
- Takamiya, R.; Takahashi, M.; Myint, T.; Park, Y.S.; Miyazawa, N.; Endo, T.; Fujiwara, N.; Sakiyama, H.; Misonou, Y.; Miyamoto, Y.; et al. Glycation proceeds faster in mutated Cu, Zn-superoxide dismutases related to familial amyotrophic lateral sclerosis. FASEB J. 2003, 17, 938–940. [Google Scholar] [CrossRef]
- Basta, G.; Lazzerini, G.; Del Turco, S.; Ratto, G.M.; Schmidt, A.M.; De Caterina, R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arter. Thromb. Vasc. Biol. 2005, 25, 1401–1407. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.; Schinzel, R.; Palm, D.; Riederer, P.; Munch, G. High molecular weight hyaluronic acid inhibits advanced glycation endproduct-induced NF-kappaB activation and cytokine expression. FEBS Lett. 1999, 453, 283–287. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.M.; Poynter, M.E.; Baeuerle, P.A. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radic. Biol. Med. 2000, 28, 1317–1327. [Google Scholar] [CrossRef]
- Jabir, N.R.; Ahmad, S.; Tabrez, S. An insight on the association of glycation with hepatocellular carcinoma. Semin. Cancer Biol. 2018, 49, 56–63. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Multiple roles of glyoxalase 1-mediated suppression of methylglyoxal glycation in cancer biology-Involvement in tumour suppression, tumour growth, multidrug resistance and target for chemotherapy. Semin. Cancer Biol. 2018, 49, 83–93. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Vander Heiden, M.G. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef]
- Sakamoto, H.; Mashima, T.; Kizaki, A.; Dan, S.; Hashimoto, Y.; Naito, M.; Tsuruo, T. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood 2000, 95, 3214–3218. [Google Scholar] [CrossRef]
- Sakamoto, H.; Mashima, T.; Sato, S.; Hashimoto, Y.; Yamori, T.; Tsuruo, T. Selective activation of apoptosis program by S-p-bromobenzylglutathione cyclopentyl diester in glyoxalase I-overexpressing human lung cancer cells. Clin. Cancer Res. 2001, 7, 2513–2518. [Google Scholar]
- Fridovich, I. The biology of oxygen radicals. Science 1978, 201, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.J.; Wondrak, G.T.; Laurean, D.C.; Jacobson, M.K.; Jacobson, E.L. DNA damage by carbonyl stress in human skin cells. Mutat. Res. 2003, 522, 45–56. [Google Scholar] [CrossRef]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Cancer malignancy is enhanced by glyceraldehyde-derived advanced glycation end-products. J. Oncol. 2010, 2010, 739852. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Akhter, F.; Shahab, U.; Rafi, Z.; Khan, M.S.; Nabi, R.; Khan, M.S.; Ahmad, K.; Ashraf, J.M.; Moinuddin. Do all roads lead to the Rome? The glycation perspective! Semin. Cancer Biol. 2018, 49, 9–19. [Google Scholar] [CrossRef]
- Li, T.; Qin, W.; Liu, Y.; Li, S.; Qin, X.; Liu, Z. Effect of RAGE gene polymorphisms and circulating sRAGE levels on susceptibility to gastric cancer: A case-control study. Cancer Cell Int. 2017, 17, 19. [Google Scholar] [CrossRef]
- Lin, J.A.; Wu, C.H.; Lu, C.C.; Hsia, S.M.; Yen, G.C. Glycative stress from advanced glycation end products (AGEs) and dicarbonyls: An emerging biological factor in cancer onset and progression. Mol. Nutr. Food Res. 2016, 60, 1850–1864. [Google Scholar] [CrossRef]
- Sakellariou, S.; Fragkou, P.; Levidou, G.; Gargalionis, A.N.; Piperi, C.; Dalagiorgou, G.; Adamopoulos, C.; Saetta, A.; Agrogiannis, G.; Theohari, I.; et al. Clinical significance of AGE-RAGE axis in colorectal cancer: Associations with glyoxalase-I, adiponectin receptor expression and prognosis. BMC Cancer 2016, 16, 174. [Google Scholar] [CrossRef]
- Nedic, O.; Rattan, S.I.; Grune, T.; Trougakos, I.P. Molecular effects of advanced glycation end products on cell signaling pathways, ageing and pathophysiology. Free Radic. Res. 2013, 47 (Suppl. S1), 28–38. [Google Scholar] [CrossRef]
- Said, G.; Guilbert, M.; Millerot-Serrurot, E.; Van Gulick, L.; Terryn, C.; Garnotel, R.; Jeannesson, P. Impact of carbamylation and glycation of collagen type I on migration of HT1080 human fibrosarcoma cells. Int. J. Oncol. 2012, 40, 1797–1804. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodriguez-Teja, M.; Gronau, J.H.; Breit, C.; Zhang, Y.Z.; Minamidate, A.; Caley, M.P.; McCarthy, A.; Cox, T.R.; Erler, J.T.; Gaughan, L.; et al. AGE-modified basement membrane cooperates with Endo180 to promote epithelial cell invasiveness and decrease prostate cancer survival. J. Pathol. 2015, 235, 581–592. [Google Scholar] [CrossRef]
- Liao, Y.F.; Yin, S.; Chen, Z.Q.; Li, F.; Zhao, B. High glucose promotes tumor cell proliferation and migration in lung adenocarcinoma via the RAGENOXs pathway. Mol. Med. Rep. 2018, 17, 8536–8541. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Negative consequences of glycation. Metabolism 2000, 49, 9–13. [Google Scholar] [CrossRef]
- Su, S.; Chien, M.; Lin, C.; Chen, M.; Yang, S. RAGE gene polymorphism and environmental factor in the risk of oral cancer. J. Dent. Res. 2015, 94, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Sanders, N.T.; Dutson, D.J.; Durrant, J.W.; Lewis, J.B.; Wilcox, S.H.; Winden, D.R.; Arroyo, J.A.; Bikman, B.T.; Reynolds, P.R. Cigarette smoke extract (CSE) induces RAGE-mediated inflammation in the Ca9-22 gingival carcinoma epithelial cell line. Arch. Oral Biol. 2017, 80, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Babtan, A.M.; Ilea, A.; Bosca, B.A.; Crisan, M.; Petrescu, N.B.; Collino, M.; Sainz, R.M.; Gerlach, J.Q.; Campian, R.S. Advanced glycation end products as biomarkers in systemic diseases: Premises and perspectives of salivary advanced glycation end products. Biomark. Med. 2019, 13, 479–495. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Yu, W.; Ma, L.; Ji, X.; Xiao, W. Expression of the receptor for advanced glycation end-products and frequency of polymorphism in lung cancer. Oncol. Lett. 2015, 10, 51–60. [Google Scholar] [CrossRef]
- Tesarova, P.; Zima, T.; Kubena, A.A.; Kalousova, M. Polymorphisms of the receptor for advanced glycation end products and glyoxalase I and long-term outcome in patients with breast cancer. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef]
- Khan, M.S.; Tabrez, S.; Al-Okail, M.S.; Shaik, G.M.; Bhat, S.A.; Rehman, T.M.; Husain, F.M.; AlAjmi, M.F. Non-enzymatic glycation of protein induces cancer cell proliferation and its inhibition by quercetin: Spectroscopic, cytotoxicity and molecular docking studies. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.L.; Tai, C.J.; Huang, C.W.; Chang, F.R.; Wang, J.Y. Efficacy of Low-Molecular-Weight Fucoidan as a Supplemental Therapy in Metastatic Colorectal Cancer Patients: A Double-Blind Randomized Controlled Trial. Mar. Drugs 2017, 15, 122. [Google Scholar] [CrossRef]
- Chen, H.; Wu, L.; Li, Y.; Meng, J.; Lin, N.; Yang, D.; Zhu, Y.; Li, X.; Li, M.; Xu, Y.; et al. Advanced glycation end products increase carbohydrate responsive element binding protein expression and promote cancer cell proliferation. Mol. Cell. Endocrinol. 2014, 395, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Piperi, C.; Adamopoulos, C.; Papavassiliou, A.G. Potential of glycative stress targeting for cancer prevention. Cancer Lett. 2017, 390, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Sasahira, T.; Akama, Y.; Fujii, K.; Kuniyasu, H. Expression of receptor for advanced glycation end products and HMGB1/amphoterin in colorectal adenomas. Virchows Arch. 2005, 446, 411–415. [Google Scholar] [CrossRef]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signaling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef]
- He, S.; Cheng, J.; Sun, L.; Wang, Y.; Wang, C.; Liu, X.; Zhang, Z.; Zhao, M.; Luo, Y.; Tian, L.; et al. HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via paracrine effect. Cell Death Dis. 2018, 9, 648. [Google Scholar] [CrossRef]
- Gugliucci, A. Alternative antiglycation mechanisms: Are spermine and fructosamine-3-kinase part of a carbonyl damage control pathway? Med. Hypotheses 2005, 64, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T.; Cervantes-Laurean, D.; Roberts, M.J.; Qasem, J.G.; Kim, M.; Jacobson, E.L.; Jacobson, M.K. Identification of alpha-dicarbonyl scavengers for cellular protection against carbonyl stress. Biochem. Pharm. 2002, 63, 361–373. [Google Scholar] [CrossRef]
- Yim, M.B.; Yim, H.S.; Lee, C.; Kang, S.O.; Chock, P.B. Protein glycation: Creation of catalytic sites for free radical generation. Ann. N. Y. Acad. Sci. 2001, 928, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Delpierre, G.; Rider, M.H.; Collard, F.; Stroobant, V.; Vanstapel, F.; Santos, H.; Van Schaftingen, E. Identification, cloning, and heterologous expression of a mammalian fructosamine-3-kinase. Diabetes 2000, 49, 1627–1634. [Google Scholar] [CrossRef]
- Szwergold, B.S.; Howell, S.; Beisswenger, P.J. Human fructosamine-3-kinase: Purification, sequencing, substrate specificity, and evidence of activity in vivo. Diabetes 2001, 50, 2139–2147. [Google Scholar] [CrossRef]
- Sanghvi, V.R.; Leibold, J.; Mina, M.; Mohan, P.; Berishaj, M.; Li, Z.; Miele, M.M.; Lailler, N.; Zhao, C.; de Stanchina, E.; et al. The Oncogenic Action of NRF2 Depends on De-glycation by Fructosamine-3-Kinase. Cell 2019, 178, 807–819.e821. [Google Scholar] [CrossRef]
- Costa, R.M.; Chigancas, V.; Galhardo Rda, S.; Carvalho, H.; Menck, C.F. The eukaryotic nucleotide excision repair pathway. Biochimie 2003, 85, 1083–1099. [Google Scholar] [CrossRef]
- Fortini, P.; Pascucci, B.; Parlanti, E.; D’Errico, M.; Simonelli, V.; Dogliotti, E. The base excision repair: Mechanisms and its relevance for cancer susceptibility. Biochimie 2003, 85, 1053–1071. [Google Scholar] [CrossRef]
- Delpierrre, G.; Vertommen, D.; Communi, D.; Rider, M.H.; Van Schaftingen, E. Identification of fructosamine residues deglycated by fructosamine-3-kinase in human hemoglobin. J. Biol. Chem. 2004, 279, 27613–27620. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.J.; Yoo, H.S.; Shin, S.; Park, Y.J.; Jeon, S.M. Dysregulation of NRF2 in Cancer: From Molecular Mechanisms to Therapeutic Opportunities. Biomol. Ther. 2018, 26, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Basak, P.; Sadhukhan, P.; Sarkar, P.; Sil, P.C. Perspectives of the Nrf2 signaling pathway in cancer progression and therapy. Toxicol. Rep. 2017, 4, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Tallman, K.A.; Vila, A.; Porter, N.A.; Marnett, L.J. Measuring electrophile stress. Curr. Protoc. Toxicol. 2009, 17, Unit17 11. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 1990, 87, 6258–6262. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Orru, C.; Giordano, S.; Columbano, A. Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases. Cancers 2020, 12, 2932. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Taguchi, K.; Masamune, A.; Yamamoto, M.; Shimosegawa, T. Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis 2017, 38, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Hyun, J.W. Oxidative Stress, Nrf2, and Epigenetic Modification Contribute to Anticancer Drug Resistance. Toxicol. Res. 2017, 33, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Cantley, L.C.; DeNicola, G.M. NRF2 Rewires Cellular Metabolism to Support the Antioxidant Response, A Master Regulator of Oxidative Stress-The Transcription Factor Nrf2; Intechopen: London, UK, 2016. [Google Scholar] [CrossRef]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Su, C.; Ren, S.; Zhou, C.; Jiang, T. Pan-cancer analysis of KEAP1 mutations as biomarkers for immunotherapy outcomes. Ann. Transl. Med. 2020, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Zanotto-Filho, A.; Masamsetti, V.P.; Loranc, E.; Tonapi, S.S.; Gorthi, A.; Bernard, X.; Goncalves, R.M.; Moreira, J.C.; Chen, Y.; Bishop, A.J. Alkylating Agent-Induced NRF2 Blocks Endoplasmic Reticulum Stress-Mediated Apoptosis via Control of Glutathione Pools and Protein Thiol Homeostasis. Mol. Cancer Ther. 2016, 15, 3000–3014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Ye, W.; Shao, Q.; Zhang, M.; Liang, J. Nrf2 is a potential therapeutic target in radioresistance in human cancer. Crit. Rev. Oncol. Hematol. 2013, 88, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Endo, T.; Hart, G.W.; Seeberger, P.H.; Wong, C.-H. Glycoscience: Biology and Medicine; Springer: Tokyo, Japan, 2015; ISBN 978-4-431-54842-3. [Google Scholar]
- Bauer, A.K.; Cho, H.Y.; Miller-Degraff, L.; Walker, C.; Helms, K.; Fostel, J.; Yamamoto, M.; Kleeberger, S.R. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS ONE 2011, 6, e26590. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Saigusa, D.; Baird, L.; Yu, L.; Rokutan, H.; Igarashi, K.; Ebina, M.; Shibata, T.; Yamamoto, M. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016, 76, 3088–3096. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013, 73, 4158–4168. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in the Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives. Biomolecules 2020, 10, 791. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Joe, Y.; Zheng, M.; Kim, H.J.; Yu, J.K.; Cho, G.J.; Chang, K.C.; Kim, H.K.; Han, J.; Ryter, S.W.; et al. Resveratrol induces hepatic mitochondrial biogenesis through the sequential activation of nitric oxide and carbon monoxide production. Antioxid. Redox Signal. 2014, 20, 2589–2605. [Google Scholar] [CrossRef] [PubMed]
- Shukla, K.; Sonowal, H.; Saxena, A.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibitor, fidarestat regulates mitochondrial biogenesis via Nrf2/HO-1/AMPK pathway in colon cancer cells. Cancer Lett. 2017, 411, 57–63. [Google Scholar] [CrossRef]
- Negrette-Guzman, M.; Huerta-Yepez, S.; Vega, M.I.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Medina-Campos, O.N.; Rodriguez, E.; Tapia, E.; Pedraza-Chaverri, J. Sulforaphane induces differential modulation of mitochondrial biogenesis and dynamics in normal cells and tumor cells. Food Chem. Toxicol. 2017, 100, 90–102. [Google Scholar] [CrossRef]
- Lim, J.K.M.; Delaidelli, A.; Minaker, S.W.; Zhang, H.F.; Colovic, M.; Yang, H.; Negri, G.L.; von Karstedt, S.; Lockwood, W.W.; Schaffer, P.; et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 116, 9433–9442. [Google Scholar] [CrossRef]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol. Cell. Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Badur, M.G.; Luebeck, J.; Magana, J.H.; Birmingham, A.; Sasik, R.; Ahn, C.S.; Ideker, T.; Metallo, C.M.; Mali, P. Combinatorial CRISPR-Cas9 Metabolic Screens Reveal Critical Redox Control Points Dependent on the KEAP1-NRF2 Regulatory Axis. Mol. Cell 2018, 69, 699–708.e697. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Effect of graded Nrf2 activation on phase-I and -II drug metabolizing enzymes and transporters in mouse liver. PLoS ONE 2012, 7, e39006. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Saddawi-Konefka, R.; Seelige, R.; Gross, E.T.; Levy, E.; Searles, S.C.; Washington, A., Jr.; Santosa, E.K.; Liu, B.; O’Sullivan, T.E.; Harismendy, O.; et al. Nrf2 Induces IL-17D to Mediate Tumor and Virus Surveillance. Cell Rep. 2016, 16, 2348–2358. [Google Scholar] [CrossRef]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329.e318. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef] [PubMed]
- DeBlasi, J.M.; DeNicola, G.M. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers 2020, 12, 3023. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef]
- Fortpied, J.; Maliekal, P.; Vertommen, D.; Van Schaftingen, E. Magnesium-dependent phosphatase-1 is a protein-fructosamine-6-phosphatase potentially involved in glycation repair. J. Biol. Chem. 2006, 281, 18378–18385. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Van Schaftingen, E.; Collard, F.; Wiame, E.; Veiga-da-Cunha, M. Enzymatic repair of Amadori products. Amino Acids 2012, 42, 1143–1150. [Google Scholar] [CrossRef]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef]
- Veiga da-Cunha, M.; Jacquemin, P.; Delpierre, G.; Godfraind, C.; Theate, I.; Vertommen, D.; Clotman, F.; Lemaigre, F.; Devuyst, O.; Van Schaftingen, E. Increased protein glycation in fructosamine 3-kinase-deficient mice. Biochem. J. 2006, 399, 257–264. [Google Scholar] [CrossRef]
- Zhang, Q.; Ames, J.M.; Smith, R.D.; Baynes, J.W.; Metz, T.O. A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: Probing the pathogenesis of chronic disease. J. Proteome Res. 2009, 8, 754–769. [Google Scholar] [CrossRef]
- Wareham, N.J.; Pfister, R. Diabetes: Glycated hemoglobin is a marker of diabetes and CVD risk. Nat. Rev. Cardiol. 2010, 7, 367–368. [Google Scholar] [CrossRef]
- Abdel-Wahab, Y.H.; O’Harte, F.P.; Barnett, C.R.; Flatt, P.R. Characterization of insulin glycation in insulin-secreting cells maintained in tissue culture. J. Endocrinol. 1997, 152, 59–67. [Google Scholar] [CrossRef]
- Anguizola, J.; Matsuda, R.; Barnaby, O.S.; Hoy, K.S.; Wa, C.; DeBolt, E.; Koke, M.; Hage, D.S. Review: Glycation of human serum albumin. Clin. Chim. Acta 2013, 425, 64–76. [Google Scholar] [CrossRef]
- Hunter, S.J.; Boyd, A.C.; O’Harte, F.P.; McKillop, A.M.; Wiggam, M.I.; Mooney, M.H.; McCluskey, J.T.; Lindsay, J.R.; Ennis, C.N.; Gamble, R.; et al. Demonstration of glycated insulin in human diabetic plasma and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in humans. Diabetes 2003, 52, 492–498. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Delplanque, J.; Delpierre, G.; Opperdoes, F.R.; Van Schaftingen, E. Tissue distribution and evolution of fructosamine 3-kinase and fructosamine 3-kinase-related protein. J. Biol. Chem. 2004, 279, 46606–46613. [Google Scholar] [CrossRef]
- Fortpied, J.; Gemayel, R.; Stroobant, V.; van Schaftingen, E. Plant ribulosamine/erythrulosamine 3-kinase, a putative protein-repair enzyme. Biochem. J. 2005, 388, 795–802. [Google Scholar] [CrossRef]
- Gemayel, R.; Fortpied, J.; Rzem, R.; Vertommen, D.; Veiga-da-Cunha, M.; Van Schaftingen, E. Many fructosamine 3-kinase homologues in bacteria are ribulosamine/erythrulosamine 3-kinases potentially involved in protein deglycation. FEBS J. 2007, 274, 4360–4374. [Google Scholar] [CrossRef]
- Kameya, M.; Sakaguchi-Mikami, A.; Ferri, S.; Tsugawa, W.; Sode, K. Advancing the development of glycated protein biosensing technology: Next-generation sensing molecules. J. Diabetes Sci. Technol. 2015, 9, 183–191. [Google Scholar] [CrossRef]
- Shrestha, S.; Katiyar, S.; Sanz-Rodriguez, C.E.; Kemppinen, N.R.; Kim, H.W.; Kadirvelraj, R.; Panagos, C.; Keyhaninejad, N.; Colonna, M.; Chopra, P.; et al. A redox-active switch in fructosamine-3-kinases expands the regulatory repertoire of the protein kinase superfamily. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Szwergold, B.S.; Beisswenger, P.J. Enzymatic deglycation--a new paradigm or an epiphenomenon? Biochem. Soc. Trans. 2003, 31, 1428–1432. [Google Scholar] [CrossRef]
- Grishok, A.; Mello, C.C. RNAi (Nematodes: Caenorhabditis elegans). Adv. Genet. 2002, 46, 339–360. [Google Scholar] [CrossRef]
- Kamath, R.S.; Fraser, A.G.; Dong, Y.; Poulin, G.; Durbin, R.; Gotta, M.; Kanapin, A.; Le Bot, N.; Moreno, S.; Sohrmann, M.; et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003, 421, 231–237. [Google Scholar] [CrossRef]
- Collard, F.; Wiame, E.; Bergans, N.; Fortpied, J.; Vertommen, D.; Vanstapel, F.; Delpierre, G.; Van Schaftingen, E. Fructosamine 3-kinase-related protein and deglycation in human erythrocytes. Biochem. J. 2004, 382, 137–143. [Google Scholar] [CrossRef]
- Collard, F.; Delpierre, G.; Stroobant, V.; Matthijs, G.; Van Schaftingen, E. A mammalian protein homologous to fructosamine-3-kinase is a ketosamine-3-kinase acting on psicosamines and ribulosamines but not on fructosamines. Diabetes 2003, 52, 2888–2895. [Google Scholar] [CrossRef]
- Wiame, E.; Delpierre, G.; Collard, F.; Van Schaftingen, E. Identification of a pathway for the utilization of the Amadori product fructoselysine in Escherichia coli. J. Biol. Chem. 2002, 277, 42523–42529. [Google Scholar] [CrossRef]
- Collard, F.; Zhang, J.; Nemet, I.; Qanungo, K.R.; Monnier, V.M.; Yee, V.C. Crystal structure of the deglycating enzyme fructosamine oxidase (amadoriase II). J. Biol. Chem. 2008, 283, 27007–27016. [Google Scholar] [CrossRef]
- Pascal, S.M.; Veiga-da-Cunha, M.; Gilon, P.; Van Schaftingen, E.; Jonas, J.C. Effects of fructosamine-3-kinase deficiency on function and survival of mouse pancreatic islets after prolonged culture in high glucose or ribose concentrations. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E586–E596. [Google Scholar] [CrossRef]
- Payne, L.S.; Brown, P.M.; Middleditch, M.; Baker, E.; Cooper, G.J.; Loomes, K.M. Mapping of the ATP-binding domain of human fructosamine 3-kinase-related protein by affinity labelling with 5’-[p-(fluorosulfonyl)benzoyl]adenosine. Biochem. J. 2008, 416, 281–288. [Google Scholar] [CrossRef]
- Szwergold, B.S.; Kappler, F.; Brown, T.R. Identification of fructose 3-phosphate in the lens of diabetic rats. Science 1990, 247, 451–454. [Google Scholar] [CrossRef]
- Szwergold, B.S.; Kappler, F.; Brown, T.R.; Pfeffer, P.; Osman, S.F. Identification of D-sorbitol 3-phosphate in the normal and diabetic mammalian lens. J. Biol. Chem. 1989, 264, 9278–9282. [Google Scholar] [CrossRef]
- Szwergold, B.; Manevich, Y.; Payne, L.; Loomes, K. Fructosamine-3-kinase-related-protein phosphorylates glucitolamines on the C-4 hydroxyl: Novel substrate specificity of an enigmatic enzyme. Biochem. Biophys. Res. Commun. 2007, 361, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.J.; Rather, P.N.; Hare, R.S.; Miller, G.H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar] [CrossRef]
- Hon, W.C.; McKay, G.A.; Thompson, P.R.; Sweet, R.M.; Yang, D.S.; Wright, G.D.; Berghuis, A.M. Structure of an enzyme required for aminoglycoside antibiotic resistance reveals homology to eukaryotic protein kinases. Cell 1997, 89, 887–895. [Google Scholar] [CrossRef]
- McKay, G.A.; Thompson, P.R.; Wright, G.D. Broad spectrum aminoglycoside phosphotransferase type III from Enterococcus: Overexpression, purification, and substrate specificity. Biochemistry 1994, 33, 6936–6944. [Google Scholar] [CrossRef]
- Inaba, S.I.; Yamaguchi-Goto, M.; Tanaka-Takanaka, K.; Yonesu, K.; Sakurai, H.; Kubota, K.; Izumi, T. Enzymatic kinetics regarding reversible metabolism of CS-0777, a sphingosine 1-phosphate receptor modulator, via phosphorylation and dephosphorylation in humans. Xenobiotica 2018, 48, 258–268. [Google Scholar] [CrossRef]
- Brownlee, M. Lilly Lecture 1993. Glycation and diabetic complications. Diabetes 1994, 43, 836–841. [Google Scholar] [CrossRef]
- Notarnicola, M.; Caruso, M.G.; Tutino, V.; Guerra, V.; Frisullo, S.; Altomare, D.F.; Misciagna, G. Reduced fructosamine-3-kinase activity and its mRNA in human distal colorectal carcinoma. Genes Nutr. 2010, 5, 257–262. [Google Scholar] [CrossRef]
- Misciagna, G.; De Michele, G.; Guerra, V.; Cisternino, A.M.; Di Leo, A.; Freudenheim, J.L.; Group, I. Serum fructosamine and colorectal adenomas. Eur. J. Epidemiol. 2004, 19, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.G.; Notarnicola, M.; Altomare, D.F.; Misciagna, G. Gene expression of fructosamine 3 kinase in patients with colorectal cancer. Oncology 2007, 73, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Fu, L.; Jung, Y.; Conte, M.L.; Lawson, J.R.; Lowther, W.T.; Sun, R.; Liu, K.; Yang, J.; Carroll, K.S. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 2018, 14, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Tsukushi, S. 3-deoxyglucosone and AGEs in uremic complications: Inactivation of glutathione peroxidase by 3-deoxyglucosone. Kidney Int. Suppl. 2001, 78, S37–S41. [Google Scholar] [CrossRef]
- Ngo, H.K.C.; Kim, D.H.; Cha, Y.N.; Na, H.K.; Surh, Y.J. Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Res. 2017, 77, 4797–4808. [Google Scholar] [CrossRef]
- Conner, J.R.; Beisswenger, P.J.; Szwergold, B.S. Some clues as to the regulation, expression, function, and distribution of fructosamine-3-kinase and fructosamine-3-kinase-related protein. Ann. N. Y. Acad. Sci. 2005, 1043, 824–836. [Google Scholar] [CrossRef]
- Ohtsuka, K.; Hata, M. Molecular chaperone function of mammalian Hsp70 and Hsp40--a review. Int. J. Hyperth. 2000, 16, 231–245. [Google Scholar] [CrossRef]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signaling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Walker-Samuel, S.; Ramasawmy, R.; Torrealdea, F.; Rega, M.; Rajkumar, V.; Johnson, S.P.; Richardson, S.; Goncalves, M.; Parkes, H.G.; Arstad, E.; et al. In vivo imaging of glucose uptake and metabolism in tumors. Nat. Med. 2013, 19, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.B.; Kiemer, L.; Brunak, S. Analysis and prediction of mammalian protein glycation. Glycobiology 2006, 16, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, J.; Aggarwal, K.; Balaram, P. Helical peptide models for protein glycation: Proximity effects in catalysis of the Amadori rearrangement. Chem. Biol. 2001, 8, 611–625. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 2016, 5. [Google Scholar] [CrossRef]
- Chatterjee, N.; North, J.A.; Dechassa, M.L.; Manohar, M.; Prasad, R.; Luger, K.; Ottesen, J.J.; Poirier, M.G.; Bartholomew, B. Histone Acetylation near the Nucleosome Dyad Axis Enhances Nucleosome Disassembly by RSC and SWI/SNF. Mol. Cell. Biol 2015, 35, 4083–4092. [Google Scholar] [CrossRef]
- Galligan, J.J.; Wepy, J.A.; Streeter, M.D.; Kingsley, P.J.; Mitchener, M.M.; Wauchope, O.R.; Beavers, W.N.; Rose, K.L.; Wang, T.; Spiegel, D.A.; et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. USA 2018, 115, 9228–9233. [Google Scholar] [CrossRef]
- Bellahcene, A.; Nokin, M.J.; Castronovo, V.; Schalkwijk, C. Methylglyoxal-derived stress: An emerging biological factor involved in the onset and progression of cancer. Semin. Cancer Biol. 2018, 49, 64–74. [Google Scholar] [CrossRef]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, T.; Joly, N.; Liebart, J.C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P.; et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 2017, 357, 208–211. [Google Scholar] [CrossRef]
- Zheng, Q.; Omans, N.D.; Leicher, R.; Osunsade, A.; Agustinus, A.S.; Finkin-Groner, E.; D’Ambrosio, H.; Liu, B.; Chandarlapaty, S.; Liu, S.; et al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat. Commun. 2019, 10, 1289. [Google Scholar] [CrossRef]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Salehiniya, H. Incidence, mortality and risk factors of cervical cancer in the world. Biomed. Res. Ther. 2017, 4, 1795–1811. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Ghoncheh, M.; Pakzad, R.; Hasanpour, H.; Salehiniya, H. Incidence and mortality of uterine cancer and relationship with Human Development Index in the world. Cukurova Med. J. 2017, 42, 233–240. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer (Dove Med. Press) 2019, 11, 151–164. [Google Scholar] [CrossRef]
- Benson, J.R.; Jatoi, I. The global breast cancer burden. Future Oncol. 2012, 8, 697–702. [Google Scholar] [CrossRef]
- Zaidi, Z.; Dib, H.A. The worldwide female breast cancer incidence and survival, 2018. Epidemiology 2019, 79. [Google Scholar] [CrossRef]
- Malvia, S.; Bagadi, S.A.; Dubey, U.S.; Saxena, S. Epidemiology of breast cancer in Indian women. Asia Pac. J. Clin. Oncol. 2017, 13, 289–295. [Google Scholar] [CrossRef]
- Saxena, S.; Chakraborty, A.; Kaushal, M.; Kotwal, S.; Bhatanager, D.; Mohil, R.S.; Chintamani, C.; Aggarwal, A.K.; Sharma, V.K.; Sharma, P.C.; et al. Contribution of germline BRCA1 and BRCA2 sequence alterations to breast cancer in Northern India. BMC Med. Genet. 2006, 7, 75. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Ji, L.; Li, H.; Gao, P.; Shang, G.; Zhang, D.D.; Zhang, N.; Jiang, T. Nrf2 pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PLoS ONE 2013, 8, e63404. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, H.J.; Bao, Q.C.; Wang, L.; Guo, T.K.; Chen, W.L.; Xu, L.L.; Zhou, H.S.; Bian, J.L.; Yang, Y.R.; et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016, 7, 73593–73606. [Google Scholar] [CrossRef]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. DPP3 in NRF2 Signaling and Breast Cancer. Free Radic. Biol. Med. 2016, 100, S132. [Google Scholar] [CrossRef]
- Zhou, X.L.; Zhu, C.Y.; Wu, Z.G.; Guo, X.; Zou, W. The oncoprotein HBXIP competitively binds KEAP1 to activate NRF2 and enhance breast cancer cell growth and metastasis. Oncogene 2019, 38, 4028–4046. [Google Scholar] [CrossRef]
- Zhang, H.S.; Zhang, Z.G.; Du, G.Y.; Sun, H.L.; Liu, H.Y.; Zhou, Z.; Gou, X.M.; Wu, X.H.; Yu, X.Y.; Huang, Y.H. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1alpha/Notch1 axis. J. Cell. Mol. Med. 2019, 23, 3451–3463. [Google Scholar] [CrossRef]
- Cong, Z.X.; Wang, H.D.; Zhou, Y.; Wang, J.W.; Pan, H.; Zhang, D.D.; Zhang, L.; Zhu, L. Temozolomide and irradiation combined treatment-induced Nrf2 activation increases chemoradiation sensitivity in human glioblastoma cells. J. Neurooncol. 2014, 116, 41–48. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Prevention of protein glycation by natural compounds. Molecules 2015, 20, 3309–3334. [Google Scholar] [CrossRef]
- Ahmad, R.; Ahmad, N.; Naqvi, A.A.; Exarchou, V.; Upadhyay, A.; Tuenter, E.; Foubert, K.; Apers, S.; Hermans, N.; Pieters, L. Antioxidant and Antiglycating Constituents from Leaves of Ziziphus oxyphylla and Cedrela serrata. Antioxidants 2016, 5, 9. [Google Scholar] [CrossRef]
- Meenatchi, P.; Purushothaman, A.; Maneemegalai, S. Antioxidant, antiglycation and insulinotrophic properties of Coccinia grandis (L.) in vitro: Possible role in prevention of diabetic complications. J. Tradit. Complement. Med. 2017, 7, 54–64. [Google Scholar] [CrossRef]
- Raghu, G.; Akileshwari, C.; Reddy, V.S.; Reddy, G.B. Attenuation of diabetic retinopathy in rats by ellagic acid through inhibition of AGE formation. J. Food Sci. Technol. 2017, 54, 2411–2421. [Google Scholar] [CrossRef]
- Sun, J.; Liu, W.; Ma, H.; Marais, J.P.J.; Khoo, C.; Dain, J.A.; Rowley, D.C.; Seeram, N.P. Effect of cranberry (Vaccinium macrocarpon) oligosaccharides on the formation of advanced glycation end-products. J. Berry Res. 2016, 6, 149–158. [Google Scholar] [CrossRef]
- Liu, W.; Wei, Z.; Ma, H.; Cai, A.; Liu, Y.; Sun, J.; DaSilva, N.A.; Johnson, S.L.; Kirschenbaum, L.J.; Cho, B.P.; et al. Anti-glycation and anti-oxidative effects of a phenolic-enriched maple syrup extract and its protective effects on normal human colon cells. Food Funct. 2017, 8, 757–766. [Google Scholar] [CrossRef]
- Gutierrez, R.M.; Baez, E.G. Evaluation of antidiabetic, antioxidant and antiglycating activities of the Eysenhardtia polystachya. Pharm. Mag. 2014, 10, S404–S418. [Google Scholar] [CrossRef] [PubMed]
- Adisakwattana, S.; Sompong, W.; Meeprom, A.; Ngamukote, S.; Yibchok-Anun, S. Cinnamic acid and its derivatives inhibit fructose-mediated protein glycation. Int. J. Mol. Sci. 2012, 13, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Dearlove, R.P.; Greenspan, P.; Hartle, D.K.; Swanson, R.B.; Hargrove, J.L. Inhibition of protein glycation by extracts of culinary herbs and spices. J. Med. Food 2008, 11, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Inada, M.; Shindo, M.; Kobayashi, K.; Sato, A.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.I. Anticancer effects of a non-narcotic opium alkaloid medicine, papaverine, in human glioblastoma cells. PLoS ONE 2019, 14, e0216358. [Google Scholar] [CrossRef]
- Takeuchi, A.; Yamamoto, Y.; Munesue, S.; Harashima, A.; Watanabe, T.; Yonekura, H.; Yamamoto, H.; Tsuchiya, H. Low molecular weight heparin suppresses receptor for advanced glycation end products-mediated expression of malignant phenotype in human fibrosarcoma cells. Cancer Sci. 2013, 104, 740–749. [Google Scholar] [CrossRef]
- Mizumoto, S.; Takahashi, J.; Sugahara, K. Receptor for advanced glycation end products (RAGE) functions as receptor for specific sulfated glycosaminoglycans, and anti-RAGE antibody or sulfated glycosaminoglycans delivered in vivo inhibit pulmonary metastasis of tumor cells. J. Biol. Chem. 2012, 287, 18985–18994. [Google Scholar] [CrossRef]
- Song, T.Y.; Yang, N.C.; Chen, C.L.; Thi, T.L.V. Protective Effects and Possible Mechanisms of Ergothioneine and Hispidin against Methylglyoxal-Induced Injuries in Rat Pheochromocytoma Cells. Oxid Med. Cell Longev 2017, 2017, 4824371. [Google Scholar] [CrossRef]
- Pellegrini, L.; Xue, J.; Larson, D.; Pastorino, S.; Jube, S.; Forest, K.H.; Saad-Jube, Z.S.; Napolitano, A.; Pagano, I.; Negi, V.S.; et al. HMGB1 targeting by ethyl pyruvate suppresses malignant phenotype of human mesothelioma. Oncotarget 2017, 8, 22649–22661. [Google Scholar] [CrossRef]
- Liu, Q.; Huo, Y.; Zheng, H.; Zhao, J.; Jia, L.; Wang, P. Ethyl pyruvate suppresses the growth, invasion and migration and induces the apoptosis of nonsmall cell lung cancer cells via the HMGB1/RAGE axis and the NFkappaB/STAT3 pathway. Oncol. Rep. 2019, 42, 817–825. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, I.Y.; Zhang, L.; Song, Y.; Liu, S.; Ren, H.; Liu, H.; Zhou, H.; Su, Y.; Yang, Y.; et al. S100B suppression alters polarization of infiltrating myeloid-derived cells in gliomas and inhibits tumor growth. Cancer Lett. 2018, 439, 91–100. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Pathways Affected by RAGEs | Regulation | Cancers | Refs. |
|---|---|---|---|
| p53 | Downregulation | Cancer cell proliferation (oral cancer, prostate cancer, pancreatic carcinoma, and erythroleukemia) | [21] |
| Beclin 1 | Upregulation | [22] | |
| JAK/STAT3 | Upregulation | [23] | |
| MAPKs | Upregulation | [23] | |
| PI3K/Akt/mTOR | Upregulation | [24] | |
| NF-kB | Upregulation | [25] | |
| VEGF | Upregulation | Angiogenesis (Breast cancer) | [25] |
| MMPs | Upregulation | Metastasis (Human breast and gastric tumors) | [13,26] |
| Nrf2 | Downregulation | Human oral cancer | [21] |
| Phytochemical | Signaling Pathways | Cancers | Refs |
|---|---|---|---|
| Papavarine | Downregulation of HMBG1, RAGE, and NF-κB | Fibrosarcoma | [20] |
| Downregulation of HMBG1 and RAGE | Glioblastoma | [224] | |
| Cinnamic acid | - | Yet to be examined against AGE-RAGE-mediated cancers (?) | [222] |
| Ellagic acid | - | [218] | |
| Maple syrup | - | [220] | |
| Thiol-amine | - | ||
| Drugs | |||
| Ergothioneine | Downregulation of AGEs, RAGE, and NF-κB | Pheochromocytoma | [227] |
| Hispidin | Pheochromocytoma | [227] | |
| Chondroitin sulfate and heparan sulfate | - | Lung metastasis | [226] |
| Duloxetine | Downregulation of S100B | Glioma cancer | [230] |
| Ethyl pyruvate | Downregulation of HMBG1, RAGE, and NF-κB | Malignant mesothelioma | [228] |
| Downregulation of the HMBG1, RAGE, NF-κB, and STAT3 Pathways | Non-small cell lung cancer | [229] | |
| Low molecular weight heparin | Downregulation of RAGE-mediated NF-kB | Fibrosarcoma | [225] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beeraka, N.M.; Bovilla, V.R.; Doreswamy, S.H.; Puttalingaiah, S.; Srinivasan, A.; Madhunapantula, S.V. The Taming of Nuclear Factor Erythroid-2-Related Factor-2 (Nrf2) Deglycation by Fructosamine-3-Kinase (FN3K)-Inhibitors-A Novel Strategy to Combat Cancers. Cancers 2021, 13, 281. https://doi.org/10.3390/cancers13020281
Beeraka NM, Bovilla VR, Doreswamy SH, Puttalingaiah S, Srinivasan A, Madhunapantula SV. The Taming of Nuclear Factor Erythroid-2-Related Factor-2 (Nrf2) Deglycation by Fructosamine-3-Kinase (FN3K)-Inhibitors-A Novel Strategy to Combat Cancers. Cancers. 2021; 13(2):281. https://doi.org/10.3390/cancers13020281
Chicago/Turabian StyleBeeraka, Narasimha M., Venugopal R. Bovilla, Shalini H. Doreswamy, Sujatha Puttalingaiah, Asha Srinivasan, and SubbaRao V. Madhunapantula. 2021. "The Taming of Nuclear Factor Erythroid-2-Related Factor-2 (Nrf2) Deglycation by Fructosamine-3-Kinase (FN3K)-Inhibitors-A Novel Strategy to Combat Cancers" Cancers 13, no. 2: 281. https://doi.org/10.3390/cancers13020281
APA StyleBeeraka, N. M., Bovilla, V. R., Doreswamy, S. H., Puttalingaiah, S., Srinivasan, A., & Madhunapantula, S. V. (2021). The Taming of Nuclear Factor Erythroid-2-Related Factor-2 (Nrf2) Deglycation by Fructosamine-3-Kinase (FN3K)-Inhibitors-A Novel Strategy to Combat Cancers. Cancers, 13(2), 281. https://doi.org/10.3390/cancers13020281