‘Educated’ Osteoblasts Reduce Osteoclastogenesis in a Bone-Tumor Mimetic Microenvironment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
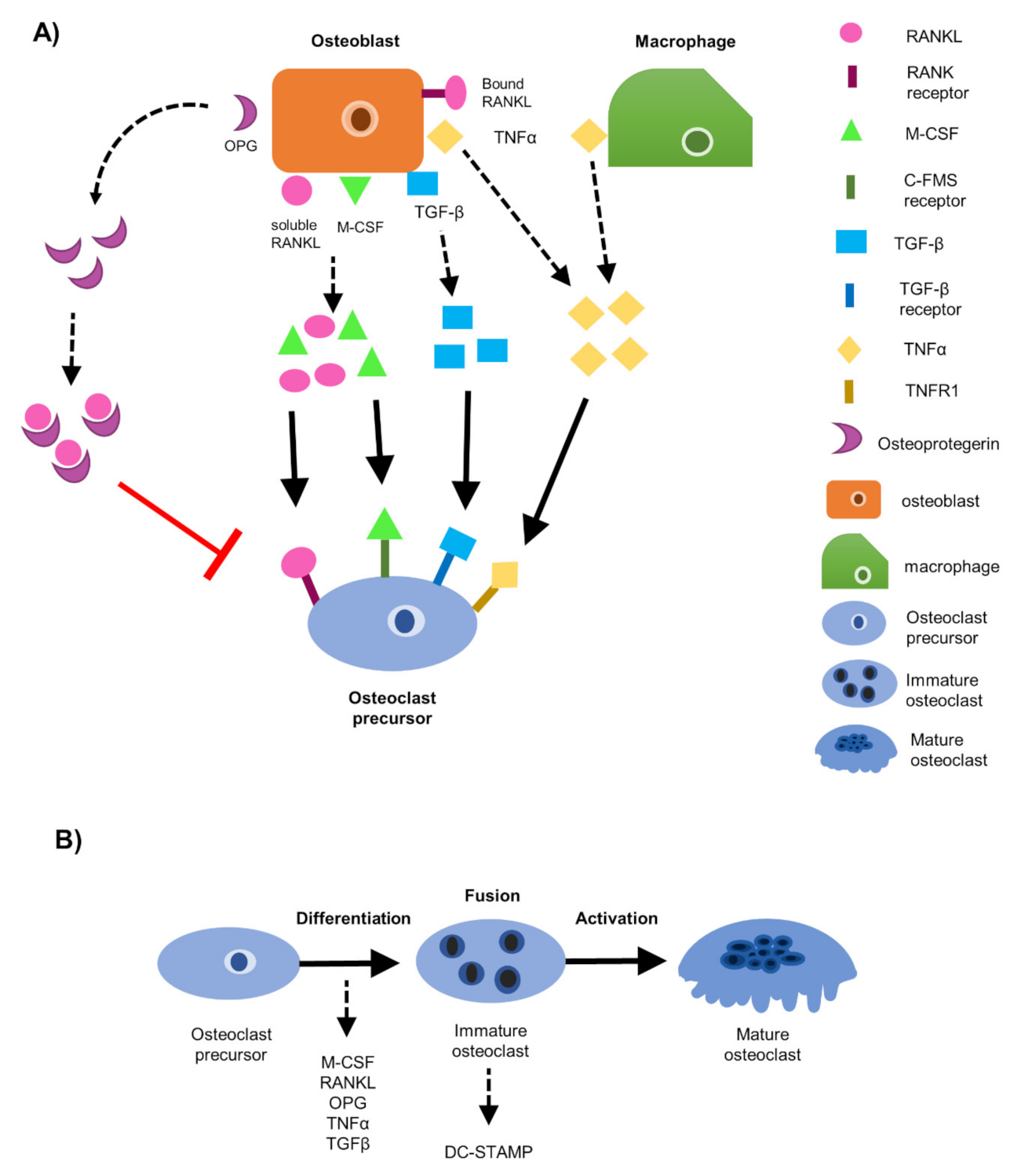
1. Introduction
2. Materials and Methods
2.1. Cells
2.1.1. Osteoblasts
2.1.2. Osteoclast Precursors
2.1.3. Breast Cancer Cell Variants
2.2. Breast Cancer Conditioned Media
2.3. Osteoblast Conditioned Media
2.4. Generation of EOs In-Vitro
2.5. EO Cell Conditioned Media
2.6. Soluble Protein Expression of Osteoclastogenic Factors RANKL, OPG, and TNFα
2.7. Separation of CD11b+ Mononuclear Cells from Murine Bone Marrow Aspirate
2.8. Identification of TRAP+ Osteoclasts in Cultures Containing CD11b+ Primary Bone Marrow Monocytes
2.9. Identification of TRAP+ Osteoclasts in Cultures Containing RAW 264.7 Pre-Osteoclasts
2.9.1. Conditioned Medium
2.9.2. Co-Culture
2.10. In-Vitro Bone Resorption Assay and Quantification on a Bone Mimetic Matrix
2.10.1. Identification and Quantification of TRAP+ Osteoclasts on a Bone Mimetic Matrix
2.10.2. Identification and Quantification of Osteoclast Resorptive Pits on Bone Mimetic Matrix
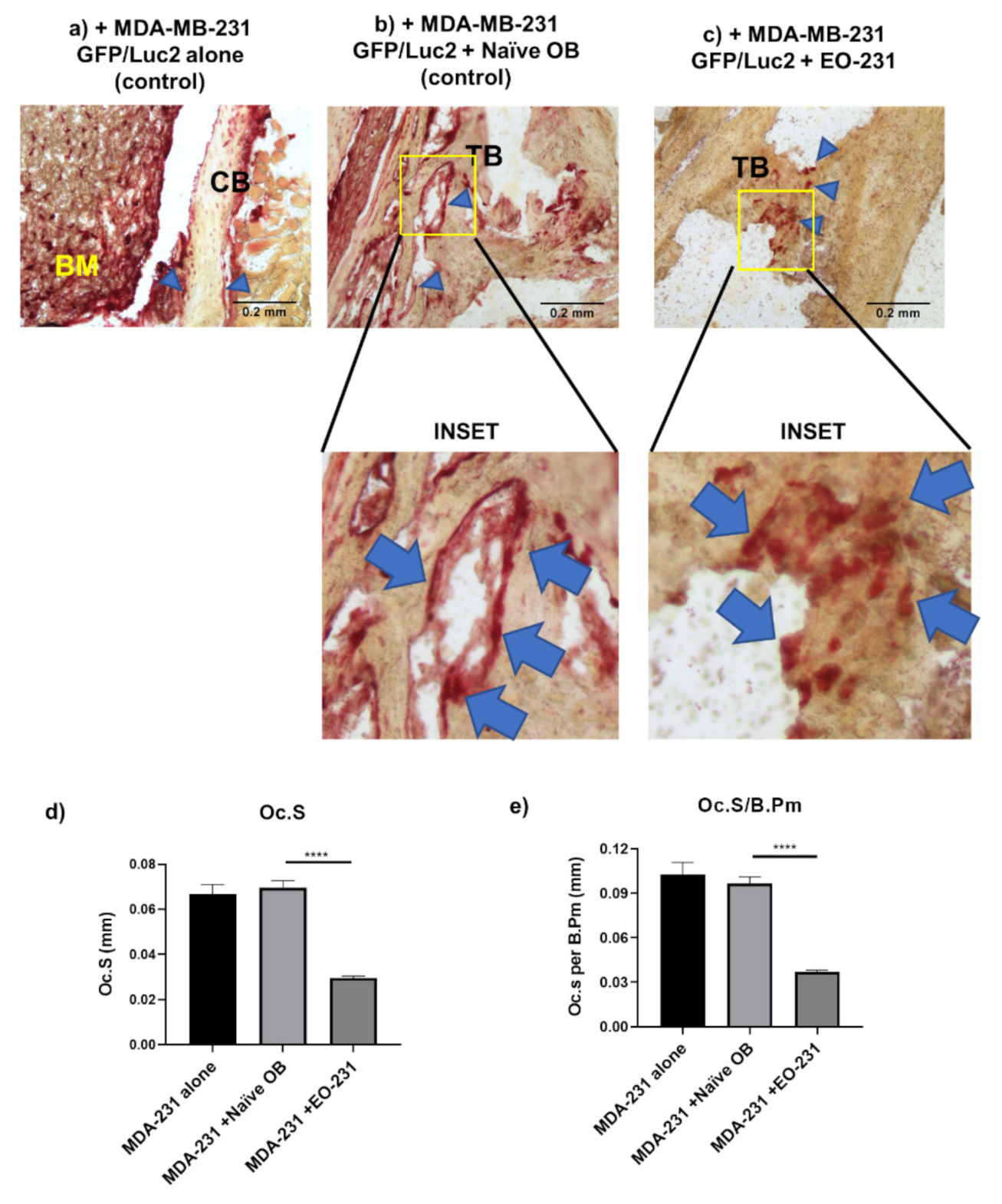
2.11. Intratibial Inoculations and TRAP Stain of Murine Bone Sections
2.11.1. Intratibial Inoculation of Murine Tibiae
2.11.2. Removal of Murine Tibiae and Bone Preparation
2.11.3. TRAP Stain on Murine Tibiae Sections
2.12. Western Blotting
2.13. TNFα Rescue and Neutralization on Pre-Osteoclasts Exposed to EO CM
2.13.1. TNFα Rescue
2.13.2. TNFα Neutralization
2.14. Statistical Analysis
3. Results
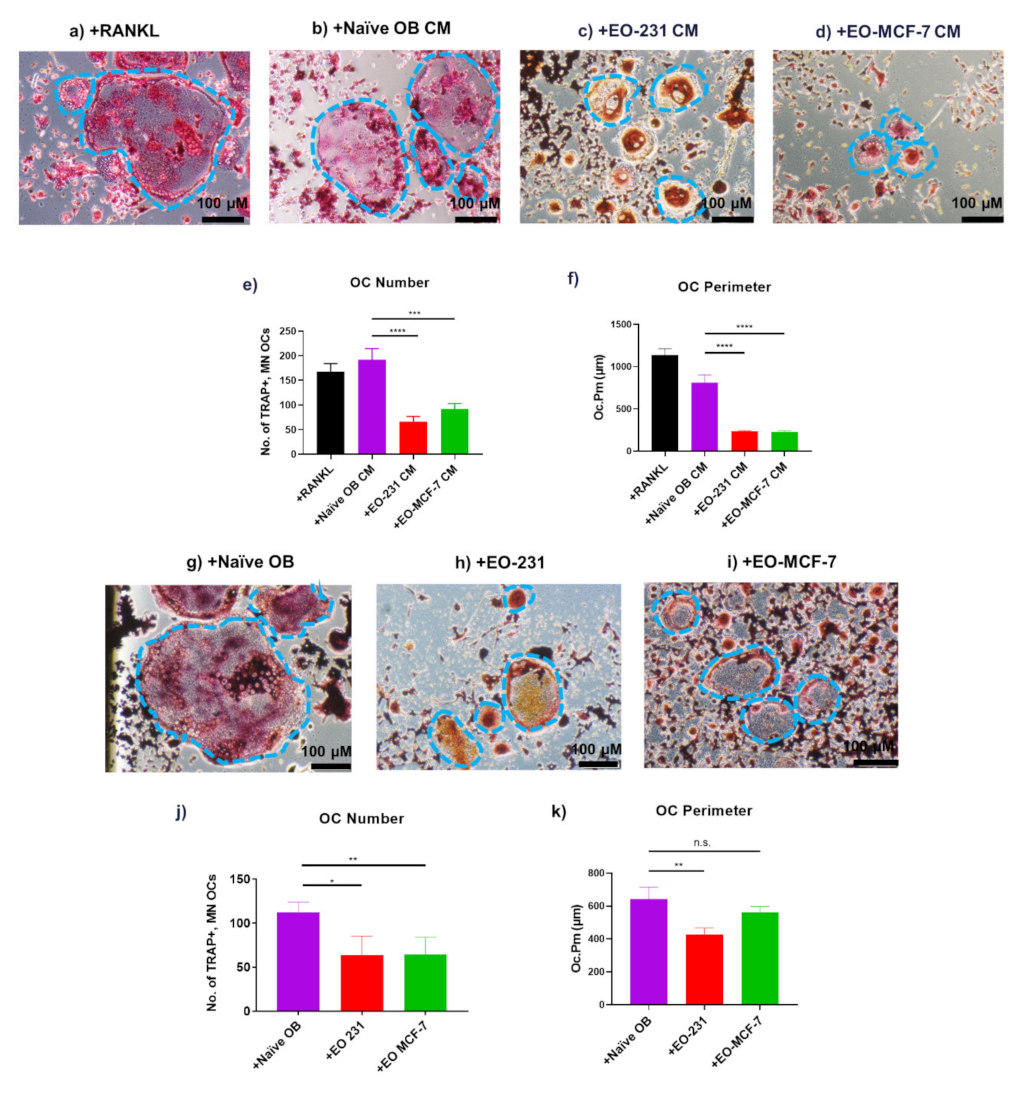
3.1. EO Cells or Their Conditioned Media Reduce Osteoclast Maturation
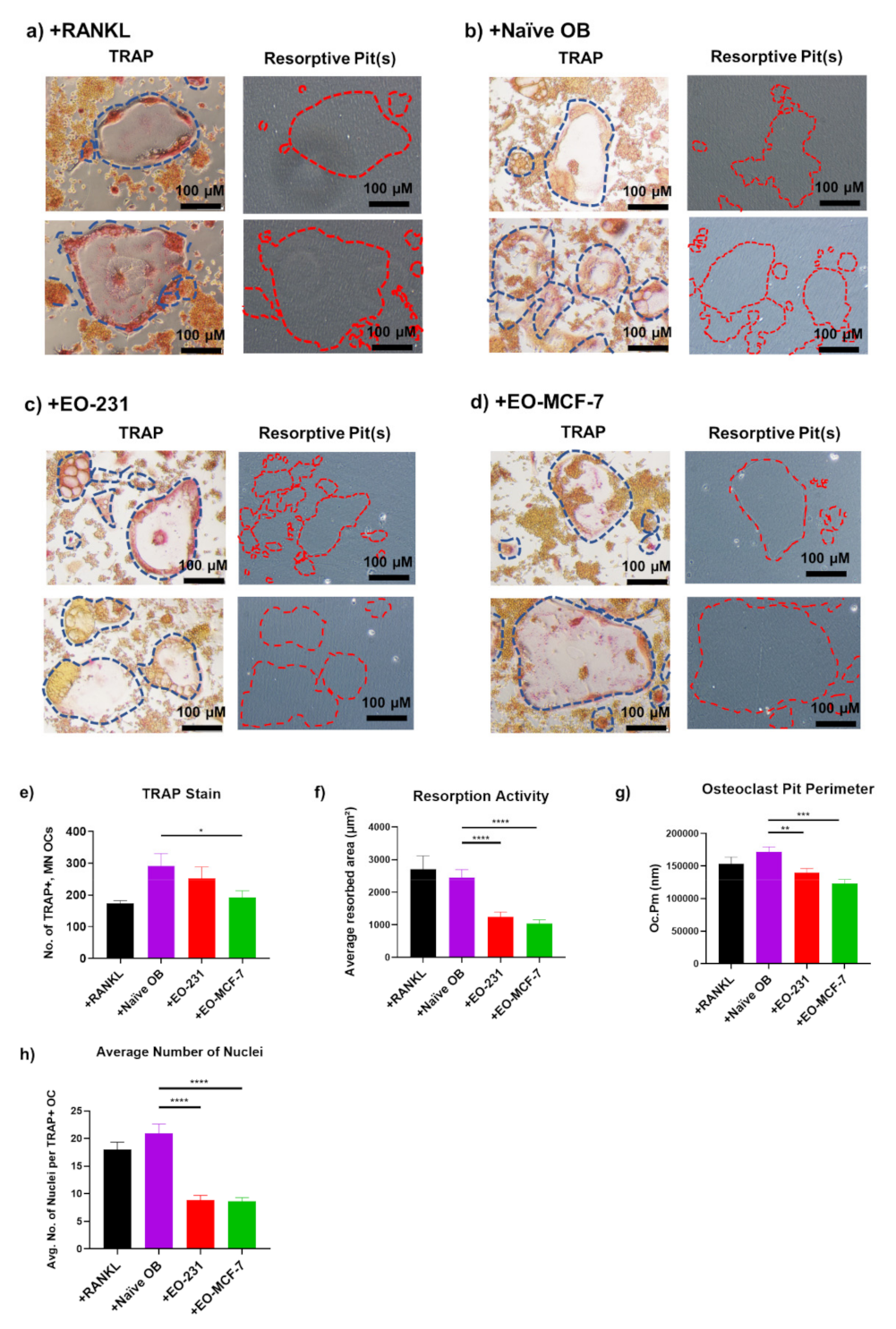
3.2. Osteoclasts Produced in the Presence of EO Cells Have Reduced Resorption
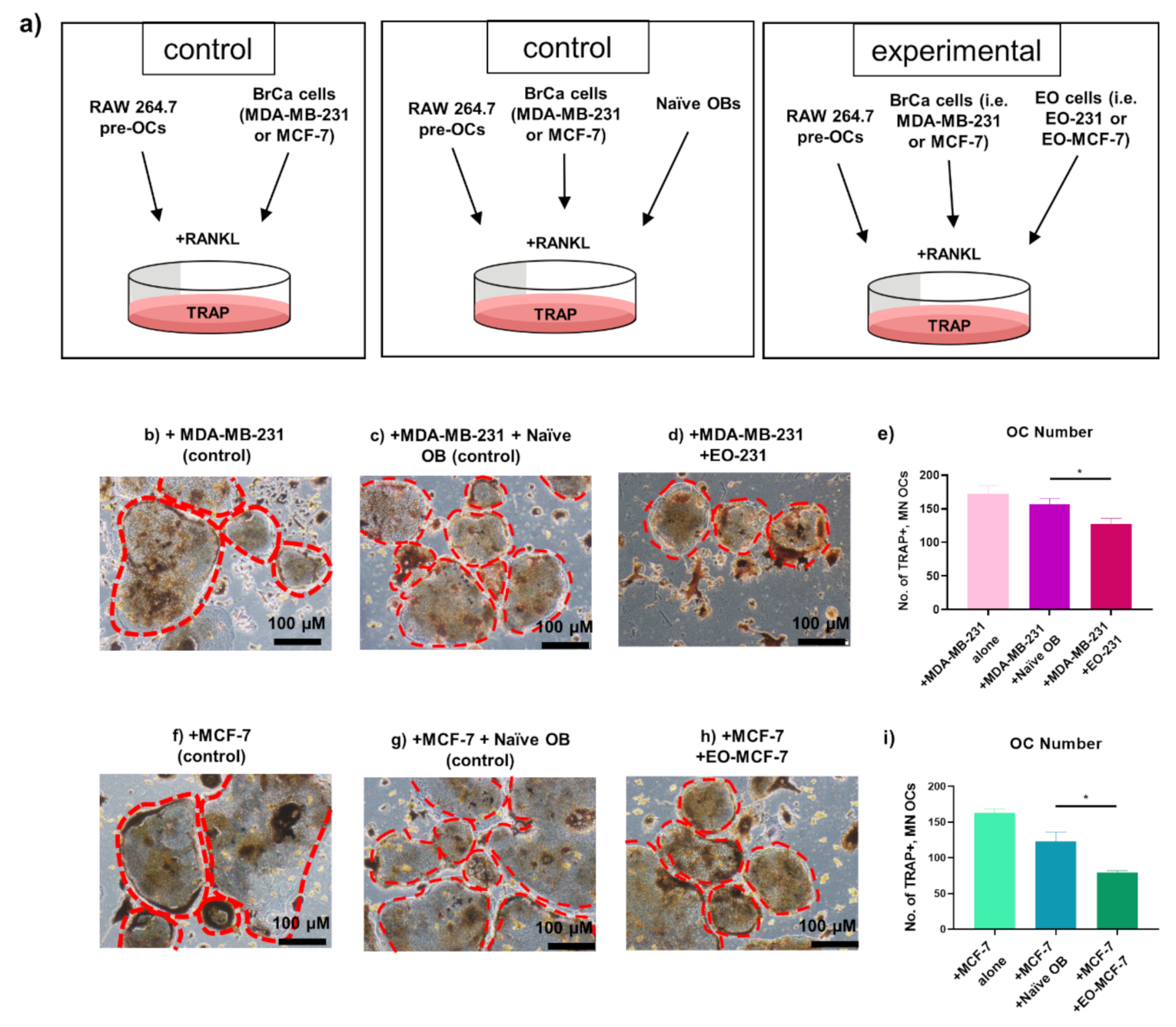
3.3. The Number of Osteoclasts Produced in the Presence of EO Cells Plus Breast Cancer Cells Are Reduced
3.4. Osteoclast Size Is Decreased When EO Cells Are Present in the Niche
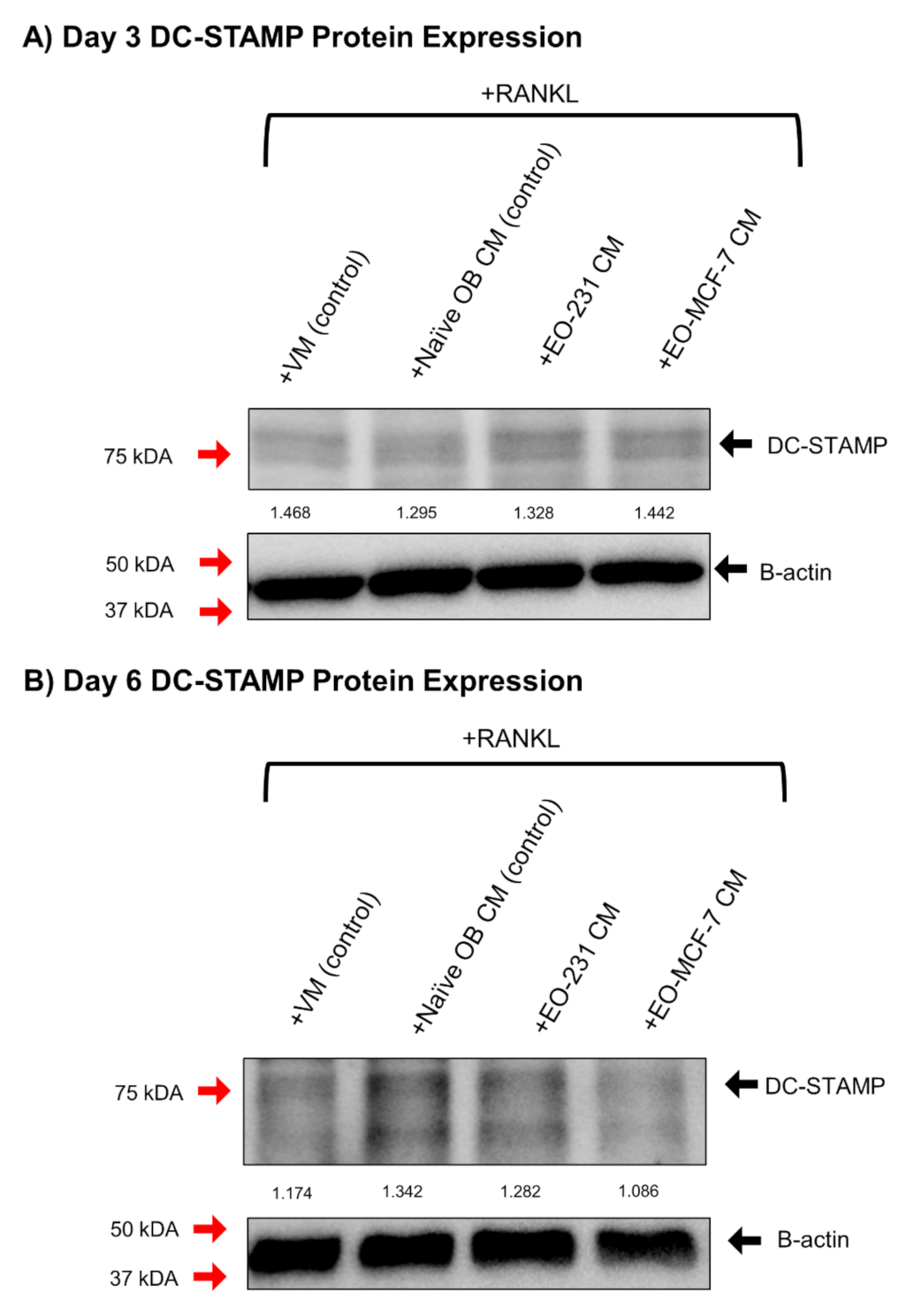
3.5. DC-STAMP Expression Is Altered in Osteoclasts Exposed to EO CM
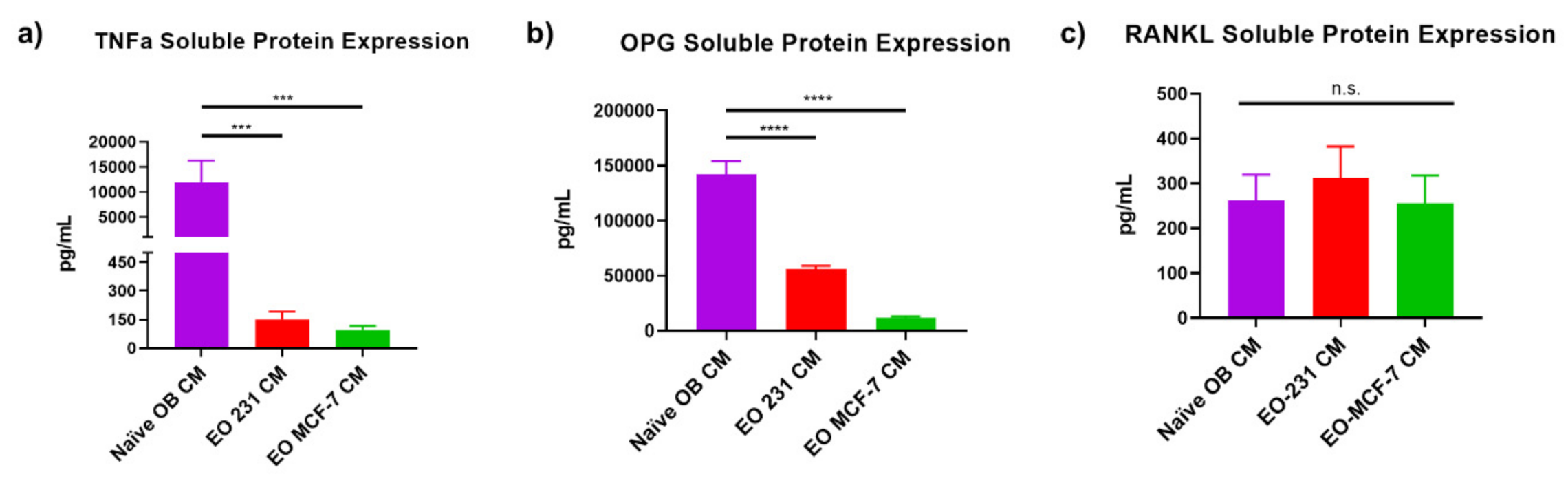
3.6. EO Cells Have Altered Expression of Osteoclast-Associated Factors, TNFα and OPG
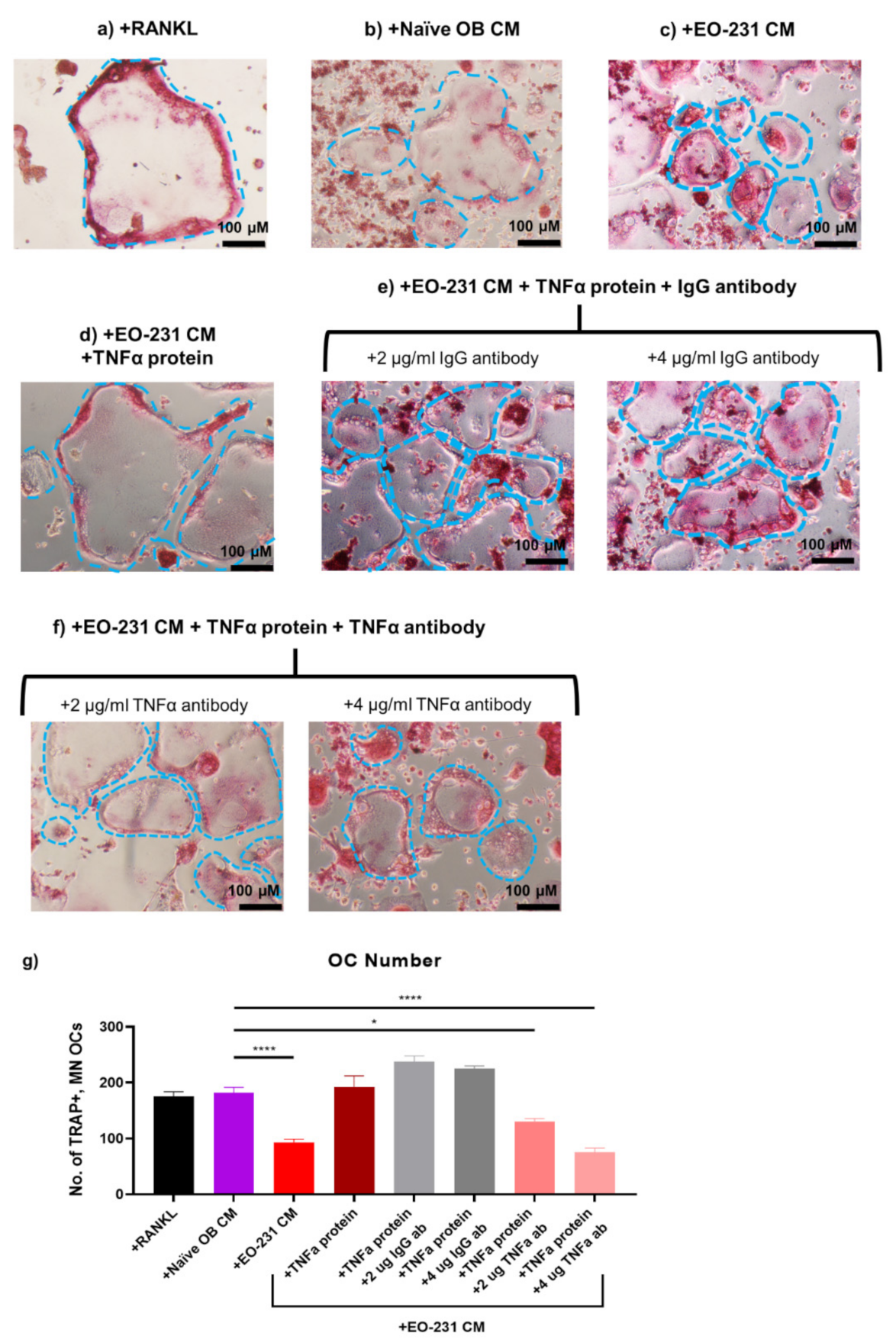
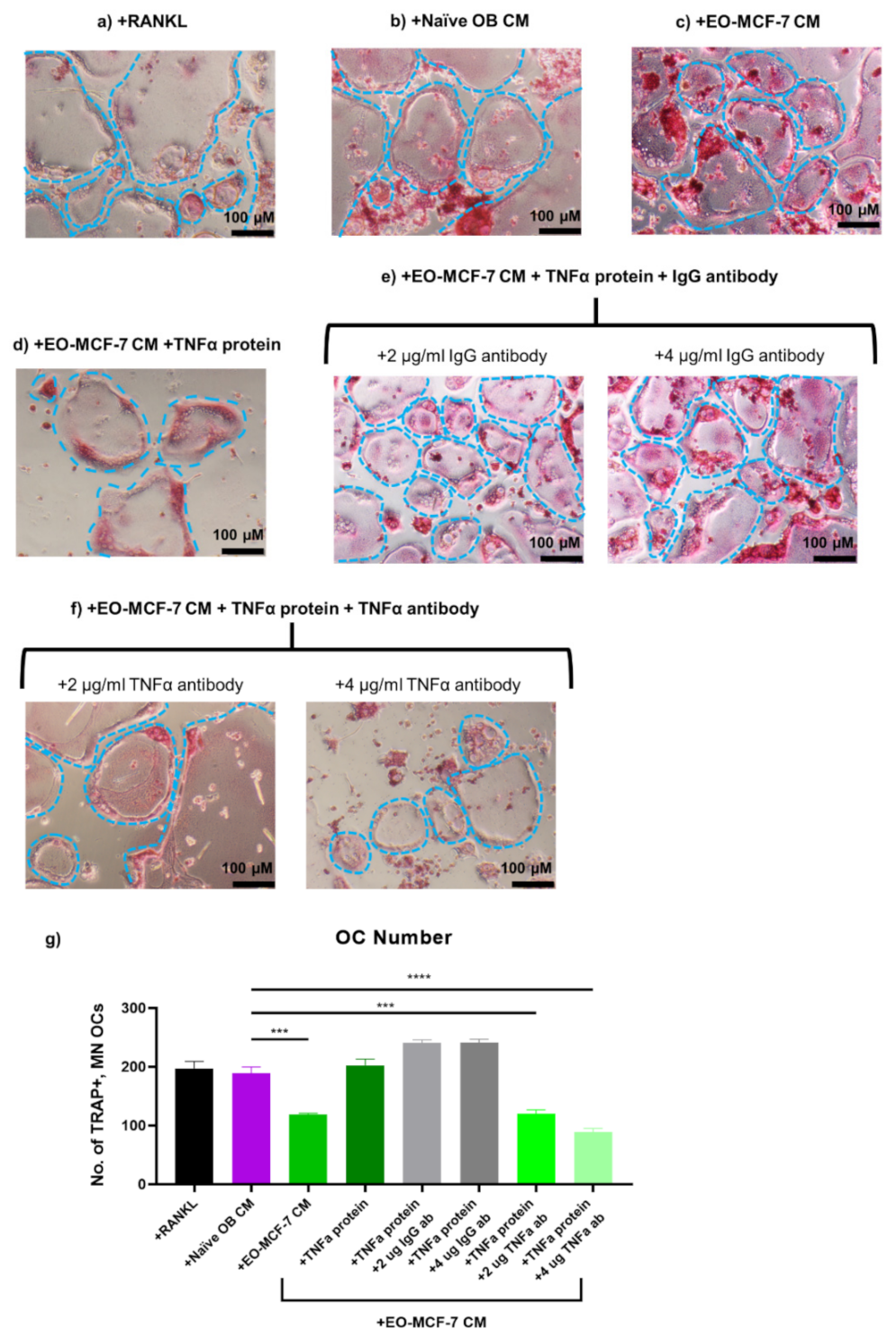
3.7. Soluble Protein TNFα Modulates Osteoclast Formation
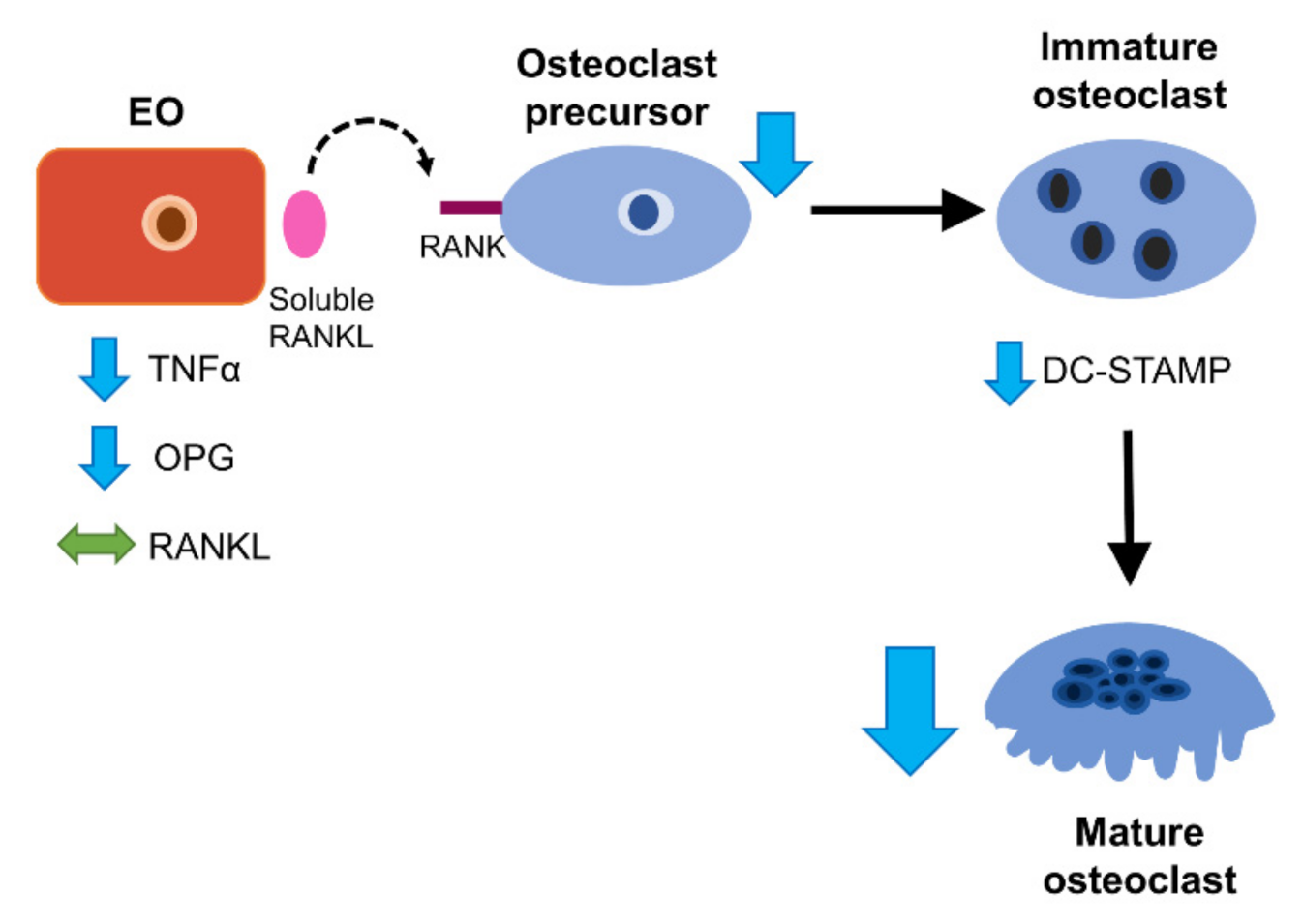
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Mariotto, A.B.; Etzioni, R.; Hurlbert, M.; Penberthy, L.; Mayer, M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol. Biomark. Prev. 2017, 26, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2007, 27, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of Bone Metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef]
- Phadke, P.A.; Mercer, R.R.; Harms, J.F.; Jia, Y.; Frost, A.R.; Jewell, J.L.; Bussard, K.M.; Nelson, S.; Moore, C.; Kappes, J.C.; et al. Kinetics of Metastatic Breast Cancer Cell Trafficking in Bone. Clin. Cancer Res. 2006, 12, 1431–1440. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Raggatt, L.J.; Partridge, N.C. Cellular and Molecular Mechanisms of Bone Remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef]
- Yoneda, T.; Sasaki, A.; Mundy, G.R. Osteolytic bone metastasis in breast cancer. Breast Cancer Res. Treat. 1994, 32, 73–84. [Google Scholar] [CrossRef]
- Yoneda, T. Cellular and molecular basis of preferential metastasis of breast cancer to bone. J. Orthop. Sci. 2000, 5, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Russell, R.G.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Body, J.-J. Rationale for the use of bisphosphonates in osteoblastic and osteolytic bone lesions. Breast 2003, 12, S37–S44. [Google Scholar] [CrossRef]
- Suen, P.K.; Qin, L. Sclerostin, an emerging therapeutic target for treating osteoporosis and osteoporotic fracture: A general review. J. Orthop. Transl. 2016, 4, 1–13. [Google Scholar] [CrossRef]
- Ominsky, M.S.; Boyd, S.K.; Varela, A.; Jolette, J.; Felx, M.; Doyle, N.; Mellal, N.; Smith, S.Y.; Locher, K.; Buntich, S.; et al. Romosozumab Improves Bone Mass and Strength While Maintaining Bone Quality in Ovariectomized Cynomolgus Monkeys. J. Bone Miner. Res. 2017, 32, 788–801. [Google Scholar] [CrossRef]
- Lau, E.M.C.; Dinavahi, R.; Woo, Y.C.; Wu, C.-H.; Guan, J.; Maddox, J.; Tolman, C.; Yang, W.; Shin, C.S. Romosozumab or alendronate for fracture prevention in East Asian patients: A subanalysis of the phase III, randomized ARCH study. Osteoporos. Int. 2020, 31, 677–685. [Google Scholar] [CrossRef]
- Ogawa, T.; Ohshika, S.; Yanagisawa, M.; Kurose, A.; Ishibashi, Y. Teriparatide may accelerate the growth of a pre-existing malignant tumor in an elderly patient with osteoporosis: A case report. Mol. Clin. Oncol. 2019, 12, 144–147. [Google Scholar] [CrossRef]
- Guise, T.A. Molecular mechanisms of osteolytic bone metastases. Cancer 2000, 88, 2892–2898. [Google Scholar] [CrossRef]
- A Guise, T.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL–RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006, 12, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nat. Cell Biol. 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Yasuda, H.; Mizuno, A.; Itoh, K.; Ueno, Y.; Shinki, T.; Gillespie, M.T.; Martin, T.J.; Higashio, K.; et al. Osteoprotegerin Produced by Osteoblasts Is an Important Regulator in Osteoclast Development and Function*. Endocrinol. 2000, 141, 3478–3484. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef]
- Nakayamada, S.; Okada, Y. Osteoblasts and Osteoclasts in Bone Remodeling and Inflammation. Curr. Drug Target Inflamm. Allergy 2005, 4, 325–328. [Google Scholar] [CrossRef]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, A.J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor Necrosis Factor α Stimulates Osteoclast Differentiation by a Mechanism Independent of the Odf/Rankl–Rank Interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- Itonaga, I.; Sabokbar, A.; Sun, S.; Kudo, O.; Danks, L.; Ferguson, D.J.P.; Fujikawa, Y.; Athanasou, N. Transforming growth factor-β induces osteoclast formation in the absence of RANKL. Bone 2004, 34, 57–64. [Google Scholar] [CrossRef]
- Abu-Amer, Y. Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J. Biol. Chem. 2000, 275, 27307–27310. [Google Scholar] [CrossRef]
- Eosta, B.; Ebenedetti, G.; Miossec, P. Classical and Paradoxical Effects of TNF-α on Bone Homeostasis. Front. Immunol. 2014, 5, 48. [Google Scholar] [CrossRef]
- Zhang, C.; E Dou, C.; Xu, J.; Dong, S. DC-STAMP, the Key Fusion-Mediating Molecule in Osteoclastogenesis. J. Cell. Physiol. 2014, 229, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.G.; Ritchlin, C.T. DC-STAMP: A Key Regulator in Osteoclast Differentiation. J. Cell. Physiol. 2016, 231, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastognesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Nat. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Mundy, G.R. Cancer and bone. Endocr. Rev. 1998, 19, 18–54. [Google Scholar]
- Wu, X.; Li, F.; Dang, L.; Liang, C.; Lu, A.; Zhang, G. RANKL/RANK System-Based Mechanism for Breast Cancer Bone Metastasis and Related Therapeutic Strategies. Front. Cell Dev. Biol. 2020, 8, 76. [Google Scholar] [CrossRef]
- Kolb, A.D.; Bussard, K.M. The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases. Cancers 2019, 11, 1020. [Google Scholar] [CrossRef]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Shupp, A.B.; Kolb, A.D.; Mukhopadhyay, D.; Bussard, K.M. Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers 2018, 10, 182. [Google Scholar] [CrossRef]
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Marini, F.C.; Bussard, K.M. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res. 2019, 21, 31. [Google Scholar] [CrossRef]
- Thomas, R.J.; Guise, T.A.; Yin, J.J.; Elliott, J.; Horwood, N.J.; Martin, T.J.; Gillespie, M.T. Breast Cancer Cells Interact with Osteoblasts to Support Osteoclast Formation1. Endocrinology 1999, 140, 4451–4458. [Google Scholar] [CrossRef]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Khoo, W.H.; Terry, R.L.; Down, J.M.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef]
- Ralph, P.; Nakoinz, I. Antibody-dependent killing of erythrocyte and tumor targets by macrophage-related cell lines: Enhancement by PPD and LPS. J. Immunol. 1977, 119, 950–954. [Google Scholar] [PubMed]
- Raschke, W.; Baird, S.; Ralph, P.; Nakoinz, I. Functional macrophage cell lines transformed by abelson leukemia virus. Cell 1978, 15, 261–267. [Google Scholar] [CrossRef]
- Takahashi, N.; Udagawa, N.; Kobayashi, Y.; Suda, T. Generation of Osteoclasts In Vitro, and Assay of Osteoclast Activity. Mol. Cardiol. 2007, 135, 285–301. [Google Scholar] [CrossRef]
- Islam, S.; Hassan, F.; Tumurkhuu, G.; Dagvadorj, J.; Koide, N.; Naiki, Y.; Yoshida, T.; Yokochi, T. Receptor activator of nuclear factor-kappa B ligand induces osteoclast formation in RAW 264.7 macrophage cells via augmented production of macrophage-colony-stimulating factor. Microbiol. Immunol. 2008, 52, 585–590. [Google Scholar] [CrossRef]
- Wei, S.; Teitelbaum, S.L.; Wang, M.W.-H.; Ross, F.P. Receptor Activator of Nuclear Factor-κB Ligand Activates Nuclear Factor-κB in Osteoclast Precursors*. Endocrinology 2001, 142, 1290–1295. [Google Scholar] [CrossRef]
- Cailleau, R.; Olivé, M.; Cruciger, Q.V.J. Long-term human breast carcinoma cell lines of metastatic origin: Preliminary characterization. In Vitro Cell. Dev. Biol. Anim. 1978, 14, 911–915. [Google Scholar] [CrossRef]
- Qian, C.; Worrede, A.; Shen, F.; DiNatale, A.; Kaur, R.; Zhang, Q.; Cristofanilli, M.; Meucci, O.; Fatatis, A. Impeding Circulating Tumor Cell Reseeding Decelerates Metastatic Progression and Potentiates Chemotherapy. Mol. Cancer Res. 2018, 16, 1844–1854. [Google Scholar] [CrossRef]
- Soule, H.D.; Vazquez, J.; Long, A.; Albert, S.; Brennan, M. A Human Cell Line From a Pleural Effusion Derived From a Breast Carcinoma. J. Natl. Cancer Inst. 1973, 51, 1409–1416. [Google Scholar] [CrossRef]
- Ishizuka, H.; García-Palacios, V.; Lu, G.; A Subler, M.; Zhang, H.; Boykin, C.S.; Choi, S.J.; Zhao, L.; Patrene, K.; Galson, D.L.; et al. ADAM8 enhances osteoclast precursor fusion and osteoclast formation in vitro and in vivo. J. Bone Miner. Res. 2010, 26, 169–181. [Google Scholar] [CrossRef]
- Minkin, C. Bone acid phosphatase: Tartrate-resistant acid phosphatase as a marker of osteoclast function. Calcif. Tissue Int. 1982, 34, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.E.; Ottewell, P.D.; Rucci, N.; Peyruchaud, O.; Pagnotti, G.M.; Chiechi, A.; Buijs, J.T.; Sterling, J.A. Murine models of breast cancer bone metastasis. BoneKEy Rep. 2016, 5, 804. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Lacey, D.L.; Dunstan, C.R.; Solovyev, I.; Colombero, A.; Timms, E.; Tan, H.-L.; Elliott, G.; Kelley, M.J.; Sarosi, I.; et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. USA 1999, 96, 3540–3545. [Google Scholar] [CrossRef]
- Collin-Osdoby, P.; Osdoby, P. RANKL-Mediated Osteoclast Formation from Murine RAW 264.7 cells. In Bone Research Protocols; Helfrich, M.H., Ralston, S.H., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 187–202. [Google Scholar] [CrossRef]
- Bernhardt, A.; Koperski, K.; Schumacher, M.; Gelinsky, M. Relevance of osteoclast-specific enzyme activities in cell-based in vitro resorption assays. Eur. Cells Mater. 2017, 33, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Solberg, L.B.; Brorson, S.-H.; Stordalen, G.A.; Baekkevold, E.S.; Andersson, G.; Reinholt, F.P. Increased Tartrate-Resistant Acid Phosphatase Expression in Osteoblasts and Osteocytes in Experimental Osteoporosis in Rats. Calcif. Tissue Int. 2014, 94, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Sud, S.; Pienta, K.J. Prostate cancer promotes CD11b positive cells to differentiate into osteoclasts. J. Cell. Biochem. 2009, 106, 563–569. [Google Scholar] [CrossRef]
- Hayashi, H.; Nakahama, K.-I.; Sato, T.; Tuchiya, T.; Asakawa, Y.; Maemura, T.; Tanaka, M.; Morita, M.; Morita, I. The role of Mac-1 (CD11b/CD18) in osteoclast differentiation induced by receptor activator of nuclear factor-κB ligand. FEBS Lett. 2008, 582, 3243–3248. [Google Scholar] [CrossRef]
- Smith, C.W. 3. Adhesion molecules and receptors. J. Allergy Clin. Immunol. 2008, 121, S375–S379. [Google Scholar] [CrossRef]
- Xiao, Y.; Palomero, J.; Grabowska, J.; Wang, L.; De Rink, I.; Van Helvert, L.; Borst, J. Macrophages and osteoclasts stem from a bipotent progenitor downstream of a macrophage/osteoclast/dendritic cell progenitor. Blood Adv. 2017, 1, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Berghaus, L.J.; Moore, J.N.; Hurley, D.J.; Vandenplas, M.L.; Fortes, B.P.; Wolfert, M.A.; Boons, G.-J. Innate immune responses of primary murine macrophage-lineage cells and RAW 264.7 cells to ligands of Toll-like receptors 2, 3, and 4. Comp. Immunol. Microbiol. Infect. Dis. 2010, 33, 443–454. [Google Scholar] [CrossRef]
- Yang, L.; Edwards, C.M.; Mundy, G.R. Gr-1+CD11b+ myeloid-derived suppressor cells: Formidable partners in tumor metastasis. J. Bone Miner. Res. 2010, 25, 1701–1706. [Google Scholar] [CrossRef]
- Jacome-Galarza, C.E.; Lee, S.-K.; Lorenzo, J.A.; Aguila, H.L. Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J. Bone Miner. Res. 2012, 28, 1203–1213. [Google Scholar] [CrossRef]
- Maurizi, A.; Rucci, N. The Osteoclast in Bone Metastasis: Player and Target. Cancers 2018, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Palokangas, H.; Mulari, M.; Vaananen, H.K. Endocytotic pathway from the basal plasma membrane to the ruffled border membrane in bone-resorbing osteoclasts. J. Cell. Sci. 1997, 110, 1767–1780. [Google Scholar]
- Boyde, A.; Ali, N.N.; Jones, S.J. Resorption of dentine by isolated osteoclasts in vitro. Br. Dent. J. 1984, 156, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.d.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Rumpler, M.; Wurger, T.; Roschger, P.; Zwettler, E.; Sturmlechner, I.; Altmann, P.; Fratzl, P.; Rogers, M.J.; Klaushofer, K. Osteoclasts on Bone and Dentin In Vitro: Mechanism of Trail Formation and Comparision of Resorption Behavior. Calcif. Tissue Int. 2013, 93, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Mastro, A.M.; Gay, C.V.; Welch, D.R.; Donahue, H.J.; Jewell, J.; Mercer, R.; DiGirolamo, D.; Chislock, E.M.; Guttridge, K. Breast cancer cells induce osteoblast apoptosis: A possible contributor to bone degradation. J. Cell. Biochem. 2004, 91, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Dempster, D.; E Compston, J.; Drezner, M.K.; Glorieux, F.H.; A Kanis, J.; Malluche, H.; Meunier, P.J.; Ott, S.M.; Recker, R.R.; Parfitt, A.M. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2012, 28, 2–17. [Google Scholar] [CrossRef]
- Chiu, Y.G.; Mensah, K.A.; Schwarz, E.M.; Ju, Y.; Takahata, M.; Feng, C.; McMahon, L.A.; Hicks, D.G.; Panepento, B.; Keng, P.C.; et al. Regulation of human osteoclast development by dendritic cell-specific transmembrane protein (DC-STAMP). J. Bone Miner. Res. 2011, 27, 79–92. [Google Scholar] [CrossRef]
- Suda, T.; Kobayashi, K.; Jimi, E.; Udagawa, N.; Takahashi, N. The molecular basis of osteoclast differentiation and activation. Novartis Found. Symp. 2001, 232, 235–250. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam. Med. J. 2016, 52, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.; Timms, E.; Tan, H.-L.; Kelley, M.; Dunstan, C.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin Ligand Is a Cytokine that Regulates Osteoclast Differentiation and Activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef]
- Fuller, K.; Murphy, C.; Kirstein, B.; Fox, S.W.; Chambers, T.J. TNFα Potently Activates Osteoclasts, through a Direct Action Independent of and Strongly Synergistic with RANKL. Endocrinology 2002, 143, 1108–1118. [Google Scholar] [CrossRef]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef]
- Luo, G.; Li, F.; Li, X.; Wang, Z.; Zhang, B. TNF-α and RANKL promote osteoclastogenesis by upregulating RANK via the NF-κB pathway. Mol. Med. Rep. 2018, 17, 6605–6611. [Google Scholar] [CrossRef]
- Steffen, U.; Schett, G.; Bozec, A. How Autoantibodies Regulate Osteoclast Induced Bone Loss in Rheumatoid Arthritis. Front. Immunol. 2019, 10, 1483. [Google Scholar] [CrossRef]
- Walsh, M.C.; Choi, Y. Biology of the RANKL–RANK–OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef]
- Luo, J.; Yang, Z.; Ma, Y.; Yue, Z.; Lin, H.; Qu, G.; Huang, J.; Dai, W.; Li, C.; Zheng, C.; et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat. Med. 2016, 22, 539–546. [Google Scholar] [CrossRef]
- Sakurai, H.; Miyoshi, H.; Toriumi, W.; Sugita, T. Functional Interactions of Transforming Growth Factor β-activated Kinase 1 with IκB Kinases to Stimulate NF-κB Activation. J. Biol. Chem. 1999, 274, 10641–10648. [Google Scholar] [CrossRef]
- Chamberlain, L.M.; Godek, M.L.; Gonzalez-Juarrero, M.; Grainger, D.D. Phenotypic non-equivalence of murine (monocyte-) macrophage cells in biomaterial and inflammatory models. J. Biomed. Mater. Res. Part A 2009, 88, 858–871. [Google Scholar] [CrossRef]
- Ng, A.Y.; Tu, C.; Shen, S.; Xu, D.; Oursler, M.J.; Qu, J.; Yang, S. Comparative Characterization of Osteoclasts Derived From Murine Bone Marrow Macrophages and RAW 264.7 Cells Using Quantitative Proteomics. JBMR Plus 2018, 2, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Cuetara, B.L.V.; Crotti, T.N.; O’Donoghue, A.J.; McHugh, K.P. Cloning and characterization of osteoclast precursors from the raw264.7 cell line. In Vitro Cell. Dev. Biol. Anim. 2006, 42, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.C.; Takada, Y.; Shishodia, S.; Aggarwal, B.B. Evidence That Receptor Activator of Nuclear Factor (NF)-κB Ligand Can Suppress Cell Proliferation and Induce Apoptosis through Activation of a NF-κB-independent and TRAF6-dependent Mechanism. J. Biol. Chem. 2004, 279, 6065–6076. [Google Scholar] [CrossRef]
- Shore, S.K.; Tantravahi, R.V.; Reddy, E.P. Transforming pathways activated by the v-Abl tyrosine kinase. Oncogene 2002, 21, 8568–8576. [Google Scholar] [CrossRef] [PubMed]
- Hattersley, G.; Owens, J.; Flanagan, A.; Chambers, T. Macrophage colony stimulating factor (M-CSF) is essential for osteoclast formation in vitro. Biochem. Biophys. Res. Commun. 1991, 177, 526–531. [Google Scholar] [CrossRef]
- Zhuang, J.; Zhang, J.; Lwin, S.T.; Edwards, J.R.; Edwards, C.M.; Mundy, G.R.; Yang, X. Osteoclasts in Multiple Myeloma Are Derived from Gr-1+CD11b+Myeloid-Derived Suppressor Cells. PLoS ONE 2012, 7, e48871. [Google Scholar] [CrossRef]
- Piper, K.; Boyde, A.; Jones, S.J. The relationship between the number of nuclei of an osteoclast and its resorptive capability in vitro. Brain Struct. Funct. 1992, 186, 291–299. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.-F.; Giuliano, M.; Trivedi, M.V.; Schiff, R.; Osborne, C.K. Metastasis Dormancy in Estrogen Receptor–Positive Breast Cancer. Clin. Cancer Res. 2013, 19, 6389–6397. [Google Scholar] [CrossRef]
- Karrison, T.G.; Ferguson, D.J.; Meier, P. Dormancy of Mammary Carcinoma After Mastectomy. J. Natl. Cancer Inst. 1999, 91, 80–85. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Yagi, M.; Miyamoto, T.; Sawatani, Y.; Iwamoto, K.; Hosogane, N.; Fujita, N.; Morita, K.; Ninomiya, K.; Suzuki, T.; Miyamoto, K.; et al. DC-STAMP is essential for cell–cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 2005, 202, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Akchurin, T.; Aissiou, T.; Kemeny, N.; Prosk, E.; Nigam, N.; Komarova, S.V. Complex Dynamics of Osteoclast Formation and Death in Long-Term Cultures. PLoS ONE 2008, 3, e2104. [Google Scholar] [CrossRef]
- Tanakas, S.; Miyazaki, T.; Fukuda, A.; Akiyama, T.; Kadono, Y.; Wakeyama, H.; Kono, S.; Hoshikawa, S.; Nakamura, M.; Ohshima, Y.; et al. Molecular Mechanism of the Life and Death of the Osteoclast. Ann. N. Y. Acad. Sci. 2006, 1068, 180–186. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9, S1. [Google Scholar] [CrossRef]
- Azuma, Y.; Kaji, K.; Katogi, R.; Takeshita, S.; Kudo, A. Tumor Necrosis Factor-a Induces Differentiation of and Bone Resorption by Osteoclasts. J. Biol. Chem. 2000, 275, 4858–4864. [Google Scholar] [CrossRef]
- Martínez-Reza, I.; Díaz, L.; García-Becerra, R. Preclinical and clinical aspects of TNF-α and its receptors TNFR1 and TNFR2 in breast cancer. J. Biomed. Sci. 2017, 24, 90. [Google Scholar] [CrossRef] [PubMed]
- Bozcuk, H.; Uslu, G.; Samur, M.; Yıldız, M.; Ozben, T.; Ozdogan, M.; Artac, M.; Altunbas, H.; Akan, I.; Savas, B. Tumour necrosis factor-alpha, interleukin-6, and fasting serum insulin correlate with clinical outcome in metastatic breast cancer patients treated with chemotherapy. Cytokine 2004, 27, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Wakabayashi, H.; Matsumine, A.; Sudo, A.; Uchida, A. TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem. Biophys. Res. Commun. 2011, 407, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Madhusudan, S.; Foster, M.; Muthuramalingam, S.R.; Braybrooke, J.P.; Wilner, S.; Kaur, K.; Han, C.; Hoare, S.; Balkwill, F.R.; Talbot, D.C.; et al. A Phase II Study of Etanercept (Enbrel), a Tumor Necrosis Factor α Inhibitor in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2004, 10, 6528–6534. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T. Cellular and molecular mechanisms of breast and prostate cancer metastasis to bone. Eur. J. Cancer 1998, 34, 240–245. [Google Scholar] [CrossRef]
- Le Pape, F.; Vargas, G.; Clézardin, P. The role of osteoclasts in breast cancer bone metastasis. J. Bone Oncol. 2016, 5, 93–95. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolb, A.D.; Dai, J.; Keller, E.T.; Bussard, K.M. ‘Educated’ Osteoblasts Reduce Osteoclastogenesis in a Bone-Tumor Mimetic Microenvironment. Cancers 2021, 13, 263. https://doi.org/10.3390/cancers13020263
Kolb AD, Dai J, Keller ET, Bussard KM. ‘Educated’ Osteoblasts Reduce Osteoclastogenesis in a Bone-Tumor Mimetic Microenvironment. Cancers. 2021; 13(2):263. https://doi.org/10.3390/cancers13020263
Chicago/Turabian StyleKolb, Alexus D., Jinlu Dai, Evan T. Keller, and Karen M. Bussard. 2021. "‘Educated’ Osteoblasts Reduce Osteoclastogenesis in a Bone-Tumor Mimetic Microenvironment" Cancers 13, no. 2: 263. https://doi.org/10.3390/cancers13020263
APA StyleKolb, A. D., Dai, J., Keller, E. T., & Bussard, K. M. (2021). ‘Educated’ Osteoblasts Reduce Osteoclastogenesis in a Bone-Tumor Mimetic Microenvironment. Cancers, 13(2), 263. https://doi.org/10.3390/cancers13020263