
Heterogeneity in Pancreatic Cancer Fibroblasts—TGFβ as a Master Regulator?


{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. TGFβ Signalling Pathway in PDAC
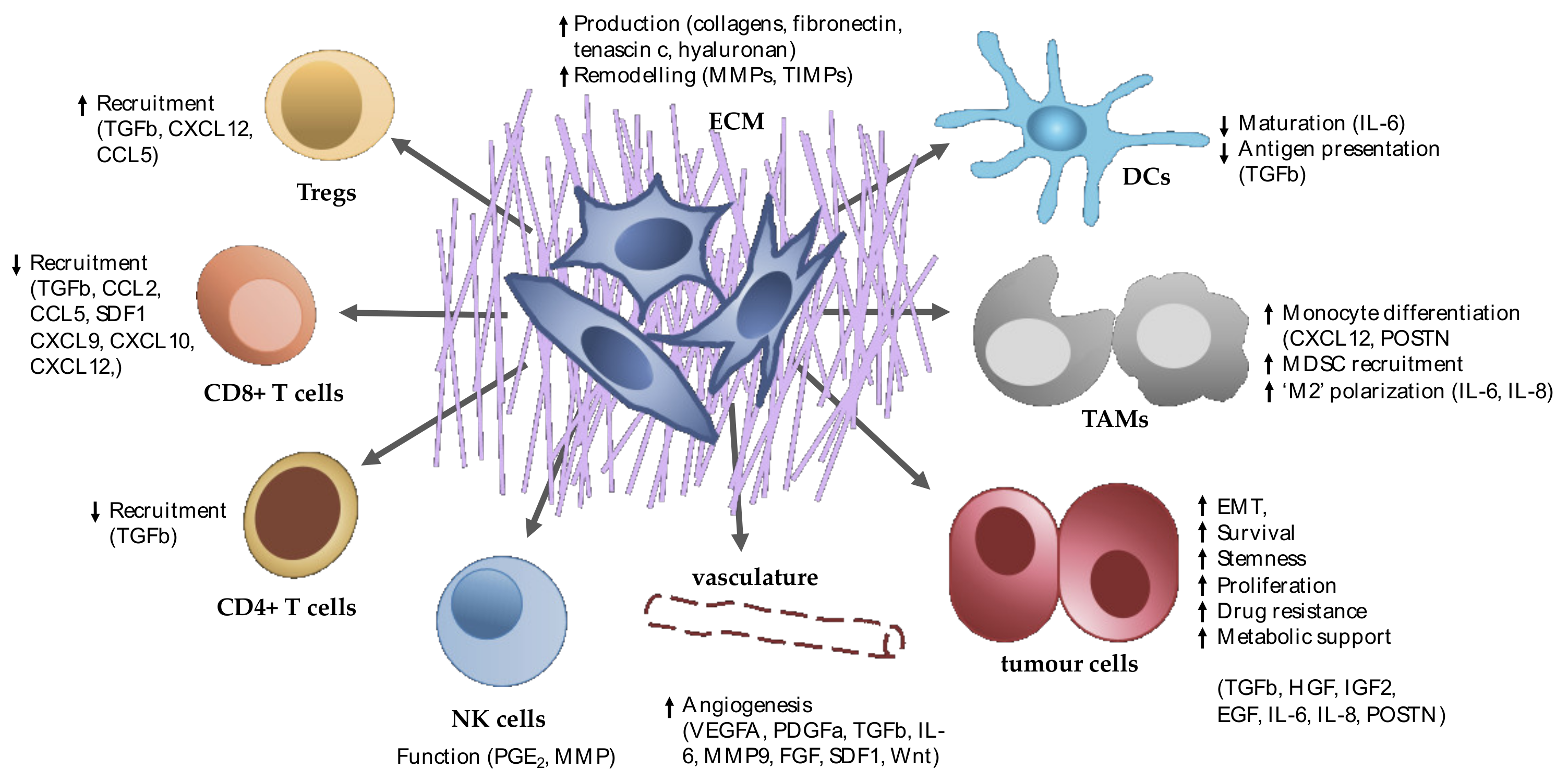
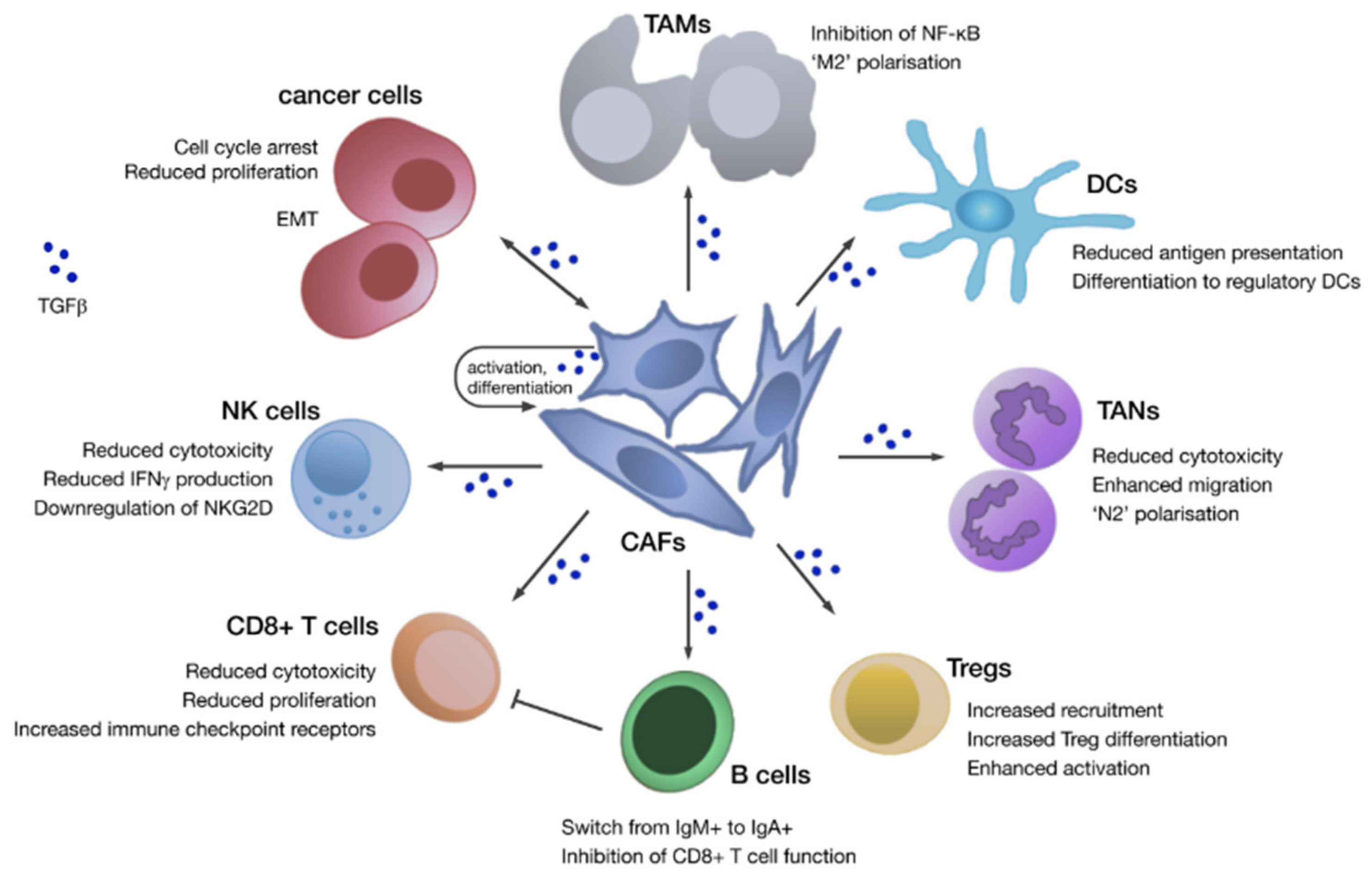
3. Fibroblasts within PDAC
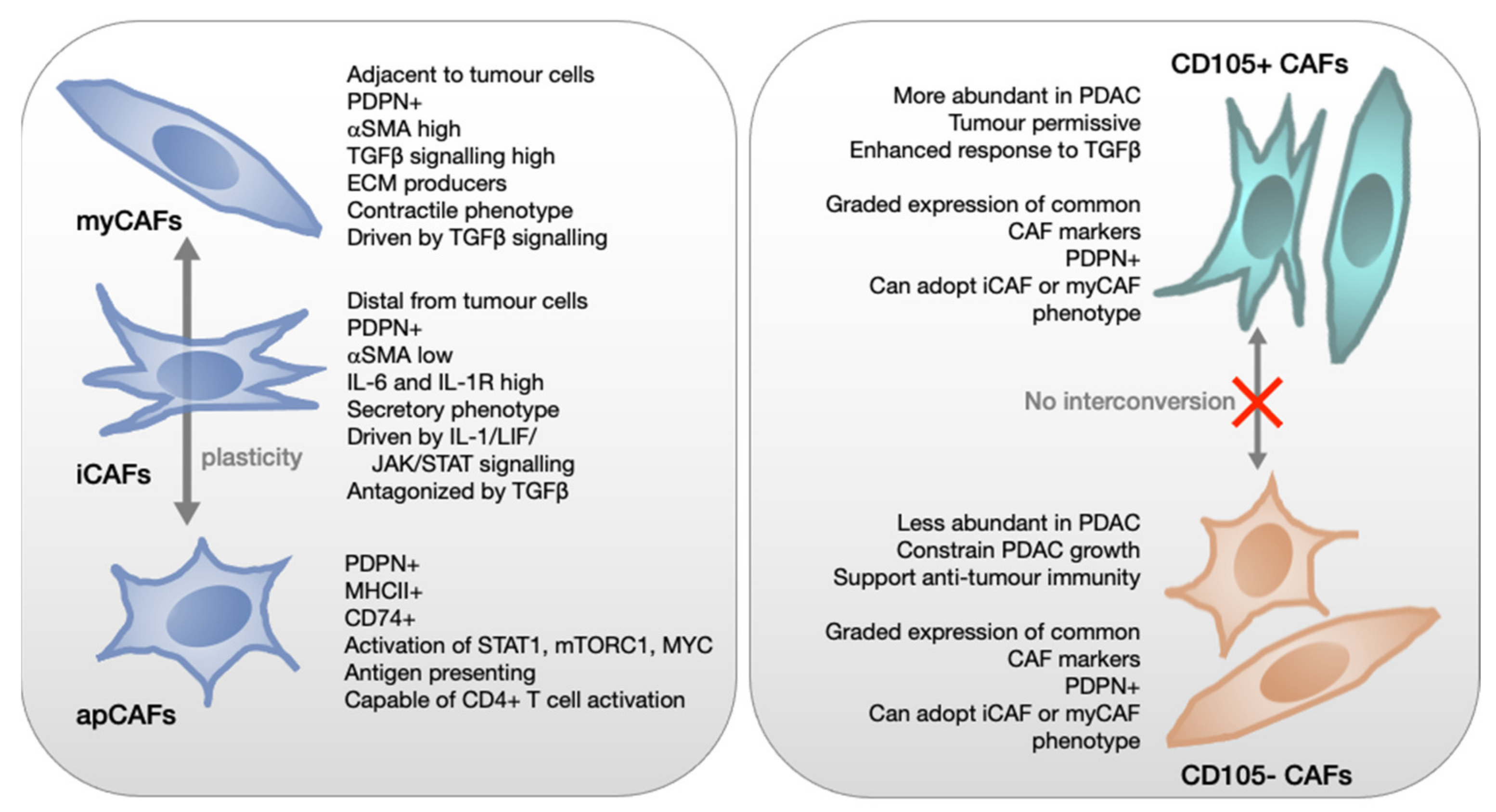
4. Inter- and Intra-Tumoural Fibroblast Heterogeneity
5. Genetics of PDAC Influence CAF Education
6. Therapeutic Intervention
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Madiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus Gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Hruban, R.H.; Raphael, B.J.; Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.D.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–U140. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Upstill-Goddard, R.; Paulus-Hock, V.; Paris, C.; Lampraki, E.M.; Dray, E.; Serrels, B.; Caligiuri, G.; Rebus, S.; Plenker, D.; et al. Targeting DNA damage response and replication stress in pancreatic cancer. Gastroenterology 2021, 160, 362–377.e13. [Google Scholar] [CrossRef]
- Whittle, M.C.; Hingorani, S.R. Fibroblasts in pancreatic ductal adenocarcinoma: Biological mechanisms and therapeutic targets. Gastroenterology 2019, 156, 2085–2096. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Huang, H.C.; Wang, Z.N.; Parmar, K.; Du, W.T.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.E.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massague, J. Transforming growth factor-beta signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Kojima, K.; Vickers, S.M.; Adsay, N.V.; Jhala, N.C.; Kim, H.G.; Schoeb, T.R.; Grizzle, W.E.; Klug, C.A. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res. 2007, 67, 8121–8130. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef]
- Cave, D.D.; Di Guida, M.; Costa, V.; Sevillano, M.; Ferrante, L.; Heeschen, C.; Corona, M.; Cucciardi, A.; Lonardo, E. TGF-beta1 secreted by pancreatic stellate cells promotes stemness and tumourigenicity in pancreatic cancer cells through L1CAM downregulation. Oncogene 2020, 39, 4271–4285. [Google Scholar] [CrossRef]
- Bergmann, F.; Wandschneider, F.; Sipos, B.; Moldenhauer, G.; Schniewind, B.; Welsch, T.; Schirrmacher, P.; Kloppel, G.; Altevogt, P.; Schafer, H.; et al. Elevated L1CAM expression in precursor lesions and primary and metastastic tissues of pancreatic ductal adenocarcinoma. Oncol. Rep. 2010, 24, 909–915. [Google Scholar] [CrossRef]
- Na’ara, S.; Amit, M.; Gil, Z. L1CAM induces perineural invasion of pancreas cancer cells by upregulation of metalloproteinase expression. Oncogene 2019, 38, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, S.; Morohashi, S.; Kudo, Y.; Akasaka, H.; Ogasawara, H.; Ono, M.; Takasugi, K.; Ishido, K.; Hakamada, K.; Kijima, H. L1 Cell adhesion molecule (L1CAM) expression at the cancer invasive front is a novel prognostic marker of pancreatic ductal adenocarcinoma. J. Surg. Oncol. 2011, 103, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Gabitova-Cornell, L.; Surumbayeva, A.; Peri, S.; Franco-Barraza, J.; Restifo, D.; Weitz, N.; Ogier, C.; Goldman, A.R.; Hartman, T.R.; Francescone, R.; et al. Cholesterol pathway inhibition induces TGF-beta signaling to promote basal differentiation in pancreatic cancer. Cancer Cell 2020, 38, 567–583.e11. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 10. [Google Scholar] [CrossRef]
- Lohr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Muller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta 1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.X.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Gu, D.L.; Feng, L.L.; Curriden, S.; Arnush, M.; Krahl, T.; Gurushanthaiah, D.; Wilson, C.; Loskutoff, D.L.; Fox, H.; et al. Accumulation of Extracellular-Matrix and Developmental Dysregulation in the Pancreas by Transgenic Production of Transforming Growth-Factor-Beta-1. Am. J. Pathol. 1995, 147, 42–52. [Google Scholar]
- Principe, D.R.; DeCant, B.; Mascarinas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.Y.; et al. TGF beta signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef]
- Ostapoff, K.T.; Cenik, B.K.; Wang, M.; Ye, R.S.; Xu, X.H.; Nugent, D.; Hagopian, M.M.; Topalovski, M.; Rivera, L.B.; Carroll, K.D.; et al. Neutralizing murine TGF beta R2 promotes a differentiated tumor cell phenotype and inhibits pancreatic cancer metastasis. Cancer Res. 2014, 74, 4996–5007. [Google Scholar] [CrossRef]
- Stylianou, A.; Gkretsi, V.; Stylianopoulos, T. Transforming growth factor-beta modulates pancreatic cancer associated fibroblasts cell shape, stiffness and invasion. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1537–1546. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT signaling is antagonized by TGF beta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H.Y. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.X.; Clauser, K.R.; Ohlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef]
- Collins, M.A.; Bednar, F.; Zhang, Y.Q.; Brisset, J.C.; Galban, S.; Galban, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer—Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Junliang, L.; Lili, W.; Xiaolong, L.; Xuguang, L.; Huanwen, W.; Zhiyong, L. High-molecular-weight hyaluronan produced by activated pancreatic stellate cells promotes pancreatic cancer cell migration via paracrine signaling. Biochem. Biophys. Res. Commun. 2019, 515, 493–498. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Pasca Di Magliano, M.; Welling, T.H.; et al. GM-CSF mediates mesenchymal-epithelial cross-talk in pancreatic cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef]
- Iwamoto, C.; Ohuchida, K.; Shinkawa, T.; Okuda, S.; Otsubo, Y.; Okumura, T.; Sagara, A.; Koikawa, K.; Ando, Y.; Shindo, K.; et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett. 2021, 512, 15–27. [Google Scholar] [CrossRef]
- Garcia, P.E.; Adoumie, M.; Kim, E.C.; Zhang, Y.Q.; Scales, M.K.; El-Tawi, Y.S.; Shaikh, A.Z.; Wen, H.J.; Bednar, F.; Allen, B.; et al. Differential contribution of pancreatic fibroblast subsets to the pancreatic cancer stroma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 581–599. [Google Scholar] [CrossRef]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef]
- Fan, Q.; He, M.; Sheng, T.; Zhang, X.; Sinha, M.; Luxon, B.; Zhao, X.; Xie, J. Requirement of TGFbeta signaling for SMO-mediated carcinogenesis. J. Biol. Chem. 2010, 285, 36570–36576. [Google Scholar] [CrossRef]
- Cigna, N.; Farrokhi Moshai, E.; Brayer, S.; Marchal-Somme, J.; Wemeau-Stervinou, L.; Fabre, A.; Mal, H.; Leseche, G.; Dehoux, M.; Soler, P.; et al. The hedgehog system machinery controls transforming growth factor-beta-dependent myofibroblastic differentiation in humans: Involvement in idiopathic pulmonary fibrosis. Am. J. Pathol. 2012, 181, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; McMillan, R.H.; Chang, Y.T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct interactions with cancer-associated fibroblasts lead to enhanced pancreatic cancer stem cell function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef]
- Honselmann, K.C.; Finetti, P.; Birnbaum, D.J.; Monsalve, C.S.; Wellner, U.F.; Begg, S.K.S.; Nakagawa, A.; Hank, T.; Li, A.; Goldsworthy, M.A.; et al. Neoplastic-stromal cell cross-talk regulates matrisome expression in pancreatic cancer. Mol. Cancer Res. 2020, 18, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef]
- Maurer, C.; Holmstrom, S.R.; He, J.; Laise, P.; Su, T.; Ahmed, A.; Hibshoosh, H.; Chabot, J.A.; Oberstein, P.E.; Sepulveda, A.R.; et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 2019, 68, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, D.J.; Begg, S.K.S.; Finetti, P.; Vanderburg, C.; Kulkarni, A.S.; Neyaz, A.; Hank, T.; Tai, E.; Deshpande, V.; Bertucci, F.; et al. Transcriptomic analysis of laser capture microdissected tumors reveals cancer- and stromal-specific molecular subtypes of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2021, 27, 2314–2325. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Somerville, T.D.; Biffi, G.; Dassler-Plenker, J.; Hur, S.K.; He, X.Y.; Vance, K.E.; Miyabayashi, K.; Xu, Y.; Maia-Silva, D.; Klingbeil, O.; et al. Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.Y.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.L.; Huang, D.; Jiang, J.L.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Muller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15(+) myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Ohuchida, K.; Mizumoto, K.; Shindo, K.; Eguchi, D.; Kozono, S.; Ikenaga, N.; Ohtsuka, T.; Takahata, S.; Aishima, S.; et al. CD271(+) subpopulation of pancreatic stellate cells correlates with prognosis of pancreatic cancer and is regulated by interaction with cancer cells. PLoS ONE 2012, 7, e52682. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.B.; Mortensen, M.B.; Sorensen, M.D.; Wirenfeldt, M.; Kristensen, B.W.; Schroder, H.D.; Pfeiffer, P.; Detlefsen, S. Spatial and phenotypic characterization of pancreatic cancer-associated fibroblasts after neoadjuvant treatment. Histol. Histopathol. 2020, 35, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, K.; Maurer, C.; Peschke, K.; Teller, S.; Schuck, K.; Steiger, K.; Engleitner, T.; Ollinger, R.; Nomura, A.; Wirges, N.; et al. Mesenchymal Plasticity Regulated by Prrx1 Drives Aggressive Pancreatic Cancer Biology. Gastroenterology 2021, 160, 346–361.e24. [Google Scholar] [CrossRef]
- Takano, S.; Reichert, M.; Bakir, B.; Das, K.K.; Nishida, T.; Miyazaki, M.; Heeg, S.; Collins, M.A.; Marchand, B.; Hicks, P.D.; et al. Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev. 2016, 30, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Pinho, A.V.; Van Bulck, M.; Chantrill, L.; Arshi, M.; Sklyarova, T.; Herrmann, D.; Vennin, C.; Gallego-Ortega, D.; Mawson, A.; Giry-Laterriere, M.; et al. ROBO2 is a stroma suppressor gene in the pancreas and acts via TGF-beta signalling. Nat. Commun. 2018, 9, 5083. [Google Scholar] [CrossRef]
- Vennin, C.; Melenec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 22. [Google Scholar] [CrossRef]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the tumor stroma for treatment of pancreatic cancer. Gastroenterology 2017, 154, 820–838. [Google Scholar] [CrossRef]
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-associated fibroblasts in pancreatic cancer are reprogrammed by tumor-induced alterations in genomic DNA methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Eyres, M.; Lanfredini, S.; Xu, H.; Burns, A.; Blake, A.; Willenbrock, F.; Goldin, R.; Hughes, D.; Hughes, S.; Thapa, A.; et al. TET2 Drives 5hmc Marking of GATA6 and epigenetically defines pancreatic ductal adenocarcinoma transcriptional subtypes. Gastroenterology 2021, 161, 653–668.e16. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, C.; Elies, A.; Kieffer, Y.; Bourachot, B.; Ladoire, S.; Pelon, F.; Hequet, D.; Guinebretiere, J.M.; Blanchet, C.; Vincent-Salomon, A.; et al. A subset of activated fibroblasts is associated with distant relapse in early luminal breast cancer. Breast Cancer Res. 2020, 22, 76. [Google Scholar] [CrossRef] [PubMed]
- Mhaidly, R.; Mechta-Grigoriou, F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol. Rev. 2021, 302, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Buscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs Show hypomethylation of repetitive elements and the execution of an intrinsic IFN program linked to a ductal cell of origin. Cancer Discov. 2021, 11, 638–659. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersa, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Rupert, J.E.; Narasimhan, A.; Jengelley, D.H.A.; Jiang, Y.; Liu, J.; Au, E.; Silverman, L.M.; Sandusky, G.; Bonetto, A.; Cao, S.; et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Huang, H.C.; Zhang, Y.Q.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.T.; Wright, S.; Hagopian, M.; Wang, Z.N.; et al. Targeting TGF beta R2-mutant tumors exposes vulnerabilities to stromal TGF beta blockade in pancreatic cancer. EMBO Mol. Med. 2019, 11, 20. [Google Scholar] [CrossRef]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e6. [Google Scholar] [CrossRef]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: A phase 2 clinical trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065. [Google Scholar] [CrossRef]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nab-paclitaxel interrupts cancer-stromal interaction through C-X-C motif chemokine 10-mediated interleukin-6 downregulation in vitro. Cancer Sci. 2018, 109, 2509–2519. [Google Scholar] [CrossRef]
- Principe, D.R.; Narbutis, M.; Kumar, S.; Park, A.; Viswakarma, N.; Dorman, M.J.; Kamath, S.D.; Grippo, P.J.; Fishel, M.L.; Hwang, R.F.; et al. Long-term gemcitabine treatment reshapes the pancreatic tumor microenvironment and sensitizes murine carcinoma to combination immunotherapy. Cancer Res. 2020, 80, 3101–3115. [Google Scholar] [CrossRef]
- Hezel, A.F.; Deshpande, V.; Zimmerman, S.M.; Contino, G.; Alagesan, B.; O’Dell, M.R.; Rivera, L.B.; Harper, J.; Lonning, S.; Brekken, R.A.; et al. TGF-beta and alphavbeta6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer Res. 2012, 72, 4840–4845. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Mucciolo, G.; Curcio, C.; Roux, C.; Li, W.D.Y.; Capello, M.; Curto, R.; Chiarle, R.; Giordano, D.; Satolli, M.A.; Lawlor, R.; et al. IL17A critically shapes the transcriptional program of fibroblasts in pancreatic cancer and switches on their protumorigenic functions. Proc. Natl. Acad. Sci. USA 2021, 118, 9. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Momblona, X.H.; et al. TGF beta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Heery, C.R.; Schlom, J.; Madan, R.A.; Cao, L.; Kang, Z.; Lamping, E.; Marte, J.L.; Donahue, R.N.; Grenga, I.; et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFbeta, in advanced solid tumors. Clin. Cancer Res. 2018, 24, 1287–1295. [Google Scholar] [CrossRef]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFbeta blockade augments PD-1 inhibition to promote T-Cell-mediated regression of pancreatic cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, Z.; Du, X.; Chen, S.; Zhang, W.; Wang, J.; Li, H.; He, X.; Cao, J.; Wang, J. Co-inhibition of the TGF-beta pathway and the PD-L1 checkpoint by pH-responsive clustered nanoparticles for pancreatic cancer microenvironment regulation and anti-tumor immunotherapy. Biomater. Sci. 2020, 8, 5121–5132. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Moustafa, M.; Knoll, M.; Xu, C.; Furkel, J.; Lazorchak, A.; Yeung, T.L.; Hasheminasab, S.M.; Jenkins, M.H.; Meister, S.; et al. Simultaneous targeting of TGF-beta/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Hartley, J.; Abken, H. Chimeric antigen receptors designed to overcome transforming growth factor-beta-mediated repression in the adoptive T-cell therapy of solid tumors. Clin. Transl. Immunol. 2019, 8, e1064. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watt, D.M.; Morton, J.P. Heterogeneity in Pancreatic Cancer Fibroblasts—TGFβ as a Master Regulator? Cancers 2021, 13, 4984. https://doi.org/10.3390/cancers13194984
Watt DM, Morton JP. Heterogeneity in Pancreatic Cancer Fibroblasts—TGFβ as a Master Regulator? Cancers. 2021; 13(19):4984. https://doi.org/10.3390/cancers13194984
Chicago/Turabian StyleWatt, Dale M., and Jennifer P. Morton. 2021. "Heterogeneity in Pancreatic Cancer Fibroblasts—TGFβ as a Master Regulator?" Cancers 13, no. 19: 4984. https://doi.org/10.3390/cancers13194984
APA StyleWatt, D. M., & Morton, J. P. (2021). Heterogeneity in Pancreatic Cancer Fibroblasts—TGFβ as a Master Regulator? Cancers, 13(19), 4984. https://doi.org/10.3390/cancers13194984