
HSP90 Inhibition Synergizes with Cisplatin to Eliminate Basal-like Pancreatic Ductal Adenocarcinoma Cells

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Transfections
2.3. Immunofluoresence
2.4. Immunoblot Analysis
2.5. Viability Assays and Determination of Drug Synergism
2.6. RNA Isolation and RT-PCR
2.7. Chromosome Spreads
2.8. Immuno-Cytological Assay for Pt-(GpG) Adducts in DNA
2.9. In Vivo Study Using a Syngeneic Orthotopic Mouse Model
2.10. Immunofluorescence Staining of Tissue Slices
2.11. RNA-Sequencing Data Analysis
2.12. Quantification and Statistical Analysis
2.13. Statistical Analysis
3. Results
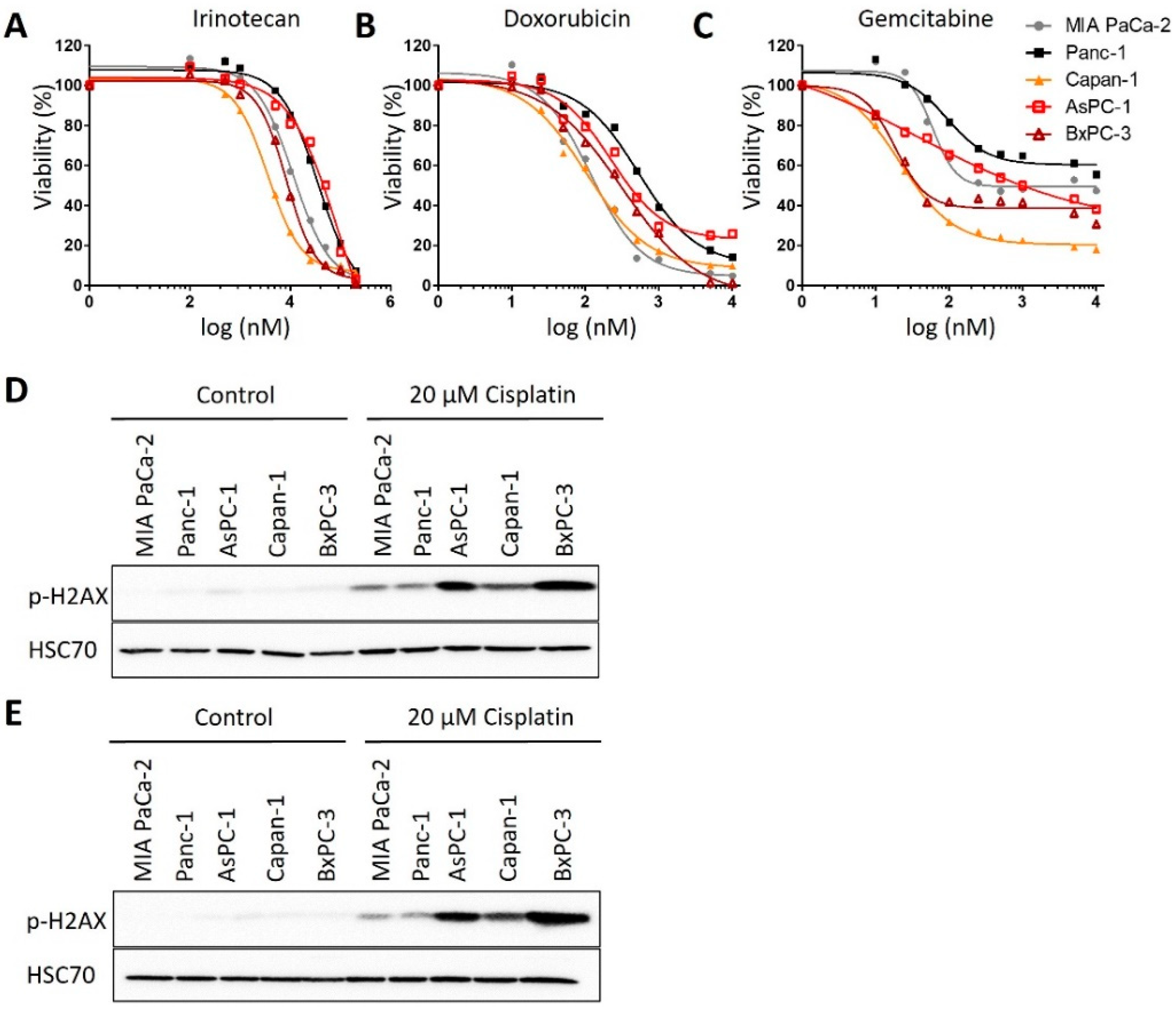
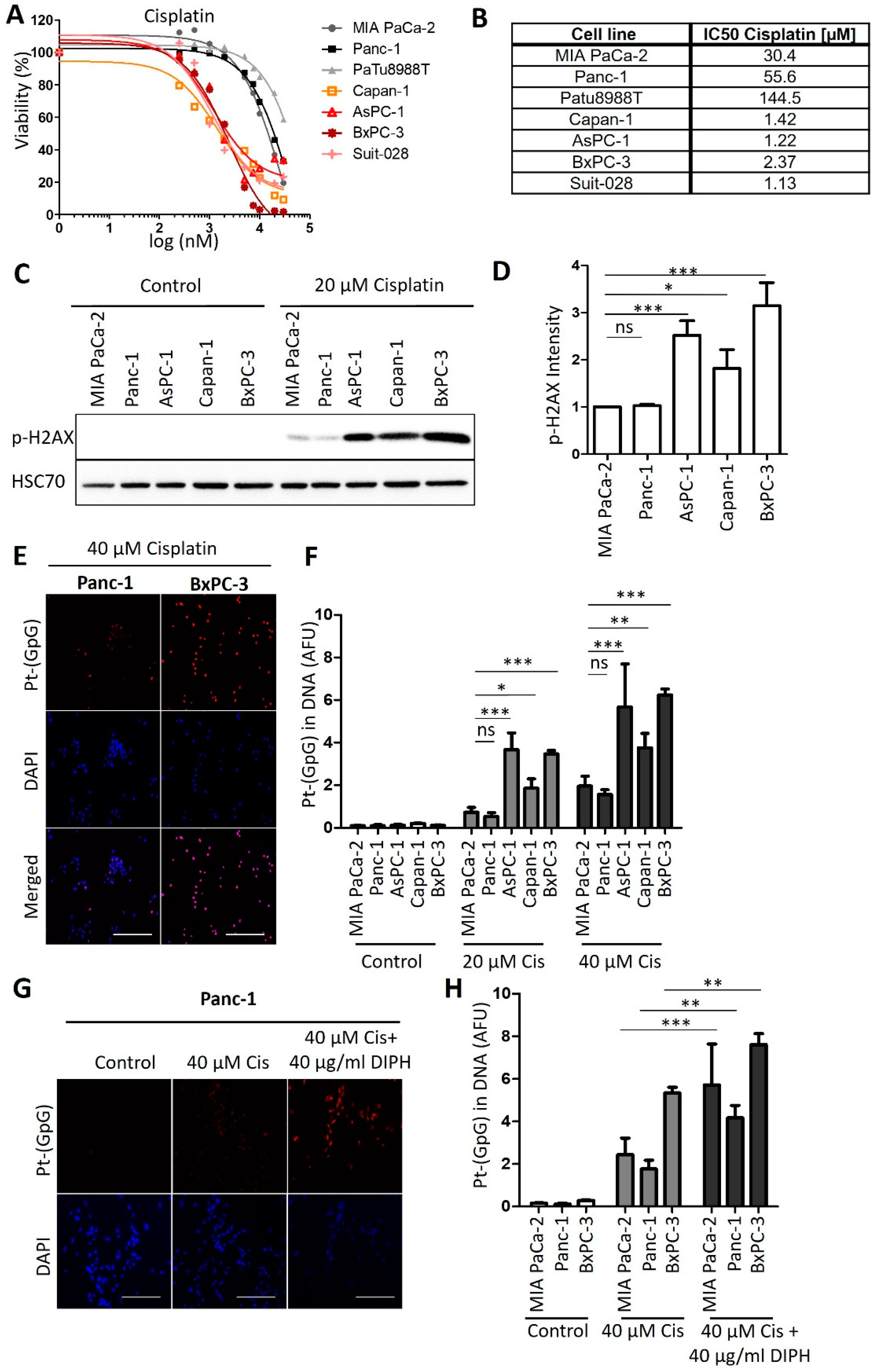
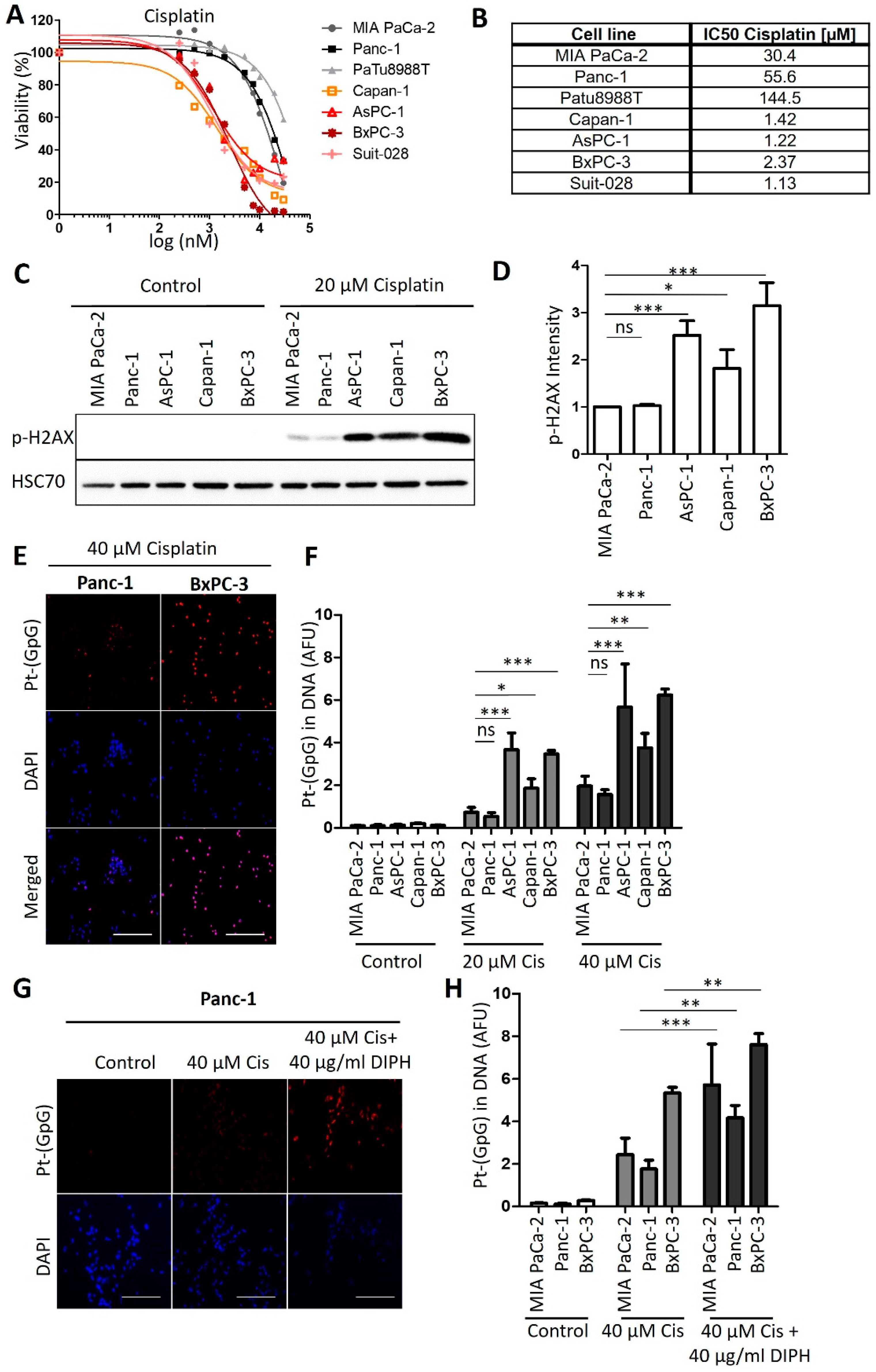
3.1. Human PDAC-Derived Cell Lines Segregate in Two Distinct Groups Regarding Their Sensitivities towards Cisplatin
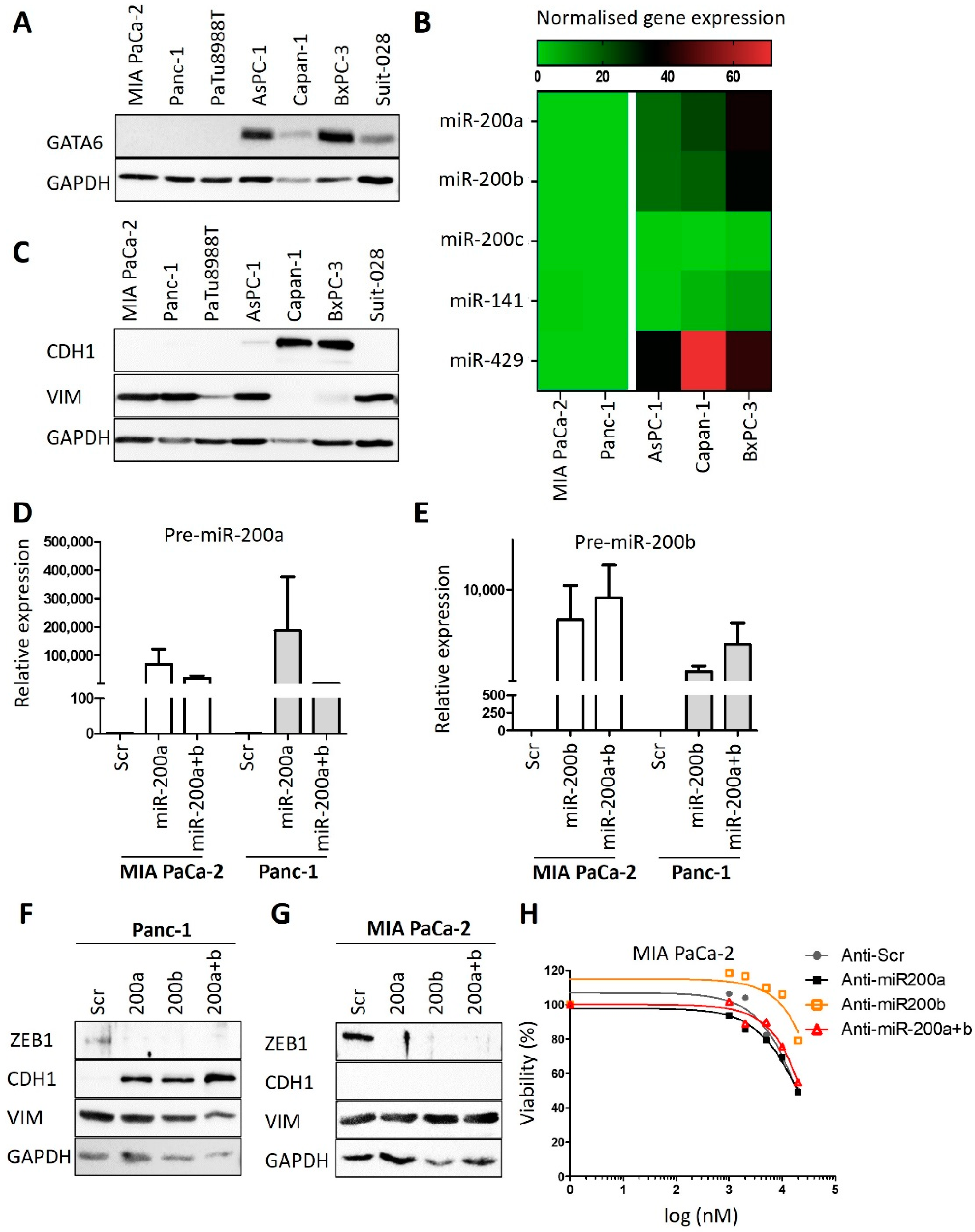
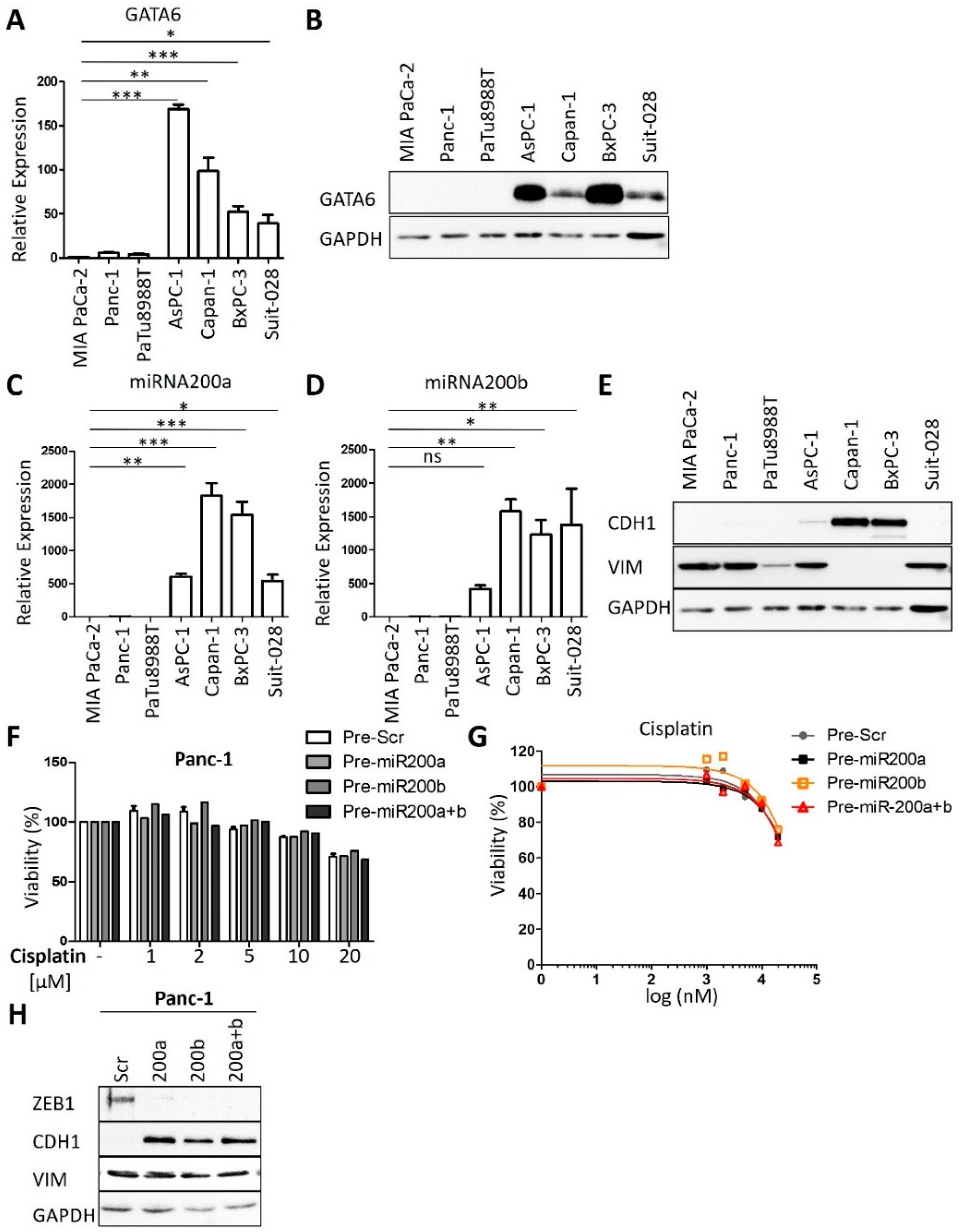
3.2. Expression Levels of GATA6 and microRNAs of the Class 200 Predict Cisplatin Sensitivity
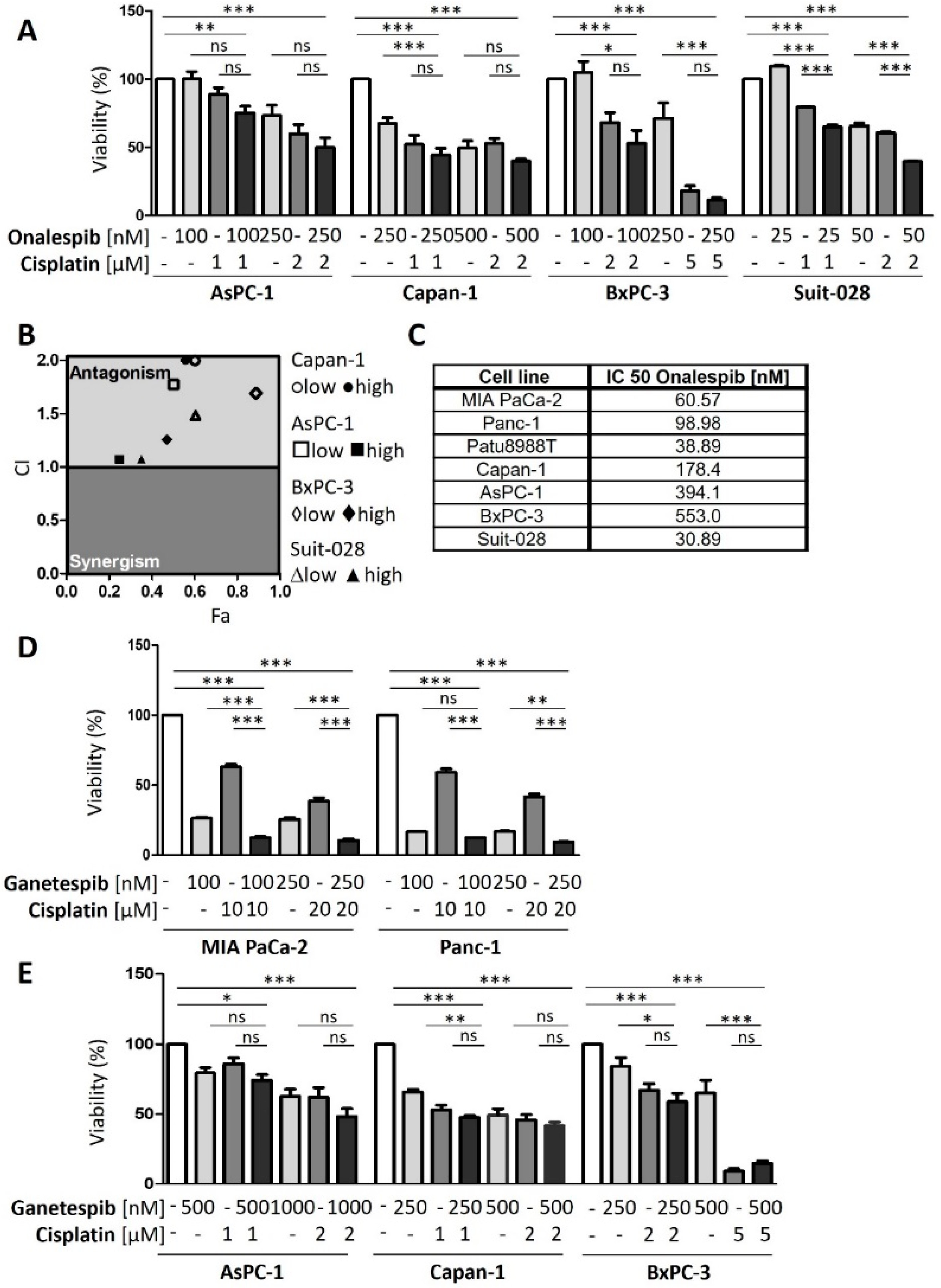
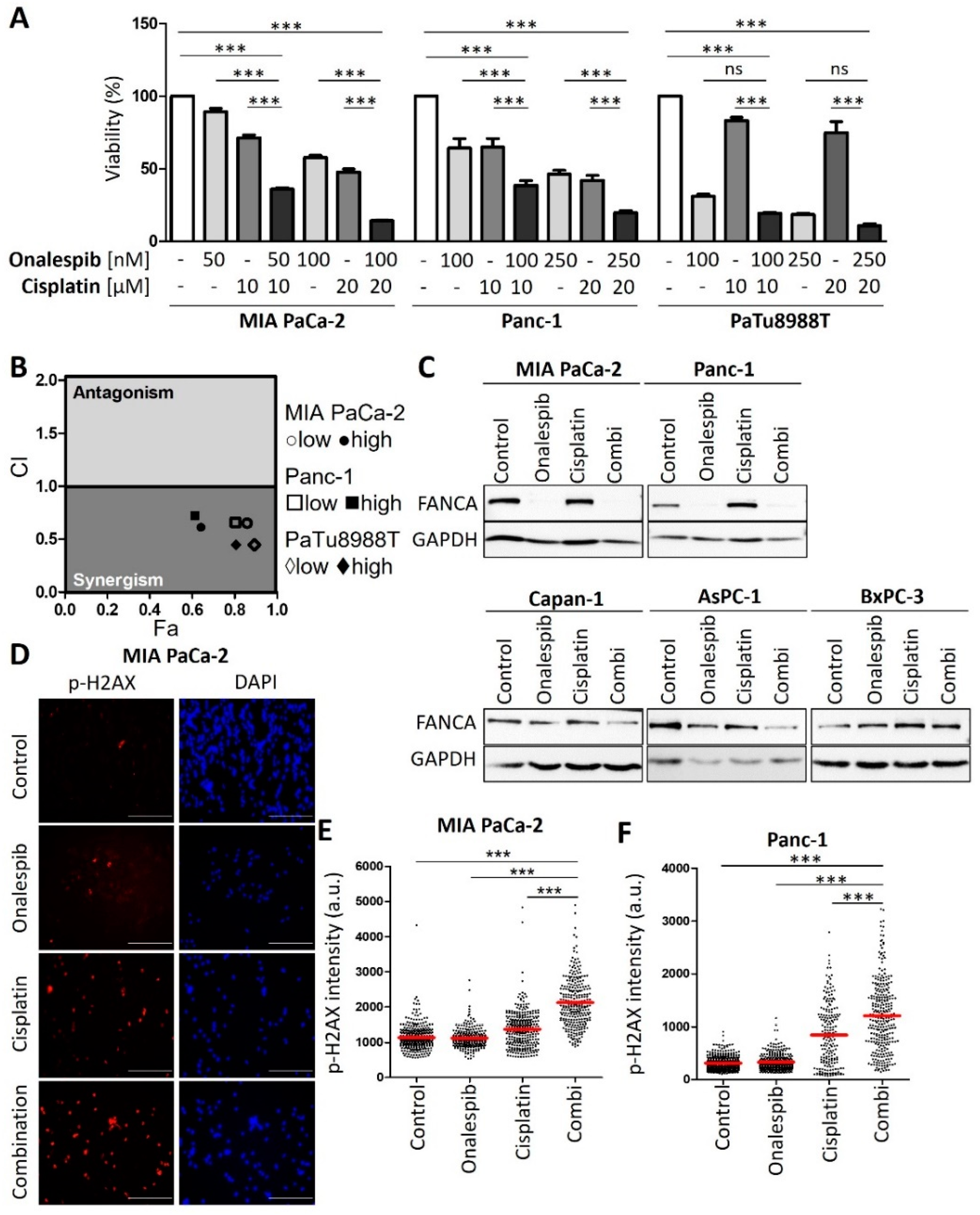
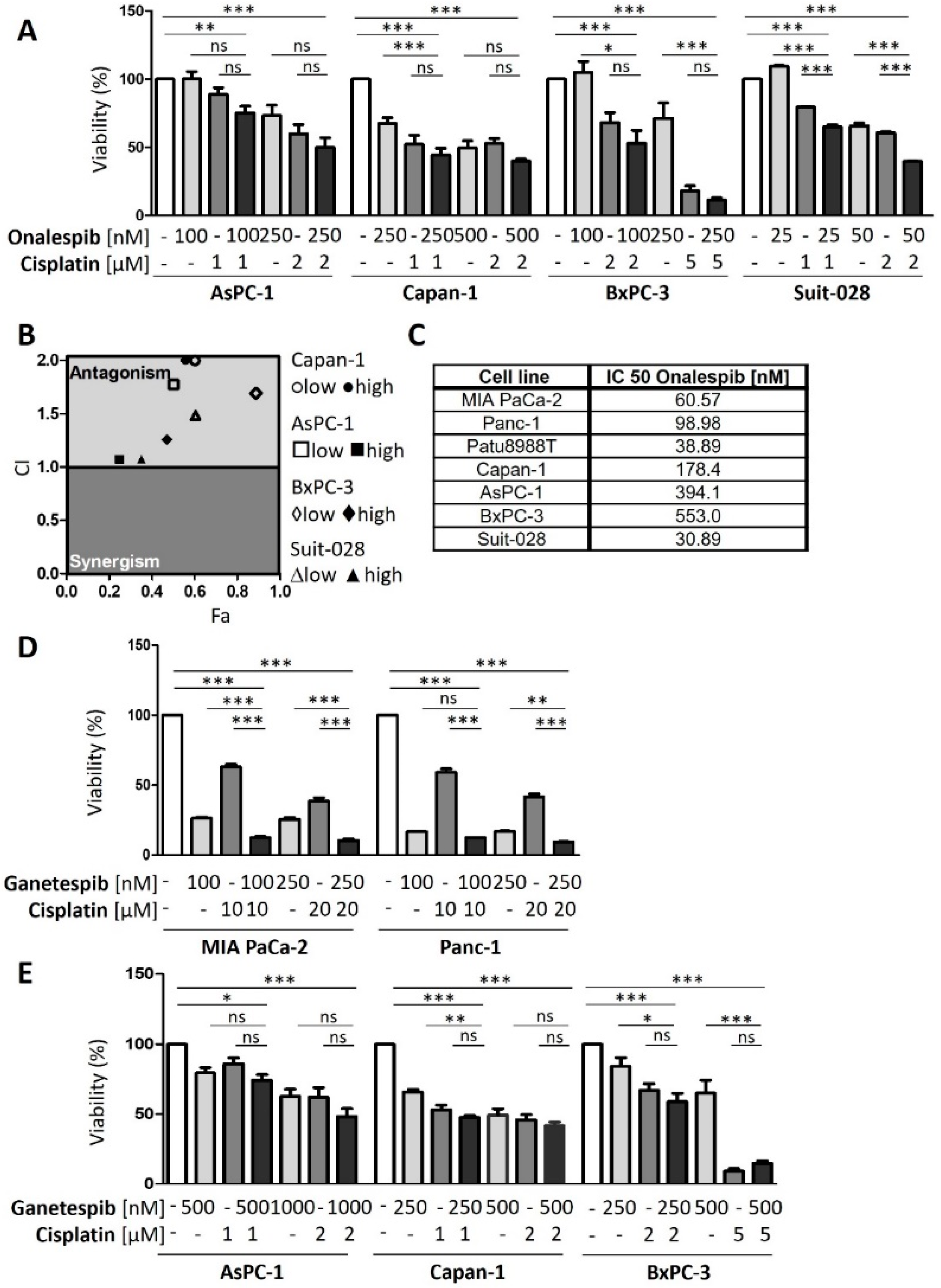
3.3. HSP90 Inhibitors Synergize with Cisplatin, Reduce Fanconi Anemia Pathway Mediators, and Sustain DNA Damage
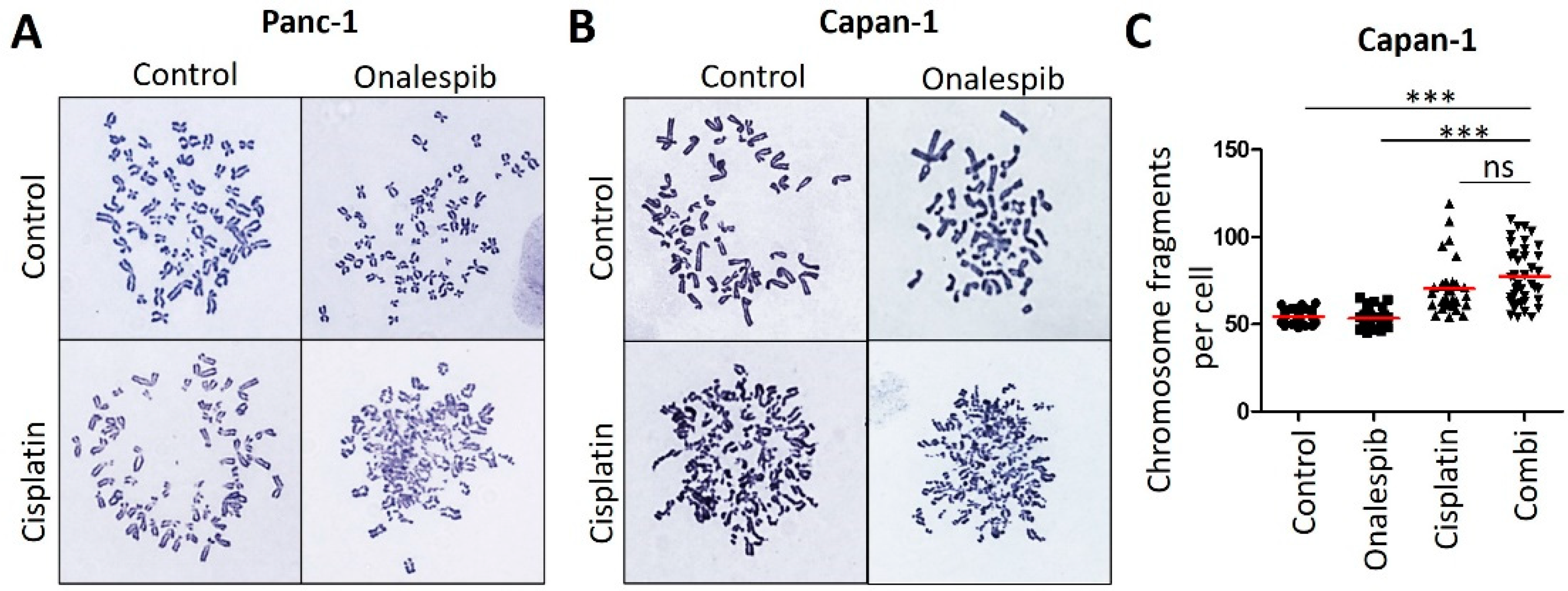
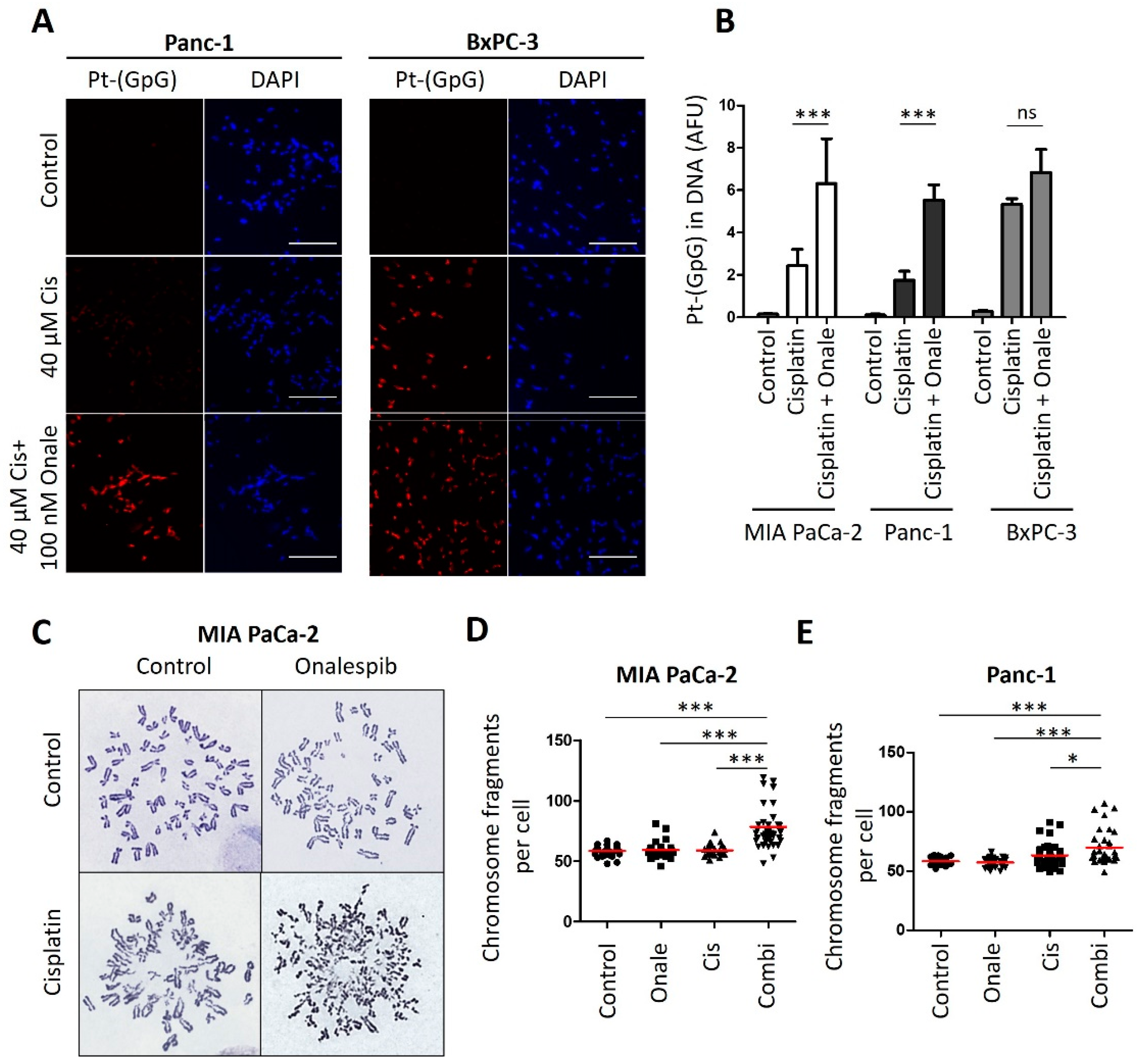
3.4. Onalespib Increases Cisplatin-DNA Adduct Levels and Chromosome Fragmentation
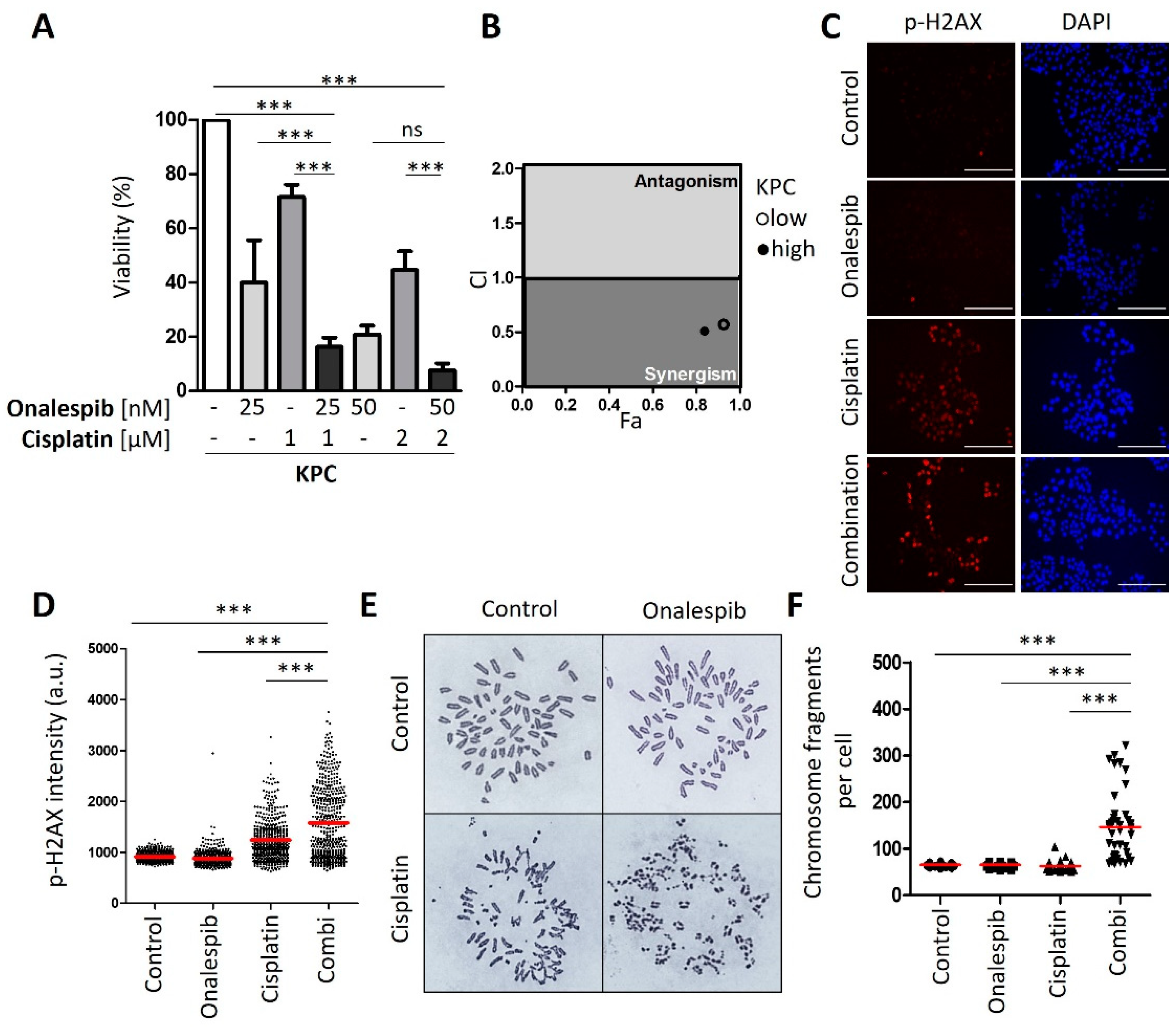
3.5. Cisplatin and HSP90 Inhibition Synergistically Induce DNA Damage, Chromosome Fragmentation, and Death in Cells Derived from the Murine PDAC Model KPC
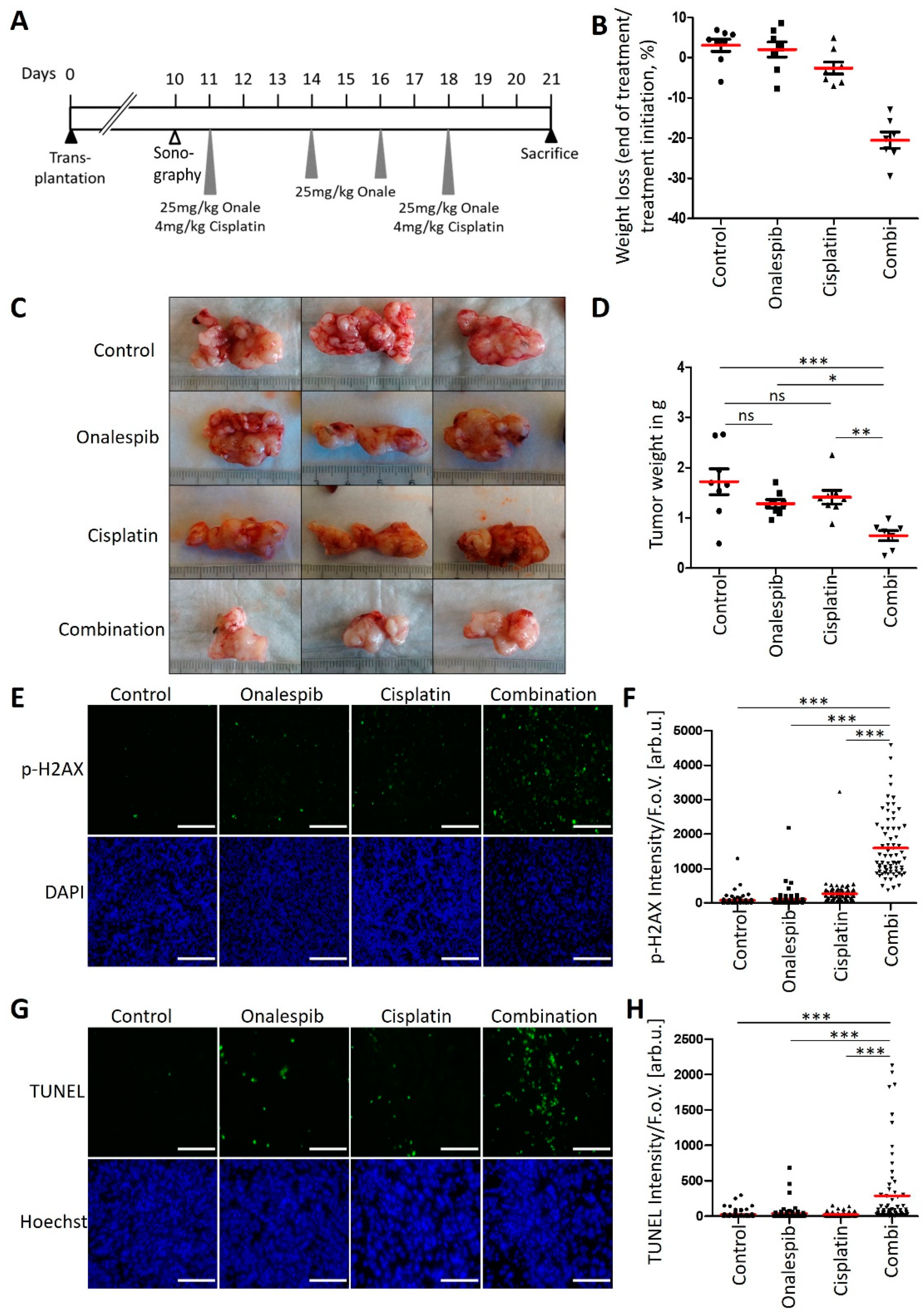
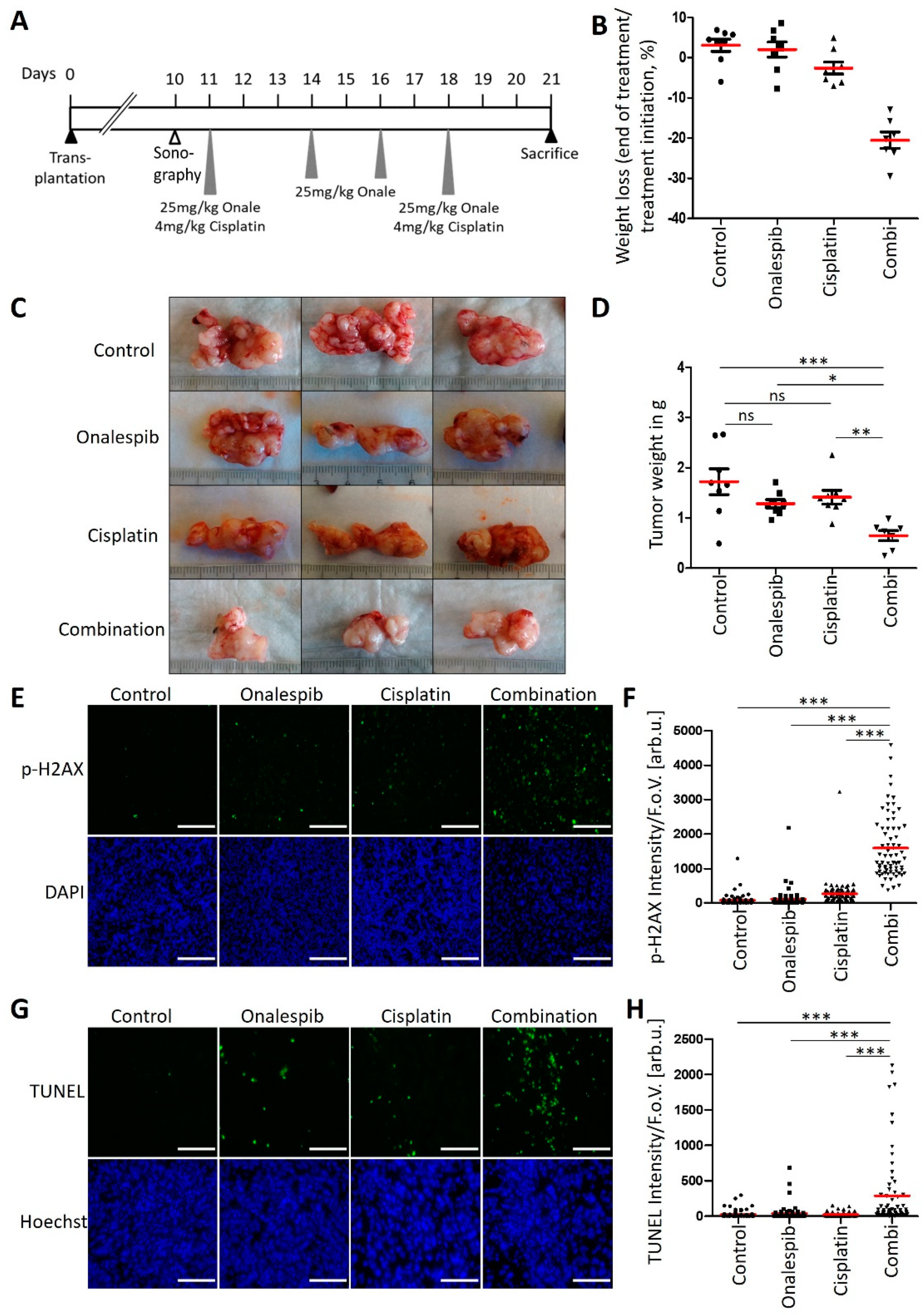
3.6. An HSP90 Inhibitor and Cisplatin Cooperate to Counteract the Growth of KPC Tumors in an Orthotopic and Syngeneic PDAC Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Jameson, G.S.; Borazanci, E.; Babiker, H.M.; Poplin, E.; Niewiarowska, A.A.; Gordon, M.S.; Barrett, M.T.; Rosenthal, A.; Stoll-D’Astice, A.; Crowley, J.; et al. Response Rate Following Albumin-Bound Paclitaxel Plus Gemcitabine Plus Cisplatin Treatment Among Patients with Advanced Pancreatic Cancer: A Phase 1b/2 Pilot Clinical Trial. JAMA Oncol. 2020, 6, 125–132. [Google Scholar] [CrossRef]
- Aiello, N.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.L.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013. [Google Scholar] [CrossRef] [Green Version]
- Rashid, N.U.; Peng, X.L.; Jin, C.; Moffitt, R.A.; Volmar, K.E.; Belt, B.A.; Panni, R.Z.; Nywening, T.M.; Herrera, S.G.; Moore, K.J.; et al. Purity Independent Subtyping of Tumors (PurIST), A Clinically Robust, Single-sample Classifier for Tumor Subtyping in Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 82–92. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.-H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef] [Green Version]
- Kramer, D.; Stark, N.; Schulz-Heddergott, R.; Erytch, N.; Edmunds, S.; Roßmann, L.; Bastians, H.; Concin, N.; Moll, U.M.; Dobbelstein, M. Strong antitumor synergy between DNA crosslinking and HSP90 inhibition causes massive premitotic DNA fragmentation in ovarian cancer cells. Cell Death Differ. 2017, 24, 300–316. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.; Talalay, P. Quantitative Dose-Effect Relationships: The Combined Effects of Multiple. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Melnikova, M.; Thomale, J. Visualization and Quantitative Measurement of Drug-Induced Platinum Adducts in the Nuclear DNA of Individual Cells by an Immuno-Cytological Assay; Schulz, W.A., Hoffmann, M.J., Niegisch, G., Eds.; Springer: New York, NY, USA, 2018; Volume 1655, pp. 351–358. [Google Scholar] [CrossRef]
- Liedert, B.; Plium, D.; Schellens, J.; Thomale, J. Adduct-specific monoclonal antibodies for the measurement of cisplatin-induced DNA lesions in individual cell nuclei. Nucleic Acids Res. 2006, 34, e47. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Goetze, R.-G.; Buchholz, S.M.; Patil, S.; Petzold, G.; Ellenrieder, V.; Hessmann, E.; Neesse, A. Utilizing High Resolution Ultrasound to Monitor Tumor Onset and Growth in Genetically Engineered Pancreatic Cancer Models. J. Vis. Exp. 2018, 2018, e56979. [Google Scholar] [CrossRef]
- Diaferia, G.R.; Balestrieri, C.; Prosperini, E.; Nicoli, P.; Spaggiari, P.; Zerbi, A.; Natoli, G. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J. 2016, 35, 595–617. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Melnikova, M.; Wauer, U.S.; Mendus, D.; Hilger, R.A.; Oliver, T.G.; Mercer, K.; Gohlke, B.O.; Erdmann, K.; Niederacher, D.; Neubauer, H.; et al. Diphenhydramine increases the therapeutic window for platinum drugs by simultaneously sensitizing tumor cells and protecting normal cells. Mol. Oncol. 2020, 14, 686–703. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Somerville, T.D.; Xu, Y.; Miyabayashi, K.; Tiriac, H.; Cleary, C.R.; Maia-Silva, D.; Milazzo, J.P.; Tuveson, D.A.; Vakoc, C.R. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep. 2018, 25, 1741–1755. [Google Scholar] [CrossRef] [Green Version]
- Candido, J.B.; Morton, J.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R+ Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Lu, P.; Wu, Y.; Wang, D.-D.; Zhou, S.; Yang, S.-J.; Shen, H.-Y.; Zhang, X.-H.; Zhao, J.-H.; Tang, J.-H. MiRNAs-mediated cisplatin resistance in breast cancer. Tumor Biol. 2016, 37, 12905–12913. [Google Scholar] [CrossRef]
- Brozovic, A.; Duran, G.E.; Wang, Y.C.; Francisco, E.B.; Sikic, B.I. The miR-200 family differentially regulates sensitivity to paclitaxel and carboplatin in human ovarian carcinoma OVCAR-3 and MES-OV cells. Mol. Oncol. 2015, 9, 1678–1693. [Google Scholar] [CrossRef]
- Dijk, F.; Veenstra, V.L.; Soer, E.C.; Dings, M.P.; Zhao, L.; Halfwerk, J.B.; Hooijer, G.K.; Damhofer, H.; Marzano, M.; Steins, A.; et al. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell-intrinsic mesenchymal features and high concordance between existing classification systems. Sci. Rep. 2020, 10, 337. [Google Scholar] [CrossRef]
- Hamdan, F.H.; Johnsen, S.A. DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc. Natl. Acad. Sci. USA 2018, 115, E12343–E12352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Materna, V.; Liedert, B.; Thomale, J.; Lage, H. Protection of platinum-DNA adduct formation and reversal of cisplatin resistance by anti-MRP2 hammerhead ribozymes in human cancer cells. Int. J. Cancer 2005, 115, 393–402. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Moll, U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat. Rev. Drug Discov. 2014, 13, 179–196. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Capo-Chichi, C.D.; Cai, K.Q.; Testa, J.R.; Godwin, A.K.; Xu, X.-X. Loss of GATA6 Leads to Nuclear Deformation and Aneuploidy in Ovarian Cancer. Mol. Cell. Biol. 2009, 29, 4766–4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional Inactivation of Endogenous MDM2 and CHIP by HSP90 Causes Aberrant Stabilization of Mutant p53 in Human Cancer Cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.; Li, J.; Conradi, L.-C.; Bohnenberger, H.; Ceteci, F.; Greten, F.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemke, L.; De Oliveira, T.; Witt, D.; Winkler, N.; Bohnenberger, H.; Bucala, R.; Conradi, L.-C.; Schulz-Heddergott, R. Hsp90-stabilized MIF supports tumor progression via macrophage recruitment and angiogenesis in colorectal cancer. Cell Death Dis. 2021, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Marchenko, N.D.; Holembowski, L.; Fingerle-Rowson, G.; Pesic, M.; Zender, L.; Dobbelstein, M.; Moll, U.M. Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression. J. Exp. Med. 2012, 209, 275–289. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Targets | Source (Catalogue Number) | Dilution |
|---|---|---|
| E-Cadherin | BD (610181) | 1:500 |
| FancA | Bethyl (A301-980A) | 1:500 |
| Phospho-H2AX (S139) | Cell Signaling (#2577) | 1:1000 |
| GAPDH | Abcam (ab8245) | 1:20,000 |
| GATA6 | R&D (AF1700) | 1:300 |
| HSC70 | Santa Cruz (#7298) | 1:15,000 |
| PARP1 | Cell Signaling (#9542) | 1:1000 |
| Vimentin | Santa Cruz (#6260) | 1:1000 |
| Zeb1 | Santa Cruz (#25388) | 1:500 |
| Gene | Gene Accession Number | Forward/Reverse | PCR Product Size |
|---|---|---|---|
| hRPLP0(36B4) | NM_053275 | 5′-GATTGGCTACCCAACTGTTG | 158 |
| 5′-CAGGGGCAGCAGCCACAAA | |||
| hGATA6 | NM_005257 | 5′-TCTACAGCAAGATGAATGGCC | 140 |
| 5′-CTCACCCTCAGCATTTCTACG | |||
| hsa-miR-200a-3p | Thermo Fisher | 000502 | |
| hsa-miR-200a-3p | Thermo Fisher | 002251 | |
| U6 snRNA | Thermo Fisher | 001973 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ewers, K.M.; Patil, S.; Kopp, W.; Thomale, J.; Quilitz, T.; Magerhans, A.; Wang, X.; Hessmann, E.; Dobbelstein, M. HSP90 Inhibition Synergizes with Cisplatin to Eliminate Basal-like Pancreatic Ductal Adenocarcinoma Cells. Cancers 2021, 13, 6163. https://doi.org/10.3390/cancers13246163
Ewers KM, Patil S, Kopp W, Thomale J, Quilitz T, Magerhans A, Wang X, Hessmann E, Dobbelstein M. HSP90 Inhibition Synergizes with Cisplatin to Eliminate Basal-like Pancreatic Ductal Adenocarcinoma Cells. Cancers. 2021; 13(24):6163. https://doi.org/10.3390/cancers13246163
Chicago/Turabian StyleEwers, Katharina M., Shilpa Patil, Waltraut Kopp, Jürgen Thomale, Tabea Quilitz, Anna Magerhans, Xin Wang, Elisabeth Hessmann, and Matthias Dobbelstein. 2021. "HSP90 Inhibition Synergizes with Cisplatin to Eliminate Basal-like Pancreatic Ductal Adenocarcinoma Cells" Cancers 13, no. 24: 6163. https://doi.org/10.3390/cancers13246163
APA StyleEwers, K. M., Patil, S., Kopp, W., Thomale, J., Quilitz, T., Magerhans, A., Wang, X., Hessmann, E., & Dobbelstein, M. (2021). HSP90 Inhibition Synergizes with Cisplatin to Eliminate Basal-like Pancreatic Ductal Adenocarcinoma Cells. Cancers, 13(24), 6163. https://doi.org/10.3390/cancers13246163