
Colorectal Tumour Mucosa Microbiome Is Enriched in Oral Pathogens and Defines Three Subtypes That Correlate with Markers of Tumour Progression

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Specimens
2.2. DNA Extraction, PCR Amplification and Sequencing of 16S rRNA Gene
2.3. Data Analysis
2.3.1. Preprocessing and Quality Control
2.3.2. Taxonomy Assignment and Metabolic Potential Prediction
2.3.3. Statistical Analysis and Data Mining
2.4. Data Access
2.5. Validation
3. Results
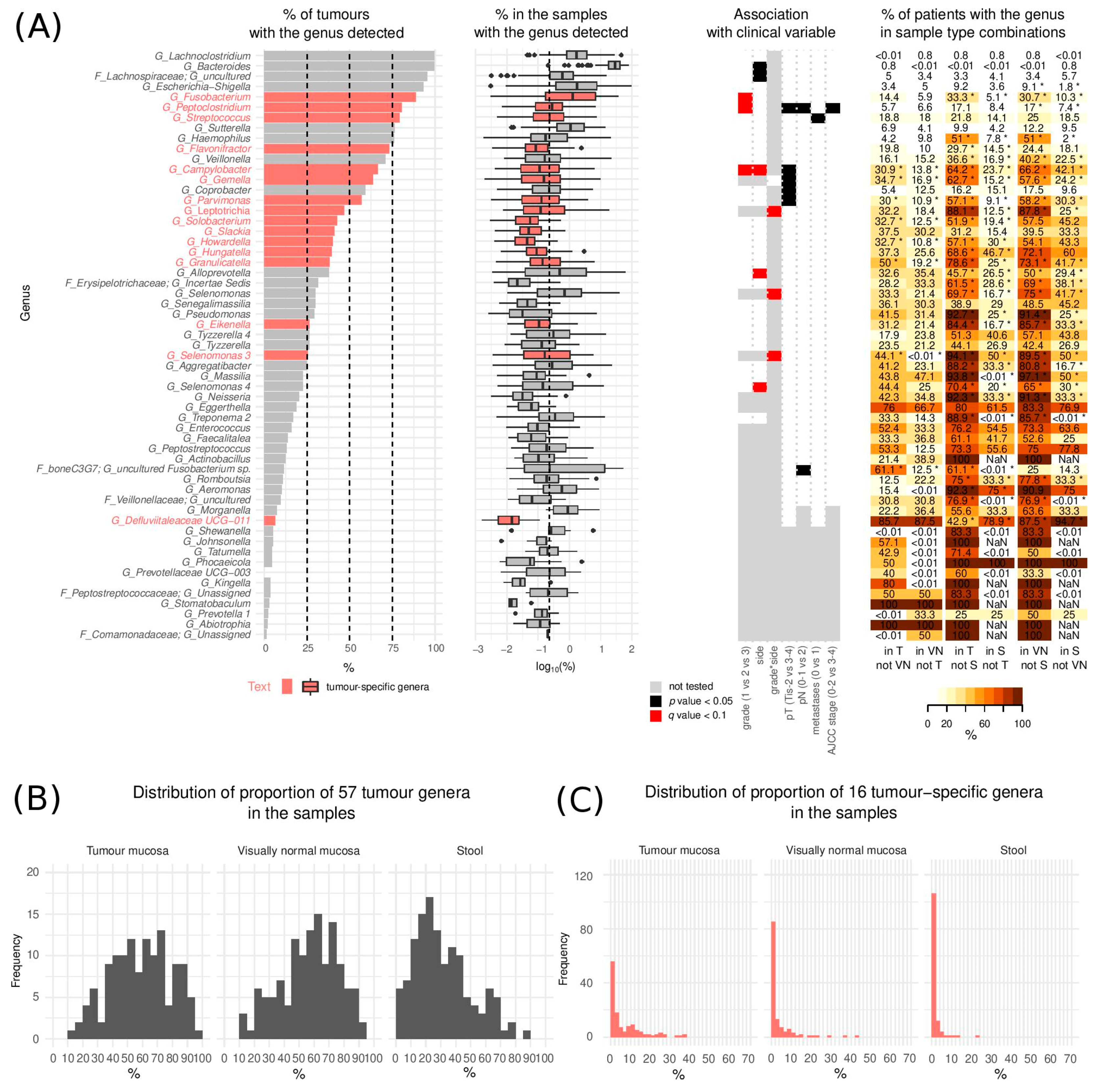
3.1. Microbial Categorisation According to Sample Type
3.2. The Landscape of CRC Tumour Microbiome
3.3. Microbiome and Clinical Variables
3.4. Tumour CRC Microbial Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Punt, C.J.A.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef]
- Van Der Jeught, K.; Xu, H.-C.; Li, Y.-J.; Lu, X.-B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Ahn, J.; Sinha, R.; Pei, Z.; Dominianni, C.; Wu, J.; Shi, J.; Goedert, J.J.; Hayes, R.; Yang, L. Human Gut Microbiome and Risk for Colorectal Cancer. J. Natl. Cancer Inst. 2013, 105, 1907–1911. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, R.; Rajendiran, E.; George, S.; Samuel, G.V.; Ramakrishna, B. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, DesulfovibrioandEnterococcus faecalisin the feces of patients with colorectal cancer. J. Gastroenterol. Hepatol. 2008, 23, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Young, S.M.; Allen, C.; Seeber, A.; Péli-Gulli, M.-P.; Panchaud, N.; Waller, A.; Ursu, O.; Yao, T.; Golden, J.E.; et al. Identification of a Small Molecule Yeast TORC1 Inhibitor with a Multiplex Screen Based on Flow Cytometry. ACS Chem. Biol. 2012, 7, 715–722. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human Intestinal Lumen and Mucosa-Associated Microbiota in Patients with Colorectal Cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef] [PubMed]
- Cipe, G.; Idiz, U.O.; Firat, D.; Bektasoglu, H. Relationship between intestinal microbiota and colorectal cancer. World J. Gastrointest. Oncol. 2015, 7, 233–240. [Google Scholar] [CrossRef]
- Kostic, A.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.; et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Zheng, J.; Hu, G.; Wang, J.; Huang, C.; Lou, L.; Wang, X.; Zeng, Y. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci. Rep. 2016, 6, 26337. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.M.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the Human Colorectal Cancer Microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Van Nhieu, J.T.; Furet, J.-P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef]
- Viljoen, K.S.; Dakshinamurthy, A.; Goldberg, P.; Blackburn, J.M. Quantitative Profiling of Colorectal Cancer-Associated Bacteria Reveals Associations between Fusobacterium spp., Enterotoxigenic Bacteroides fragilis (ETBF) and Clinicopathological Features of Colorectal Cancer. PLoS ONE 2015, 10, e0119462. [Google Scholar] [CrossRef]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2011, 6, 320–329. [Google Scholar] [CrossRef]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z.; et al. Dysbiosis Signature of Fecal Microbiota in Colorectal Cancer Patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; McDowell, A.; Kim, E.K.; Seo, H.; Lee, W.H.; Moon, C.-M.; Kym, S.-M.; Lee, D.H.; Park, Y.S.; Jee, Y.-K.; et al. Development of a colorectal cancer diagnostic model and dietary risk assessment through gut microbiome analysis. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Zackular, J.P.; Baxter, N.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The Gut Microbiome Modulates Colon Tumorigenesis. mBio 2013, 4, e00692-13. [Google Scholar] [CrossRef]
- Zackular, J.; Rogers, M.; Ruffin, M.; Schloss, P.D. The Human Gut Microbiome as a Screening Tool for Colorectal Cancer. Cancer Prev. Res. 2014, 7, 1112–1121. [Google Scholar] [CrossRef]
- Vaupel, P.; Harrison, L. Tumor Hypoxia: Causative Factors, Compensatory Mechanisms, and Cellular Response. Oncologist 2004, 9 (Suppl. S5), 4–9. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Tlaskalova-Hogenova, H.; Vannucci, L.; Klimesova, K.; Stepankova, R.; Krizan, J.; Kverka, M. Microbiome and Colorectal Carcinoma. Cancer J. 2014, 20, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Freeman, G.J. The Microsatellite Instable Subset of Colorectal Cancer Is a Particularly Good Candidate for Checkpoint Blockade Immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver–passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Genet. 2012, 10, 575–582. [Google Scholar] [CrossRef]
- Pennisi, E. Cancer Therapies Use a Little Help from Microbial Friends. Science 2013, 342, 921. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Welch, J.L.M.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Yang, Y.; Cai, Q.; Shu, X.; Steinwandel, M.D.; Blot, W.J.; Zheng, W.; Long, J. Prospective study of oral microbiome and colorectal cancer risk in low-income and African American populations. Int. J. Cancer 2019, 144, 2381–2389. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Shang, Y.; Wu, B.; Yang, E.; Luo, Y.-Y.; Li, X. Intestinal bacteria detected in cancer and adjacent tissue from patients with colorectal cancer. Oncol. Lett. 2018, 17, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.Z.C.T.; Yamamoto, K.; Honda, T.; Nakamura, M.; Yamamura, T.; Hattori, S.; Burt, A.D.; Singh, R.; Hirooka, Y.; Fujishiro, M. Microbiota profile is different for early and invasive colorectal cancer and is consistent throughout the colon. J. Gastroenterol. Hepatol. 2020, 35, 433–437. [Google Scholar] [CrossRef]
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.; Ryan, F.; Claesson, M.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef]
- Egao, Z.; Eguo, B.; Egao, R.; Ezhu, Q.; Eqin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Li, E.; Hamm, C.M.; Gulati, A.S.; Sartor, R.B.; Chen, H.; Wu, X.; Zhang, T.; Rohlf, F.J.; Zhu, W.; Gu, C.; et al. Inflammatory Bowel Diseases Phenotype, C. difficile and NOD2 Genotype Are Associated with Shifts in Human Ileum Associated Microbial Composition. PLoS ONE 2012, 7, e26284. [Google Scholar] [CrossRef]
- Han, S.; Wu, W.; Da, M.; Xu, J.; Zhuang, J.; Zhang, L.; Zhang, X.; Yang, X. Adequate Lymph Node Assessments and Investigation of Gut Microorganisms and Microbial Metabolites in Colorectal Cancer. OTT 2020, 13, 1893–1906. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, L.; Li, Q.; Wu, J.; Peng, W.; Li, H.; Chen, K.; Ren, Y.; Fu, X. Microbiota Diversity in Human Colorectal Cancer Tissues Is Associated with Clinicopathological Features. Nutr. Cancer 2019, 71, 214–222. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Aronesty, E. Comparison of Sequencing Utility Programs. TOBIOIJ 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, J. The Statistical Analysis of Compositional Data. J. R. Stat. Soc. Ser. B Methodol. 1982, 44, 139–160. [Google Scholar] [CrossRef]
- Gloor, G.B.; Wu, J.R.; Pawlowsky-Glahn, V.; Egozcue, J.J. It’s all relative: Analyzing microbiome data as compositions. Ann. Epidemiol. 2016, 26, 322–329. [Google Scholar] [CrossRef]
- Martín-Fernández, J.-A.; Hron, K.; Templ, M.; Filzmoser, P.; Palarea-Albaladejo, J. Bayesian-multiplicative treatment of count zeros in compositional data sets. Stat. Model. Int. J. 2015, 15, 134–158. [Google Scholar] [CrossRef]
- Tsilimigras, M.C.; Fodor, A.A. Compositional data analysis of the microbiome: Fundamentals, tools, and challenges. Ann. Epidemiol. 2016, 26, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Oaksen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package. 2019. Available online: http://cran.rproject.org/package=vegan (accessed on 4 July 2020).
- Comas-Cufí, M. R Package. coda.base: A Basic Set of Functions for Compositional Data Analysis. 2020. Available online: https://rdrr.io/cran/coda.base/ (accessed on 4 July 2020).
- Kloke, J.D.; McKean, J.W. Rfit: Rank-Based Estimation for Linear Models. R J. 2012, 4, 57–64. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. Gplots: Various R Programming Tools for Plotting Data; R Package. 2020. Available online: https://rdrr.io/cran/gplots/ (accessed on 4 July 2020).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: Cham, Switzerland, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Wray, C.M.; Ziogas, A.; Hinojosa, M.W.; Le, H.; Stamos, M.J.; Zell, J.A. Tumor Subsite Location Within the Colon Is Prognostic for Survival After Colon Cancer Diagnosis. Dis. Colon Rectum 2009, 52, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Schiffer, L.; Manghi, P.; Renson, A.; Obenchain, V.; Truong, D.T.; Beghini, F.; Malik, F.; Ramos, M.; Dowd, J.; et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods 2017, 14, 1023–1024. [Google Scholar] [CrossRef]
- De Almeida, C.V.; Lulli, M.; Di Pilato, V.; Schiavone, N.; Russo, E.; Nannini, G.; Baldi, S.; Borrelli, R.; Bartolucci, G.; Menicatti, M.; et al. Differential Responses of Colorectal Cancer Cell Lines to Enterococcus faecalis’ Strains Isolated from Healthy Donors and Colorectal Cancer Patients. JCM 2019, 8, 388. [Google Scholar] [CrossRef]
- Gupta, A.; Dhakan, D.B.; Maji, A.; Saxena, R.; Visnu Prasoodanan, P.K.; Mahajan, S.; Pulikkan, J.; Kurian, J.; Gomez, A.M.; Scaria, J.; et al. Association of Flavonifractor plautii, a Flavonoid-Degrading Bacterium, with the Gut Microbiome of Colorectal Cancer Patients in India. mSystems 2019, 4. [Google Scholar] [CrossRef]
- Ai, D.; Pan, H.; Li, X.; Gao, Y.; Liu, G.; Xia, L.C. Identifying Gut Microbiota Associated with Colorectal Cancer Using a Zero-Inflated Lognormal Model. Front. Microbiol. 2019, 10, 826. [Google Scholar] [CrossRef]
- Ito, M.; Kanno, S.; Nosho, K.; Sukawa, Y.; Mitsuhashi, K.; Kurihara, H.; Igarashi, H.; Takahashi, T.; Tachibana, M.; Takahashi, H.; et al. Association ofFusobacterium nucleatumwith clinical and molecular features in colorectal serrated pathway. Int. J. Cancer 2015, 137, 1258–1268. [Google Scholar] [CrossRef]
- Bahmani, S.; Azarpira, N.; Moazamian, E. Anti-colon cancer activity of Bifidobacterium metabolites on colon cancer cell line SW742. Turk. J. Gastroenterol. 2019, 30, 835–842. [Google Scholar] [CrossRef]
- Mangifesta, M.; Mancabelli, L.; Milani, C.; Gaiani, F.; De’Angelis, N.; De’Angelis, G.L.; Van Sinderen, D.; Ventura, M.; Turroni, F. Mucosal microbiota of intestinal polyps reveals putative biomarkers of colorectal cancer. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Parisa, A.; Roya, G.; Mahdi, R.; Shabnam, R.; Maryam, E.; Malihe, T. Anti-cancer effects of Bifidobacterium species in colon cancer cells and a mouse model of carcinogenesis. PLoS ONE 2020, 15, e0232930. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Koncina, E.; Haan, S.; Rauh, S.; Letellier, E. Prognostic and Predictive Molecular Biomarkers for Colorectal Cancer: Updates and Challenges. Cancers 2020, 12, 319. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Koliarakis, I.; Messaritakis, I.; Nikolouzakis, T.K.; Hamilos, G.; Souglakos, J.; Tsiaoussis, J. Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4146. [Google Scholar] [CrossRef]
- Long, X.; Wong, C.C.; Tong, L.; Chu, E.S.H.; Szeto, C.H.; Go, M.Y.Y.; Coker, O.O.; Chan, A.W.H.; Chan, F.K.; Sung, J.J.Y.; et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat. Microbiol. 2019, 4, 2319–2330. [Google Scholar] [CrossRef]
- Abed, J.; Emgård, J.E.; Zamir, G.; Faroja, M.; Almogy, G.; Grenov, A.; Sol, A.; Naor, R.; Pikarsky, E.; Atlan, K.A.; et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe 2016, 20, 215–225. [Google Scholar] [CrossRef]
- Zou, X.; Feng, B.; Dong, T.; Yan, G.; Tan, B.; Shen, H.; Huang, A.; Zhang, X.; Zhang, M.; Yang, P.; et al. Up-regulation of type I collagen during tumorigenesis of colorectal cancer revealed by quantitative proteomic analysis. J. Proteom. 2013, 94, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N. Microbial ecosystem in the oral cavity: Metabolic diversity in an ecological niche and its relationship with oral diseases. Int. Congr. Ser. 2005, 1284, 103–112. [Google Scholar] [CrossRef]
- Eley, B.M.; Cox, S.W. Proteolytic and hydrolytic enzymes from putative periodontal pathogens: Characterization, molecular genetics, effects on host defenses and tissues and detection in gingival crevice fluid. Periodontol. 2000 2003, 31, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Potempa, J.; Sroka, A.; Imamura, T.; Travis, J. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: Structure, function and assembly of multidomain protein complexes. Curr. Protein Pept. Sci. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.F.H.; Fermiano, D.; Feres, M.; Figueiredo, L.C.; Teles, F.R.P.; Mayer, M.P.A.; Faveri, M. Levels ofSelenomonasspecies in generalized aggressive periodontitis. J. Periodontal Res. 2012, 47, 711–718. [Google Scholar] [CrossRef]
- Scher, J.U.; Ubeda, C.; Equinda, M.; Khanin, R.; Buischi, Y.; Viale, A.; Lipuma, L.; Attur, M.; Pillinger, M.; Weissmann, G.; et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012, 64, 3083–3094. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.A.; Novince, C.M.; Kirkwood, K.L. Aggregatibacter actinomycetemcomitans, a potent immunoregulator of the periodontal host defense system and alveolar bone homeostasis. Mol. Oral Microbiol. 2016, 31, 207–227. [Google Scholar] [CrossRef]
- Lin, W.-R.; Chen, Y.-S.; Liu, Y.-C. Cellulitis and Bacteremia Caused by Bergeyella zoohelcum. J. Formos. Med. Assoc. 2007, 106, 573–576. [Google Scholar] [CrossRef]
- Peel, M.M.; Hornidge, K.A.; Luppino, M.; Stacpoole, A.M.; Weaver, R.E. Actinobacillus spp. and related bacteria in infected wounds of humans bitten by horses and sheep. J. Clin. Microbiol. 1991, 29, 2535–2538. [Google Scholar] [CrossRef]
- Sohn, K.M.; Huh, K.; Baek, J.-Y.; Kim, Y.-S.; Kang, C.-I.; Peck, K.R.; Lee, N.Y.; Song, J.-H.; Ko, K.S.; Chung, D.R. A new causative bacteria of infective endocarditis, Bergeyella cardium sp. nov. Diagn. Microbiol. Infect. Dis. 2015, 81, 213–216. [Google Scholar] [CrossRef]
- Friis-Møller, A.; Christensen, J.J.; Fussing, V.; Hesselbjerg, A.; Christiansen, J.; Bruun, B. Clinical Significance and Taxonomy of Actinobacillus hominis. J. Clin. Microbiol. 2001, 39, 930–935. [Google Scholar] [CrossRef]
- Zha, Z.; Lv, Y.; Tang, H.; Li, T.; Miao, Y.; Cheng, J.; Wang, G.; Tan, Y.; Zhu, Y.; Xing, X.; et al. An orally administered butyrate-releasing xylan derivative reduces inflammation in dextran sulphate sodium-induced murine colitis. Int. J. Biol. Macromol. 2020, 156, 1217–1233. [Google Scholar] [CrossRef]
- Kelly, T.N.; Bazzano, L.A.; Ajami, N.J.; He, H.; Zhao, J.; Petrosino, J.F.; Correa, A.; He, J. Gut Microbiome Associates with Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ. Res. 2016, 119, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Niederseer, D.; Bracher, I.; Stadlmayr, A.; Huber-Schönauer, U.; Plöderl, M.; Obeid, S.; Schmied, C.; Hammerl, S.; Stickel, F.; Lederer, D.; et al. Association between Cardiovascular Risk and Diabetes with Colorectal Neoplasia: A Site-Specific Analysis. J. Clin. Med. 2018, 7, 484. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.-X.; Huang, C.-L.; Luo, S.-Z.; Zhang, X.-M.; Zeng, Y.; Lu, Y.-Y. Characterization of the duodenal bacterial microbiota in patients with pancreatic head cancer vs. healthy controls. Pancreatology 2018, 18, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Kong, C.; Huang, L.; Li, H.; Qu, X.; Liu, Z.; Lan, P.; Wang, J.; Qin, H. Mucosa-associated microbiota signature in colorectal cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Yuefen, P.; Wei, W.; Quan, Q.; Jing, Z.; Jiamin, X.; Shuwen, H. Analysis of prognosis, genome, microbiome, and microbial metabolome in different sites of colorectal cancer. J. Transl. Med. 2019, 17, 1–22. [Google Scholar] [CrossRef]
- Kamphuis, J.; Mercier-Bonin, M.; Eutamène, H.; Théodorou, V. Mucus organisation is shaped by colonic content; a new view. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef]
- Luu, T.H.; Michel, C.; Bard, J.-M.; Dravet, F.; Nazih, H.; Bobin-Dubigeon, C. Intestinal Proportion ofBlautiasp. is Associated with Clinical Stage and Histoprognostic Grade in Patients with Early-Stage Breast Cancer. Nutr. Cancer 2017, 69, 267–275. [Google Scholar] [CrossRef]
- Wu, A.H.; Tseng, C.; Vigen, C.; Yu, Y.; Cozen, W.; Garcia, A.A.; Spicer, D. Gut microbiome associations with breast cancer risk factors and tumor characteristics: A pilot study. Breast Cancer Res. Treat. 2020, 182, 451–463. [Google Scholar] [CrossRef]
- Zhuang, H.; Cheng, L.; Wang, Y.; Zhang, Y.-K.; Zhao, M.-F.; Liang, G.-D.; Zhang, M.-C.; Li, Y.-G.; Zhao, J.-B.; Gao, Y.-N.; et al. Dysbiosis of the Gut Microbiome in Lung Cancer. Front. Cell. Infect. Microbiol. 2019, 9, 112. [Google Scholar] [CrossRef]
- Budinska, E.; Popovici, V.; Tejpar, S.; D’Ario, G.; Lapique, N.; Sikora, K.O.; Di Narzo, A.F.; Yan, P.; Hodgson, J.G.; Weinrich, S.; et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J. Pathol. 2013, 231, 63–76. [Google Scholar] [CrossRef]
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. Npj Biofilms Microbiomes 2017, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, C.W.; Lux, R.; Haake, S.K.; Shi, W. TheFusobacterium nucleatumouter membrane protein RadD is an arginine-inhibitable adhesin required for inter-species adherence and the structured architecture of multispecies biofilm. Mol. Microbiol. 2009, 71, 35–47. [Google Scholar] [CrossRef]
- Tomkovich, S.; Dejea, C.M.; Winglee, K.; Drewes, J.L.; Chung, L.; Housseau, F.; Pope, J.L.; Gauthier, J.; Sun, X.; Mühlbauer, M.; et al. Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J. Clin. Investig. 2019, 129, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Jorth, P.; Turner, K.H.; Gumus, P.; Nizam, N.; Buduneli, N.; Whiteley, M. Metatranscriptomics of the Human Oral Microbiome during Health and Disease. mBio 2014, 5, e01012-14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Geng, R.; Liu, L.; Jin, X.; Yan, W.; Zhao, F.; Wang, S.; Guo, X.; Ghimire, G.; Wei, Y. Gut Microbiota-Based Algorithms in the Prediction of Metachronous Adenoma in Colorectal Cancer Patients Following Surgery. Front. Microbiol. 2020, 11, 1106. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Wilson, I.D.; Teare, J.; Marchesi, J.; Nicholson, J.K.; Kinross, J. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.-S.; Tan, L.T.-H.; Law, J.W.-F.; Pusparajah, P.; Goh, B.-H.; Ab Mutalib, N.S.; Lee, L.-H. Targeting Gut Microbial Biofilms—A Key to Hinder Colon Carcinogenesis? Cancers 2020, 12, 2272. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Kim, Y.-S.; Lee, S.H.; Park, S.G.; Kim, D.H.; Cho, J.Y.; Hahm, K.B.; Hong, S.P.; Yoo, J.-H. Periodontitis is associated with an increased risk for proximal colorectal neoplasms. Sci. Rep. 2019, 9, 7528. [Google Scholar] [CrossRef]
- Horz, H.-P.; Scheer, S.; Huenger, F.; Vianna, M.E.; Conrads, G. Selective isolation of bacterial DNA from human clinical specimens. J. Microbiol. Methods 2008, 72, 98–102. [Google Scholar] [CrossRef]
- Walker, S.P.; Tangney, M.; Claesson, M. Sequence-Based Characterization of Intratumoral Bacteria—A Guide to Best Practice. Front. Oncol. 2020, 10, 179. [Google Scholar] [CrossRef]
- Heravi, F.S.; Zakrzewski, M.; Vickery, K.; Hu, H. Host DNA depletion efficiency of microbiome DNA enrichment methods in infected tissue samples. J. Microbiol. Methods 2020, 170, 105856. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Variables | Data Subset Comparison | Tumour Microbiome Subtypes | |||||
|---|---|---|---|---|---|---|---|
| All Tumours (n = 178) | Triplets (n = 127) | p-Value | TMS1 (n = 46) | TMS2 (n = 55) | TMS3 (n = 77) | p-Value | |
| age at diagnosis | Mean (SD) | Mean (SD) | 0.804 | Mean (SD) | Mean (SD) | Mean (SD) | 0.887 |
| 66.92 (10.66) | 66.61 (10.61) | - | 66.89 (9.88) | 67.47 (11.39) | 66.55 (10.69) | - | |
| gender | n (%) | n (%) | 1 | n (%) | n (%) | n (%) | 0.729 |
| male | 99 (55.6) | 70 (55.1) | - | 25 (54.3) | 33 (60.0) | 41 (53.2) | - |
| female | 79 (44.4) | 57 (44.9) | - | 21 (45.7) | 22 (40.0) | 36 (46.8) | - |
| tumour localisation | n (%) | n (%) | 0.597 | n (%) | n (%) | n (%) | <0.001 |
| right | 64 (36.0) | 48 (37.8) | - | 28 (60.9) | 11 (20.0) | 25 (32.5) | - |
| transverse | 19 (10.7) | 13 (10.2) | - | 6 (13.0) | 5 (9.1) | 8 (10.4) | - |
| left | 44 (24.7) | 36 (28.3) | - | 4 (8.7) | 17 (30.9) | 23 (29.9) | - |
| rectosigmoideum | 32 (18.0) | 23 (18.1) | - | 6 (13.0) | 10 (18.2) | 16 (20.8) | - |
| rectum | 19 (10.7) | 7 (5.5) | - | 2 (4.3) | 12 (21.8) | 5 (6.5) | - |
| grade | n (%) | n (%) | 0.998 | n (%) | n (%) | n (%) | <0.001 |
| NA, in situ | 7 (3.9) | 5 (3.9) | - | 0 (0.0) | 3 (5.5) | 4 (5.2) | - |
| 1 | 18 (10.1) | 12 (9.4) | - | 1 (2.2) | 5 (9.1) | 12 (15.6) | - |
| 2 | 102 (57.3) | 73 (57.5) | - | 18 (39.1) | 37 (67.3) | 47 (61.0) | - |
| 3 | 51 (28.7) | 37 (29.1) | - | 27 (58.7) | 10 (18.2) | 14 (18.2) | - |
| AJCC stage | n (%) | n (%) | 0.968 | n (%) | n (%) | n (%) | 0.136 |
| 0 | 8 (4.5) | 6 (4.7) | - | 0 (0.0) | 3 (5.5) | 5 (6.5) | - |
| I | 31 (17.4) | 26 (20.5) | - | 2 (4.3) | 12 (21.8) | 17 (22.1) | - |
| II | 66 (37.1) | 45 (35.4) | - | 21 (45.7) | 19 (34.5) | 26 (33.8) | - |
| III | 48 (27.0) | 34 (26.8) | - | 16 (34.8) | 12 (21.8) | 20 (26.0) | - |
| IV | 25 (14.0) | 16 (12.6) | - | 7 (15.2) | 9 (16.4) | 9 (11.7) | - |
| tumour pathologic stage | n (%) | n (%) | 0.979 | n (%) | n (%) | n (%) | 0.007 |
| pTis | 8 (4.5) | 6 (4.7) | - | 0 (0.0) | 3 (5.5) | 5 (6.5) | - |
| pT1 | 11 (6.2) | 10 (7.9) | - | 0 (0.0) | 5 (9.1) | 6 (7.8) | - |
| pT2 | 32 (18.0) | 24 (18.9) | - | 2 (4.3) | 12 (21.8) | 18 (23.4) | - |
| pT3 | 115 (64.6) | 79 (62.2) | - | 42 (91.3) | 30 (54.5) | 43 (55.8) | - |
| pT4 | 12 (6.7) | 8 (6.3) | - | 2 (4.3) | 5 (9.1) | 5 (6.5) | - |
| regional lymph nodes pathologic stage | n (%) | n (%) | 0.618 | n (%) | n (%) | n (%) | 0.041 |
| pN0 | 109 (61.2) | 79 (62.2) | - | 23 (50.0) | 36 (65.5) | 50 (64.9) | - |
| pN1 | 46 (25.8) | 36 (28.3) | - | 13 (28.3) | 10 (18.2) | 23 (29.9) | - |
| pN2 | 23 (12.9) | 12 (9.4) | - | 10 (21.7) | 9 (16.4) | 4 (5.2) | - |
| synchronous distant metastasis | n (%) | n (%) | 0.846 | n (%) | n (%) | n (%) | 0.722 |
| M0 | 153 (86.0) | 111 (87.4) | - | 39 (84.8) | 46 (83.6) | 68 (88.3) | - |
| M1 | 25 (14.0) | 16 (12.6) | - | 7 (15.2) | 9 (16.4) | 9 (11.7) | - |
| MSI/MSS | n (%) | n (%) | 1 | n (%) | n (%) | n (%) | <0.001 |
| MSI | 27 (15.2) | 19 (15.0) | - | 16 (34.8) | 4 (7.3) | 7 (9.1) | - |
| MSS | 110 (61.8) | 81 (63.8) | - | 22 (47.8) | 37 (67.3) | 51 (66.2) | - |
| NA | 41 (23.0) | 27 (21.2) | - | 8 (17.4) | 14 (25.4) | 19 (24.7) | - |
| BRAF | n (%) | n (%) | 1 | n (%) | n (%) | n (%) | 0.022 |
| BRAF wt | 77 (43.3) | 53 (41.7) | - | 17 (37.0) | 27 (49.1) | 33 (42.9) | - |
| BRAF mut | 12 (6.7) | 9 (7.1) | - | 7 (15.2) | 1 (1.8) | 4 (5.2) | - |
| NA | 89 (50.0) | 65 (51.2) | - | 22 (47.8) | 27 (49.1) | 40 (51.9) | - |
| KRAS | n (%) | n (%) | 1 | n (%) | n (%) | n (%) | 0.839 |
| KRAS wt | 24 (13.5) | 17 (13.4) | - | 7 (15.2) | 8 (14.5) | 9 (11.7) | - |
| KRAS mut | 13 (7.3) | 9 (7.1) | - | 5 (10.9) | 4 (7.3) | 4 (5.2) | - |
| NA | 141 (79.2) | 101 (79.5) | - | 34 (73.9) | 43 (78.2) | 64 (83.1) | - |
| NRAS | n (%) | n (%) | 1 | n (%) | n (%) | n (%) | 0.553 |
| NRAS wt | 37 (20.8) | 26 (20.5) | - | 11 (23.9) | 12 (21.8) | 14 (18.2) | - |
| NRAS mut | 2 (1.1) | 1 (0.8) | - | 1 (2.2) | 1 (1.8) | 0 (0.0) | - |
| NA | 139 (78.1) | 100 (78.7) | - | 34 (73.9) | 42 (76.4) | 63 (81.8) | - |
| Regression Covariate | Effect/Contrast | Tumour Mucosa | Visually Normal Mucosa | Stool |
|---|---|---|---|---|
| grade | increasing grade | ↑ Fusobacterium *,Campylobacter*, Leptotrichia, Peptoclostridium, Mogibacterium * | - | - |
| - | ↓ Unassigned genus from order Opitutae vadin HA64 | - | ||
| location | right-sided/transverse vs left-sided and rectum/rectosigmoid | ↑ Holdemania, Selenomonas 4, Clostridium sensu stricto 1, Alloprevotella | ↑ Selenomonas 3, Selenomonas, Treponema 2 | - |
| ↓ Bifidobacterium *, Christensenellaceae R-7 group2, Ruminococcaceae UCG-013, Fusicatenibacter | ↓ Lachnospira, Bifidobacterium, Coprococcus 1, Christensenellaceae R-7 group | - | ||
| right-sided/transverse vs left-sided | ↑ Campylobacter, Alloprevotella | - | - | |
| ↓ Family XIII AD3011 group, Coprococcus 1 | - | |||
| right-sided/transverse vs rectosigmoid/rectum | ↑ Oribacterium, Fretibacterium | - | - | |
| - | ↓ [Eubacterium] ventriosum group | |||
| grade*location interaction | low-graded; right-sided/transverse | ↑Ruminococcaceae UCG-010, uncultured bacterium from Clostridiales vadinBB60 group | - | ↑ Unassigned genus from order Opitutae vadin HA64, Porphyromonas |
| grade 2; left-sided | ↓Coprococcus 2, Ruminiclostridium 6, [Eubacterium] ventriosum group, Incertae Sedis from Lachnospiraceae family | ↓ Gemella, Corynebacterium 1 | ↓ Ruminiclostridium 6, Coprococcus 2 | |
| grade 2; rectosigmoid/rectum | - | ↑ Veillonella | ↑ Veillonella | |
| ↓ Methanobrevibacter, Dielma, Victivallis | ↓ Methanobrevibacter, an uncultured genus from the Peptococcaceae family | ↓ Victivallis, Ruminiclosridium 6, Lachnospiraceae UCG-005, an unassigned genus from order Mollicutes RF9 | ||
| grade 3; right-sided/transverse | ↑ Prevotella, Selenomonas, Selenomonas 3 | - | - | |
| grade 3; left-sided | - | ↑ Eisenbergiella, Leptotrichia, Escherichia-Shigella, Veillonella | ↑ Veillonella, Prevotella 7 | |
| ↓ Coprococcus 2, Ruminiclostridium 6, [Eubacterium] ventriosum group, Incertae Sedis from Lachnospiraceae family, Odoribacter | ↓ Gemella, Corynebacterium 1 | ↓ Coprococcus 2 | ||
| grade 3; rectosigmoid/rectum | ↑ Lachnospira | ↑ Veillonella | ↑ Prevotella, Prevotella 7 | |
| ↓ Methanobrevibacter, Dielma, Victivallis | ↓ Methanobrevibacter, Eisenbergiella, an uncultured genus from the Peptococcaceae family | ↓ Lachnospiraceae UCG-005, unassigned genus from order Mollicutes RF9 | ||
| AJCC stage | III–IV vs 0–II | ↑ Peptoclostridium | - | ↑ Akkermansia |
| - | ↓ Gelria | - | ||
| Tumour pathologic stage | pT 3–4 vs pTis-2 | ↑ Peptoclostridium, Gemella, Campylobacter, Parvimonas | ↑ Peptoclostridium, Escherichia-Shigella, an unassigned species from Ruminococcaceae | ↑ Escherichia-Shigella |
| ↓ Coprobacter, Intestinimonas, Ruminococcaceae UCG-009, Oscillospira, Cloacibacillus | ↓ Intestinimonas, Ruminococcaceae UCG-009, Holdemanella, Coprobacter, Gelria, an uncultured genus from the Christensenellaceae family | ↓ Prevotella 6, Ruminococcaceae UCG-011 1 | ||
| Regional lymph nodes stage | N1–2 vs N0 | ↑ Peptoclostridium | - | ↑ Peptococcus, Campylobacter, Akkermansia *, Selenomonas, Porphyromonas *, Streptococcus, Oscillospira |
| ↓ Prevotellaceae UCG-001, uncultured Fusobacterium sp. from family boneC3G7 | ↓ [Eubacterium] hallii group | ↓ Faecalibacterium, Ruminiclostridium, Dorea 1, Lachnospiraceae FCS020 group | ||
| Synchronous distant metastasis | present vs absent | ↑ Porphyromonas, Streptococcus, Ruminococcaceae UCG-005 | ↑ Akkermansia | ↑ uncultured genus from Erysipelotrichaceae family, Akkermansia, Coprococcus 1, Solobacterium |
| - | ↓ Gelria, [Eubacterium] brachy group, uncultured genera from Christensenellaceae family, Gordonibacter, Fretibacterium | ↓ Selenomonas, Ruminococcaceae UCG-004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zwinsová, B.; Petrov, V.A.; Hrivňáková, M.; Smatana, S.; Micenková, L.; Kazdová, N.; Popovici, V.; Hrstka, R.; Šefr, R.; Bencsiková, B.; et al. Colorectal Tumour Mucosa Microbiome Is Enriched in Oral Pathogens and Defines Three Subtypes That Correlate with Markers of Tumour Progression. Cancers 2021, 13, 4799. https://doi.org/10.3390/cancers13194799
Zwinsová B, Petrov VA, Hrivňáková M, Smatana S, Micenková L, Kazdová N, Popovici V, Hrstka R, Šefr R, Bencsiková B, et al. Colorectal Tumour Mucosa Microbiome Is Enriched in Oral Pathogens and Defines Three Subtypes That Correlate with Markers of Tumour Progression. Cancers. 2021; 13(19):4799. https://doi.org/10.3390/cancers13194799
Chicago/Turabian StyleZwinsová, Barbora, Vyacheslav A. Petrov, Martina Hrivňáková, Stanislav Smatana, Lenka Micenková, Natálie Kazdová, Vlad Popovici, Roman Hrstka, Roman Šefr, Beatrix Bencsiková, and et al. 2021. "Colorectal Tumour Mucosa Microbiome Is Enriched in Oral Pathogens and Defines Three Subtypes That Correlate with Markers of Tumour Progression" Cancers 13, no. 19: 4799. https://doi.org/10.3390/cancers13194799
APA StyleZwinsová, B., Petrov, V. A., Hrivňáková, M., Smatana, S., Micenková, L., Kazdová, N., Popovici, V., Hrstka, R., Šefr, R., Bencsiková, B., Zdražilová-Dubská, L., Brychtová, V., Nenutil, R., Vídeňská, P., & Budinská, E. (2021). Colorectal Tumour Mucosa Microbiome Is Enriched in Oral Pathogens and Defines Three Subtypes That Correlate with Markers of Tumour Progression. Cancers, 13(19), 4799. https://doi.org/10.3390/cancers13194799