
The Role of Bacteria in KSHV Infection and KSHV-Induced Cancers


{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Bacterial Infection and the Innate Immune Response: Effects on Cancer Pathology
3. KSHV and KSHV-Associated Cancers
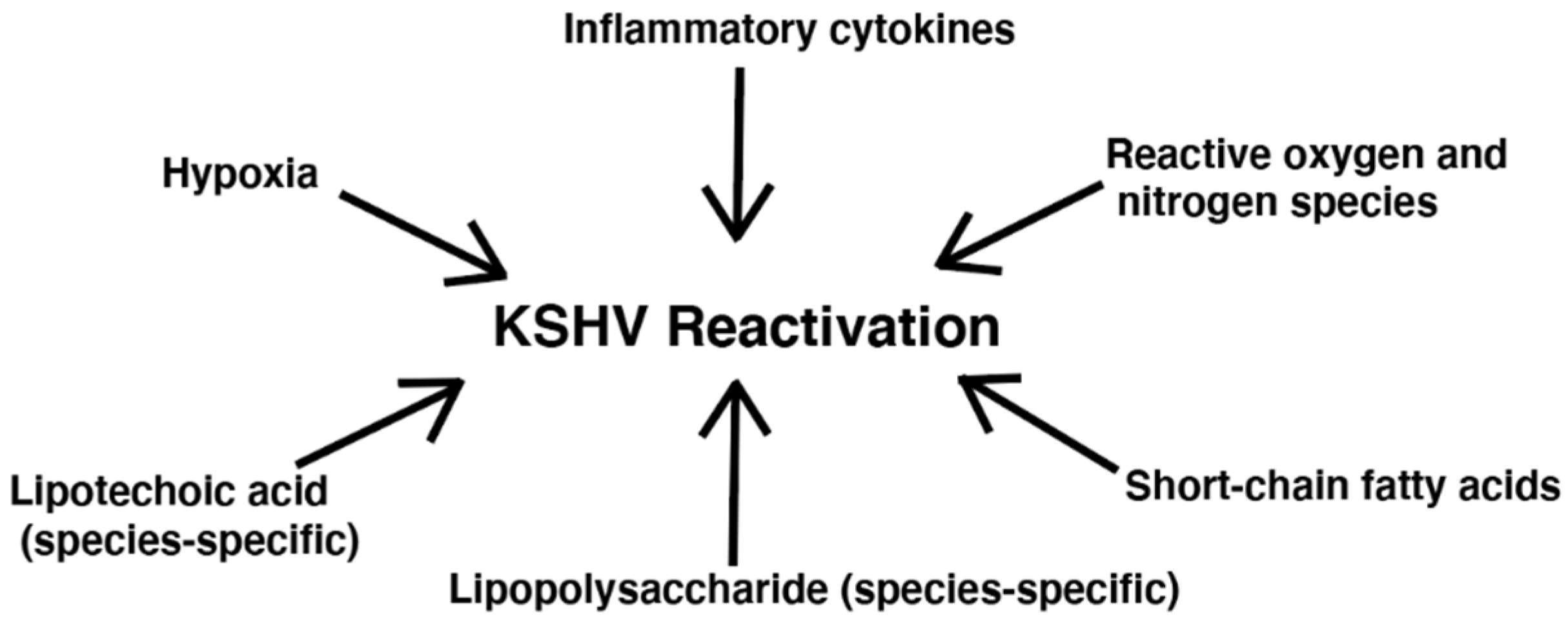
4. Impact of Bacteria on KSHV Lytic Replication
5. Impact of Bacteria on KSHV-Induced Cell Proliferation and Cellular Transformation
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef]
- van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Górska, A.; Przystupski, D.; Niemczura, M.J.; Kulbacka, J. Probiotic bacteria: A promising tool in cancer prevention and therapy. Curr. Microbiol. 2019, 76, 939–949. [Google Scholar] [CrossRef]
- Thakker, S.; Verma, S.C. Co-infections and pathogenesis of KSHV-associated malignancies. Front. Microbiol. 2016, 7, 151. [Google Scholar] [CrossRef]
- Morales-Sánchez, A.; Fuentes-Pananá, E.M. Human viruses and cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Lambris, J.D. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 2007, 171, 715–727. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Qu, X.; Tang, Y.; Hua, S. Immunological approaches towards cancer and inflammation: A cross talk. Front. Immunol. 2018, 9, 563. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Hoving, J.C.; Wilson, G.J.; Brown, G.D. Signalling C-type lectin receptors, microbial recognition and immunity. Cell Microbiol. 2014, 16, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, B.; Wang, T.; Xu, L.; He, C.; Wen, H.; Yan, J.; Su, H.; Zhu, X. Toll-like receptor 4 prompts human breast cancer cells invasiveness via lipopolysaccharide stimulation and is overexpressed in patients with lymph node metastasis. PLoS ONE 2014, 9, e109980. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, H.; Ou, Y.; Feng, Z.; Zhang, Q.; Zhu, Q.; Cai, Z. LPS-induced CXCR7 expression promotes gastric Cancer proliferation and migration via the TLR4/MD-2 pathway. Diagn. Pathol. 2019, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Kang, M.J.; Kim, Y.S.; Kim, S.M.; Lee, S.E.; Kim, C.H.; Kim, D.J.; Park, J.H. Flagellin promotes the proliferation of gastric cancer cells via the Toll-like receptor 5. Int. J. Mol. Med. 2011, 28, 115–119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, W.; Huang, Y.; Xie, W.; Guo, A.; Wu, W. Bacteria peptidoglycan promoted breast cancer cell invasiveness and adhesiveness by targeting toll-like receptor 2 in the cancer cells. PLoS ONE 2010, 5, e10850. [Google Scholar] [CrossRef]
- Jain, S.; Dash, P.; Minz, A.P.; Satpathi, S.; Samal, A.G.; Behera, P.K.; Satpathi, P.S.; Senapati, S. Lipopolysaccharide (LPS) enhances prostate cancer metastasis potentially through NF-κB activation and recurrent dexamethasone administration fails to suppress it in vivo. Prostate 2019, 79, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Fedirko, V.; Tran, H.Q.; Gewirtz, A.T.; Stepien, M.; Trichopoulou, A.; Aleksandrova, K.; Olsen, A.; Tjønneland, A.; Overvad, K.; Carbonnel, F.; et al. Exposure to bacterial products lipopolysaccharide and flagellin and hepatocellular carcinoma: A nested case-control study. BMC Med. 2017, 15, 72. [Google Scholar] [CrossRef]
- Kong, S.Y.; Tran, H.Q.; Gewirtz, A.T.; McKeown-Eyssen, G.; Fedirko, V.; Romieu, I.; Tjønneland, A.; Olsen, A.; Overvad, K.; Boutron-Ruault, M.C.; et al. Serum endotoxins and flagellin and risk of colorectal cancer in the European prospective investigation into cancer and nutrition (EPIC) cohort. Cancer Epidemiol. Biomark. Prev. 2016, 25, 291–301. [Google Scholar] [CrossRef]
- de Waal, G.M.; de Villiers, W.J.S.; Forgan, T.; Roberts, T.; Pretorius, E. Colorectal cancer is associated with increased circulating lipopolysaccharide, inflammation and hypercoagulability. Sci. Rep. 2020, 10, 8777. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Fried, S.; Tosun, S.; Troost, G.; Keil, S.; Zaenker, K.S.; Dittmar, T. Lipopolysaccharide (LPS) promotes apoptosis in human breast epithelial × breast cancer hybrids, but not in parental cells. PLoS ONE 2016, 11, e0148438. [Google Scholar] [CrossRef]
- Cai, Z.; Sanchez, A.; Shi, Z.; Zhang, T.; Liu, M.; Zhang, D. Activation of Toll-like receptor 5 on breast cancer cells by flagellin suppresses cell proliferation and tumor growth. Cancer Res. 2011, 71, 2466–2475. [Google Scholar] [CrossRef]
- Duong, M.T.; Qin, Y.; You, S.H.; Min, J.J. Bacteria-cancer interactions: Bacteria-based cancer therapy. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Shetab Boushehri, M.A.; Lamprecht, A. Challenges of using lipopolysaccharides for cancer immunotherapy and potential delivery-based solutions thereto. Ther. Deliv. 2019, 10, 165–187. [Google Scholar] [CrossRef]
- Jouhi, L.; Renkonen, S.; Atula, T.; Mäkitie, A.; Haglund, C.; Hagström, J. Different toll-like receptor expression patterns in progression toward cancer. Front. Immunol. 2014, 5, 638. [Google Scholar] [CrossRef]
- Khan, A.A.; Khan, Z.; Warnakulasuriya, S. Cancer-associated toll-like receptor modulation and insinuation in infection susceptibility: Association or coincidence? Ann. Oncol. 2016, 27, 984–997. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Cheng, F.; da Silva, S.R.; Tan, B.; Sorel, O.; Gruffaz, M.; Li, T.; Gao, S.J. Molecular biology of KSHV in relation to HIV/AIDS-associated oncogenesis. Cancer Treat. Res. 2019, 177, 23–62. [Google Scholar] [CrossRef]
- Osmond, D.H.; Buchbinder, S.; Cheng, A.; Graves, A.; Vittinghoff, E.; Cossen, C.K.; Forghani, B.; Martin, J.N. Prevalence of Kaposi sarcoma-associated herpesvirus infection in homosexual men at beginning of and during the HIV epidemic. JAMA 2002, 287, 221–225. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef]
- Bhutani, M.; Polizzotto, M.N.; Uldrick, T.S.; Yarchoan, R. Kaposi sarcoma-associated herpesvirus-associated malignancies: Epidemiology, pathogenesis, and advances in treatment. Semin. Oncol. 2015, 42, 223–246. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Ye, F.; Lei, X.; Gao, S.J. Mechanisms of Kaposi’s sarcoma-associated herpesvirus latency and reactivation. Adv. Virol. 2011, 2011, 193860. [Google Scholar] [CrossRef]
- Okoye, A.A.; Picker, L.J. CD4(+) T-cell depletion in HIV infection: Mechanisms of immunological failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef]
- Aneja, K.K.; Yuan, Y. Reactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus: An update. Front. Microbiol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Yan, Q.; Shen, C.; Qin, J.; Li, W.; Hu, M.; Lu, H.; Qin, D.; Zhu, J.; Gao, S.J.; Lu, C. HIV-1 Vpr inhibits Kaposi’s sarcoma-associated herpesvirus lytic replication by inducing microRNA miR-942-5p and activating NF-κB signaling. J. Virol. 2016, 90, 8739–8753. [Google Scholar] [CrossRef]
- Yao, S.; Hu, M.; Hao, T.; Li, W.; Xue, X.; Xue, M.; Zhu, X.; Zhou, F.; Qin, D.; Yan, Q.; et al. MiRNA-891a-5p mediates HIV-1 Tat and KSHV Orf-K1 synergistic induction of angiogenesis by activating NF-κB signaling. Nucleic Acids Res. 2019, 47, 2700. [Google Scholar] [CrossRef]
- Yan, Q.; Zhao, R.; Shen, C.; Wang, F.; Li, W.; Gao, S.J.; Lu, C. Upregulation of microRNA 711 mediates HIV-1 Vpr promotion of Kaposi’s sarcoma-associated herpesvirus latency and induction of pro-proliferation and pro-survival cytokines by targeting the Notch/NF-κB-signaling axis. J. Virol. 2018, 92, e00580-18. [Google Scholar] [CrossRef]
- Zhou, F.; Xue, M.; Qin, D.; Zhu, X.; Wang, C.; Zhu, J.; Hao, T.; Cheng, L.; Chen, X.; Bai, Z.; et al. HIV-1 Tat promotes Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6-induced angiogenesis and tumorigenesis by regulating PI3K/PTEN/AKT/GSK-3β signaling pathway. PLoS ONE 2013, 8, e53145. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, X.; Huang, Z.; Cheng, L.; Yao, S.; Qin, D.; Chen, X.; Tang, Q.; Lv, Z.; Zhang, L.; et al. Intracellular Tat of human immunodeficiency virus type 1 activates lytic cycle replication of Kaposi’s sarcoma-associated herpesvirus: Role of JAK/STAT signaling. J. Virol. 2007, 81, 2401–2417. [Google Scholar] [CrossRef]
- Xue, M.; Yao, S.; Hu, M.; Li, W.; Hao, T.; Zhou, F.; Zhu, X.; Lu, H.; Qin, D.; Yan, Q.; et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res. 2014, 42, 9862–9879. [Google Scholar] [CrossRef]
- Annavajhala, M.K.; Khan, S.D.; Sullivan, S.B.; Shah, J.; Pass, L.; Kister, K.; Kunen, H.; Chiang, V.; Monnot, G.C.; Ricupero, C.L.; et al. Oral and gut microbial diversity and immune regulation in patients with HIV on antiretroviral therapy. mSphere 2020, 5, e00798-19. [Google Scholar] [CrossRef]
- Taur, Y.; Pamer, E.G. The intestinal microbiota and susceptibility to infection in immunocompromised patients. Curr. Opin. Infect. Dis. 2013, 26, 332–337. [Google Scholar] [CrossRef]
- Gruffaz, M.; Zhang, T.; Marshall, V.; Gonçalves, P.; Ramaswami, R.; Labo, N.; Whitby, D.; Uldrick, T.S.; Yarchoan, R.; Huang, Y.; et al. Signatures of oral microbiome in HIV-infected individuals with oral Kaposi’s sarcoma and cell-associated KSHV DNA. PLoS Pathog. 2020, 16, e1008114. [Google Scholar] [CrossRef]
- Contreras, A.; Slots, J. Herpesviruses in human periodontal disease. J. Periodontal. Res. 2000, 35, 3–16. [Google Scholar] [CrossRef]
- Yu, X.; Shahir, A.M.; Sha, J.; Feng, Z.; Eapen, B.; Nithianantham, S.; Das, B.; Karn, J.; Weinberg, A.; Bissada, N.F.; et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi’s sarcoma-associated herpesvirus replication. J. Virol. 2014, 88, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Barrett, L.; Plaisance-Bonstaff, K.; Post, S.R.; Qin, Z. Porphyromonas gingivalis coinfects with KSHV in oral cavities of HIV+ patients and induces viral lytic reactivation. J. Med. Virol. 2020, 92, 3862–3867. [Google Scholar] [CrossRef]
- Morris, T.L.; Arnold, R.R.; Webster-Cyriaque, J. Signaling cascades triggered by bacterial metabolic end products during reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 6032–6042. [Google Scholar] [CrossRef]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485s–2493s. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Xie, J.; Ye, F.; Gao, S.J. Modulation of Kaposi’s sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 2006, 80, 5371–5382. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Bai, Z.; Ye, F.; Xie, J.; Kim, C.G.; Huang, Y.; Gao, S.J. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 2010, 12, 193–199. [Google Scholar] [CrossRef]
- Xie, J.; Pan, H.; Yoo, S.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 2005, 79, 15027–15037. [Google Scholar] [CrossRef]
- Xie, J.; Ajibade, A.O.; Ye, F.; Kuhne, K.; Gao, S.J. Reactivation of Kaposi’s sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 2008, 371, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Gao, S.J. A novel role of hydrogen peroxide in Kaposi sarcoma-associated herpesvirus reactivation. Cell Cycle 2011, 10, 3237–3238. [Google Scholar] [CrossRef][Green Version]
- Erttmann, S.F.; Gekara, N.O. Hydrogen peroxide release by bacteria suppresses inflammasome-dependent innate immunity. Nat. Commun. 2019, 10, 3493. [Google Scholar] [CrossRef]
- Dai, L.; DeFee, M.R.; Cao, Y.; Wen, J.; Wen, X.; Noverr, M.C.; Qin, Z. Lipoteichoic acid (LTA) and lipopolysaccharides (LPS) from periodontal pathogenic bacteria facilitate oncogenic herpesvirus infection within primary oral cells. PLoS ONE 2014, 9, e101326. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Chakraborty, S.; Chandran, B. Reactive oxygen species are induced by Kaposi’s sarcoma-associated herpesvirus early during primary infection of endothelial cells to promote virus entry. J. Virol. 2013, 87, 1733–1749. [Google Scholar] [CrossRef]
- Sharma-Walia, N.; Krishnan, H.H.; Naranatt, P.P.; Zeng, L.; Smith, M.S.; Chandran, B. ERK1/2 and MEK1/2 induced by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 2005, 79, 10308–10329. [Google Scholar] [CrossRef] [PubMed]
- Sadagopan, S.; Sharma-Walia, N.; Veettil, M.V.; Raghu, H.; Sivakumar, R.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 2007, 81, 3949–3968. [Google Scholar] [CrossRef]
- Dai, L.; Qiao, J.; Yin, J.; Goldstein, A.; Lin, H.Y.; Post, S.R.; Qin, Z. Kaposi sarcoma-associated herpesvirus and Staphylococcus aureus coinfection in oral cavities of HIV-positive patients: A unique niche for oncogenic virus lytic reactivation. J. Infect. Dis. 2020, 221, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.I.; Jewtuchowicz, V.; Brusca, M.I.; Nastri, M.L.; Rosa, A.C. Prevalence of Staphylococcus spp. and Candida spp. in the oral cavity and periodontal pockets of periodontal disease patients. Acta Odontol. Lat. 2010, 23, 20–26. [Google Scholar]
- Ferreira Dde, C.; Silva, G.R.; Cavalcante, F.S.; Carmo, F.L.; Fernandes, L.A.; Moreira, S.; Passos, M.R.; Colombo, A.P.; Santos, K.R. Methicillin-resistant Staphylococcus aureus in HIV patients: Risk factors associated with colonization and/or infection and methods for characterization of isolates—A systematic review. Clinics 2014, 69, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Tribble, G.D.; Kerr, J.E.; Wang, B.Y. Genetic diversity in the oral pathogen Porphyromonas gingivalis: Molecular mechanisms and biological consequences. Future Microbiol. 2013, 8, 607–620. [Google Scholar] [CrossRef]
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Giffin, L.; Damania, B. KSHV: Pathways to tumorigenesis and persistent infection. Adv. Virus Res. 2014, 88, 111–159. [Google Scholar] [CrossRef]
- Campbell, M.; Yang, W.S.; Yeh, W.W.; Kao, C.H.; Chang, P.C. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency. Front. Microbiol. 2020, 11, 850. [Google Scholar] [CrossRef]
- Punjabi, A.S.; Carroll, P.A.; Chen, L.; Lagunoff, M. Persistent activation of STAT3 by latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 2007, 81, 2449–2458. [Google Scholar] [CrossRef]
- King, C.A.; Li, X.; Barbachano-Guerrero, A.; Bhaduri-McIntosh, S. STAT3 regulates lytic activation of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2015, 89, 11347–11355. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Jones, T.; Song, D.Y.; Jang, J.H.; Jung, J.U.; Gao, S.J. Exploitation of the complement system by oncogenic Kaposi’s sarcoma-associated herpesvirus for cell survival and persistent infection. PLoS Pathog. 2014, 10, e1004412. [Google Scholar] [CrossRef] [PubMed]
- Gruffaz, M.; Vasan, K.; Tan, B.; Ramos da Silva, S.; Gao, S.J. TLR4-mediated inflammation promotes KSHV-induced cellular transformation and tumorigenesis by activating the STAT3 pathway. Cancer Res. 2017, 77, 7094–7108. [Google Scholar] [CrossRef] [PubMed]
- Markazi, A.; Bracci, P.M.; McGrath, M.; Gao, S.J. Pseudomonas aeruginosa stimulates inflammation and enhances Kaposi’s sarcoma herpesvirus-induced cell proliferation and cellular transformation through both lipopolysaccharide and flagellin. mBio 2020, 11, e02843-20. [Google Scholar] [CrossRef] [PubMed]
- Sadikot, R.T.; Blackwell, T.S.; Christman, J.W.; Prince, A.S. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 1209–1223. [Google Scholar] [CrossRef]
- Jones, T.; Ye, F.; Bedolla, R.; Huang, Y.; Meng, J.; Qian, L.; Pan, H.; Zhou, F.; Moody, R.; Wagner, B.; et al. Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J. Clin. Investig. 2012, 122, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Broussard, G.; Damania, B. KSHV: Immune modulation and immunotherapy. Front. Immunol. 2020, 10, 3084. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed]
- Wynendaele, E.; Verbeke, F.; D’Hondt, M.; Hendrix, A.; Van De Wiele, C.; Burvenich, C.; Peremans, K.; De Wever, O.; Bracke, M.; De Spiegeleer, B. Crosstalk between the microbiome and cancer cells by quorum sensing peptides. Peptides 2015, 64, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Grabiner, M.A.; Fu, Z.; Wu, T.; Barry, K.C.; Schwarzer, C.; Machen, T.E. Pseudomonas aeruginosa quorum-sensing molecule homoserine lactone modulates inflammatory signaling through PERK and eI-F2α. J. Immunol. 2014, 193, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Cao, Y.; Zabaleta, J.; Yang, L.; Dai, L.; Qin, Z. Regulation of virus-associated lymphoma growth and gene expression by bacterial quorum-sensing molecules. J. Virol. 2018, 92, e00478-18. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markazi, A.; Meng, W.; Bracci, P.M.; McGrath, M.S.; Gao, S.-J. The Role of Bacteria in KSHV Infection and KSHV-Induced Cancers. Cancers 2021, 13, 4269. https://doi.org/10.3390/cancers13174269
Markazi A, Meng W, Bracci PM, McGrath MS, Gao S-J. The Role of Bacteria in KSHV Infection and KSHV-Induced Cancers. Cancers. 2021; 13(17):4269. https://doi.org/10.3390/cancers13174269
Chicago/Turabian StyleMarkazi, Ashley, Wen Meng, Paige M. Bracci, Michael S. McGrath, and Shou-Jiang Gao. 2021. "The Role of Bacteria in KSHV Infection and KSHV-Induced Cancers" Cancers 13, no. 17: 4269. https://doi.org/10.3390/cancers13174269
APA StyleMarkazi, A., Meng, W., Bracci, P. M., McGrath, M. S., & Gao, S.-J. (2021). The Role of Bacteria in KSHV Infection and KSHV-Induced Cancers. Cancers, 13(17), 4269. https://doi.org/10.3390/cancers13174269