
The Breast Tumor Microenvironment: A Key Player in Metastatic Spread

 and
and {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Breast Cancer: Current Challenges
1.2. Breast Cancer Subtypes
1.3. The Tumor Microenvironment
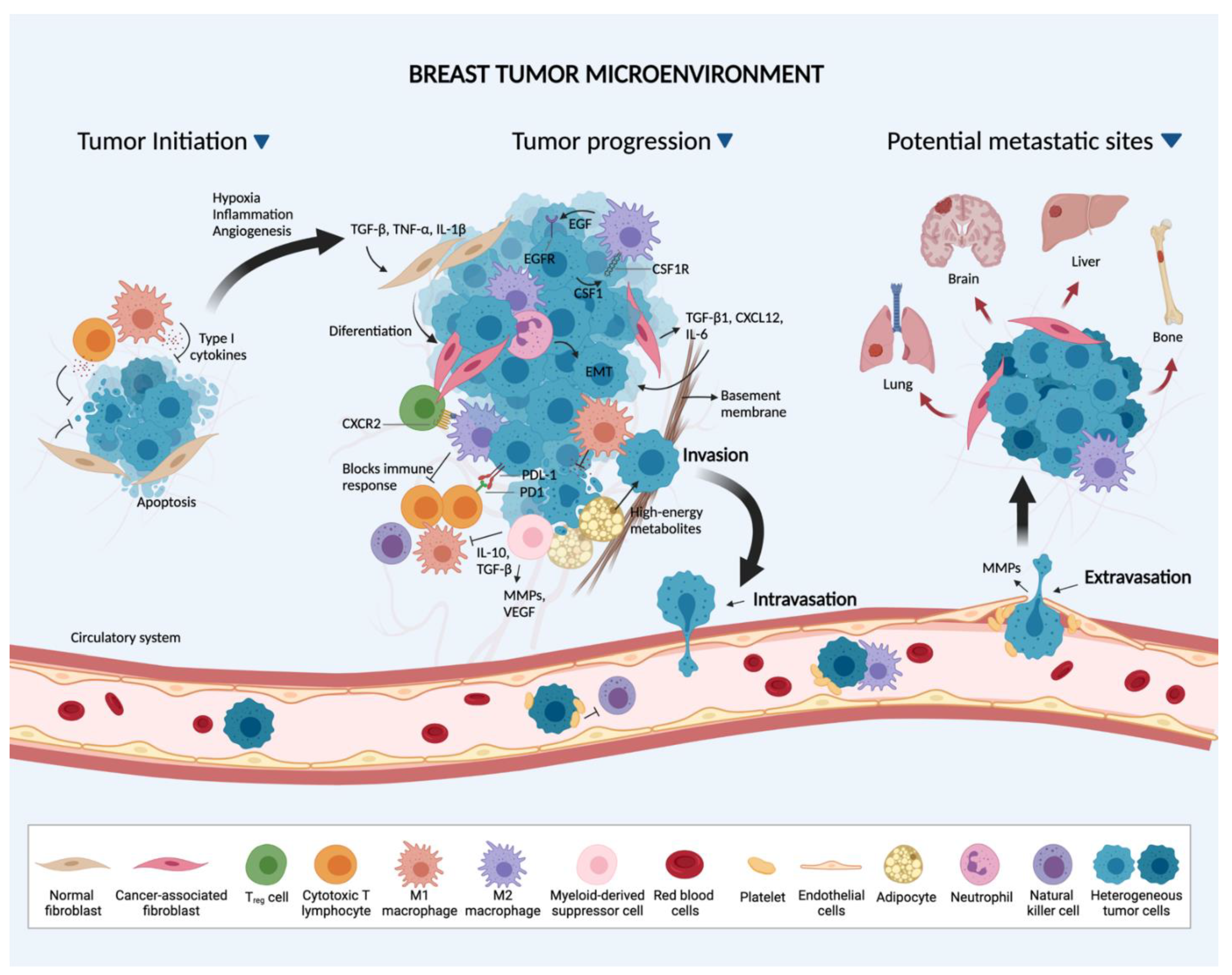
2. Breast Cancer Cell-Stromal Interactions
2.1. Primary Site
2.1.1. Tunneling Nanotubes
2.1.2. Fibroblasts
2.1.3. Endothelial Cells
2.1.4. Adipocytes
2.1.5. Immune Cells
2.2. The Metastatic Process and Preferred BC Metastatic Sites
2.2.1. Metastatic Process
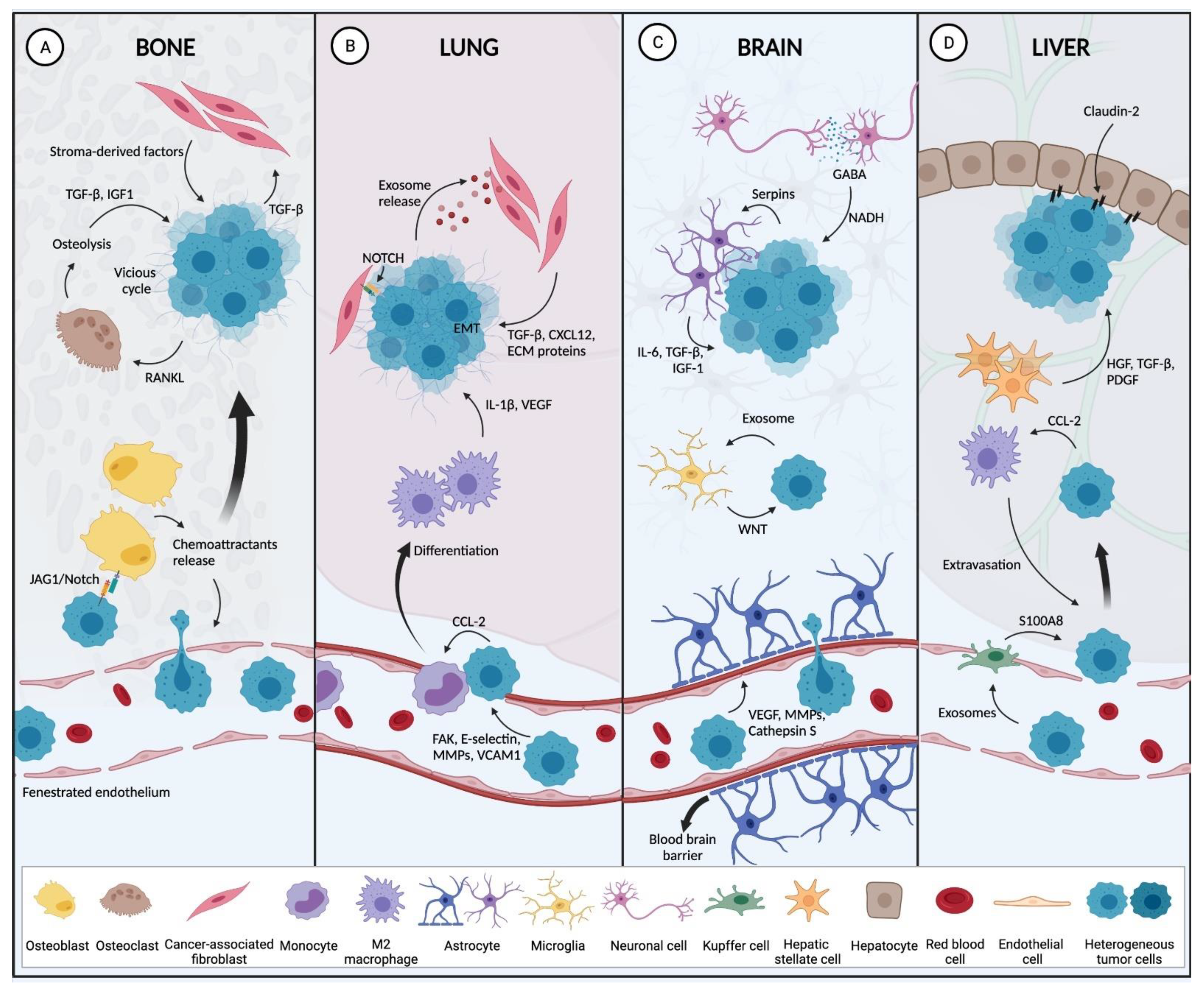
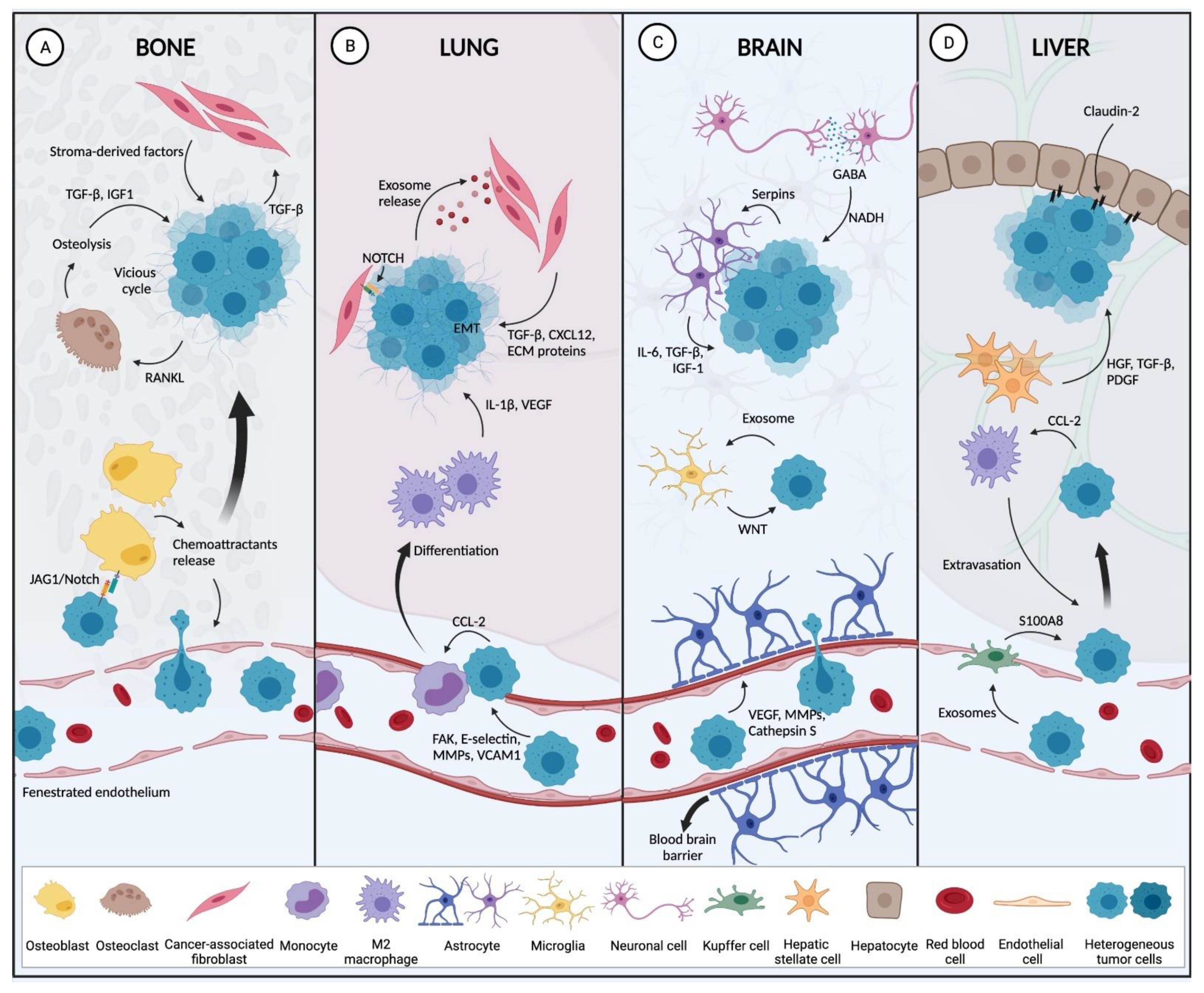
2.2.2. Preferred BC Metastatic Sites
Bone
Lung
Liver
Brain
3. Breast Cancer Cell-Stromal Interactions: Implications for Prognosis
4. Breast Cancer Cell-Stromal Interactions: Implications for Oncotherapy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALDH1 | Aldehyde Dehydrogenase 1 |
| AKT | Protein Kinase B |
| BBB | Blood Brain Barrier |
| BC | Breast Cancer |
| BCAFs | Breast Cancer Associated Fibroblasts |
| BMPs | Bone Morphogenetic Proteins |
| CAA | Cancer Associated Adipocyte |
| CAF | Cancer Associated Fibroblast |
| CCL2 | Chemokine C-C Motif Ligand 2 |
| CCL3 | Chemokine C-C Motif Ligand 3 |
| CCL5 | Chemokine C-C Motif Ligand 5 |
| COX-2 | Cyclooxygenase 2 |
| CTCs | Circulating Tumor Cells |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 |
| DSP | Digital Spatial Profiling |
| EC | Endothelial Cells |
| ECM | Extracellular Matrix |
| EGF-1 | Epidermal Growth Factor-1 |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial-Mesenchymal Transition |
| ER | Estrogen Receptor |
| FAK | focal adhesion kinase |
| FAPα | Fibroblast Activation Protein Alfa |
| GABA | Gamma Aminobutyric Acid |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HGF | Hepatocyte Growth Factor |
| IGF1 | Insulin-like Growth Factor 1 |
| IL-10 | Interleukin-10 |
| IL-1β | Interleukin-1 Beta |
| IL-3 | Interleukin 3 |
| IL-6 | Interleukin-6 |
| JAG1 | Notch Ligand Jagger1 |
| JAK/STAT3 | Janus Kinase/Signal Transducer and Activator of Transcription 3 |
| M1 | Classically Activated Macrophages |
| M2 | Alternatively activated Macrophages |
| MDSCs | Myeloid Derived Suppressor Cells |
| MMPs | Matrix Metalloproteinase |
| NADH | Reduced Nicotinamide Adenine Dinucleotide |
| NF-kB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| PD-1 | Programmed Cell Death-1 |
| PDGF | Platelet-Derived Growth Factor |
| PDGFR-α | Platelet-Derived Growth Factor Receptor A |
| PD-L1 | Programmed Cell Death-1 Ligand |
| PI3K | Phosphatidylinositol 3-Kinase |
| PR | Progesterone Receptor |
| PTEN | Phosphatase and Tensin Homolog |
| RANKL | Receptor Activator of Nuclear Factor Kappa-Β Ligand |
| S100A4 | S100 Calcium Binding Protein A4 |
| scRNA-seq | Single Cell RNA Sequencing |
| STING | Stimulator of Interferon Genes |
| TAM | Tumor Associated Macrophages |
| TEC | Tumor endothelial cells |
| TGF-β1 | Transforming Growth Factor-Beta 1 |
| TME | Tumor Microenvironment |
| TNBC | Triple-Negative Breast Cancer |
| TNFRSF4 | Tumor Necrosis Factor Receptor Superfamily, Member 4 |
| TNF-α | Tumor Necrosis Factor-Alpha |
| VEGF | Vascular Endothelial Growth Factor |
| Wnt | Wingless-related Integration Site |
References
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Cancer Facts & Figures 2019|American Cancer Society. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html (accessed on 12 January 2020).
- Vanni, G.; Pellicciaro, M.; Materazzo, M.; Pedini, D.; Portarena, I.; Buonomo, C.; Perretta, T.; Rizza, S.; Pistolese, C.A.; Buonomo, O.C. Advanced Stages and Increased Need for Adjuvant Treatments in Breast Cancer Patients: The Effect of the One-year COVID-19 Pandemic. Anticancer Res. 2021, 41, 2689–2696. [Google Scholar] [CrossRef]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological types of breast cancer: How special are they? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.; Sørlie, T.; Eisen, M.; Van De Rijn, M.; Jeffrey, S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Gonzalez-Angulo, A.-M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.-S.; Fridlyand, J.; Sahin, A.A.; Agarwal, R.; Joy, C.; et al. Characterization of a Naturally Occurring Breast Cancer Subset Enriched in Epithelial-to-Mesenchymal Transition and Stem Cell Characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef] [Green Version]
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sørlie, T. Re-Definition of claudin-low as a breast cancer phenotype. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [Green Version]
- Dawson, S.-J.; Rueda, O.M.; Aparicio, S.; Caldas, C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013, 32, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Lehmann, B.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Edechi, C.A.; Ikeogu, N.M.; Terceiro, L.E.L.; Uzonna, J.E.; Myal, Y. Metastasis: A Bane of Breast Cancer Therapy. Eur. Med. J. 2020, 5, 55–62. [Google Scholar] [CrossRef]
- Wu, S.-G.; Sun, J.-Y.; Yang, L.-C.; Tang, L.-Y.; Wang, X.; Chen, X.-T.; Liu, G.-H.; Lin, H.-X.; Lin, Q.; He, Z.-Y. Patterns of distant metastasis in Chinese women according to breast cancer subtypes. Oncotarget 2016, 7, 47975–47984. [Google Scholar] [CrossRef]
- Buonomo, O.C.; Caredda, E.; Portarena, I.; Vanni, G.; Orlandi, A.; Bagni, C.; Petrella, G.; Palombi, L.; Orsaria, P. New insights into the metastatic behavior after breast cancer surgery, according to well-established clinicopathological variables and molecular subtypes. PLoS ONE 2017, 12, e0184680. [Google Scholar] [CrossRef] [Green Version]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-Associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J.; et al. Cancer-Associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Bado, I.; Wang, H.; Zhang, W.; Rosen, J.M.; Zhang, X.H.-F. Metastasis Organotropism: Redefining the Congenial Soil. Dev. Cell 2019, 49, 375–391. [Google Scholar] [CrossRef]
- Hill, B.S.; Sarnella, A.; D’Avino, G.; Zannetti, A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin. Cancer Biol. 2020, 60, 202–213. [Google Scholar] [CrossRef]
- Davis, D.M.; Sowinski, S. Membrane nanotubes: Dynamic long-distance connections between animal cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 431–436. [Google Scholar] [CrossRef]
- Vignais, M.-L.; Caicedo, A.; Brondello, J.-M.; Jorgensen, C. Cell Connections by Tunneling Nanotubes: Effects of Mitochondrial Trafficking on Target Cell Metabolism, Homeostasis, and Response to Therapy. Stem Cells Int. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gurke, S.; Barroso, J.F.V.; Gerdes, H.-H. The art of cellular communication: Tunneling nanotubes bridge the divide. Histochem. Cell Biol. 2008, 129, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A.S. Tunneling Nanotubes Provide a Unique Conduit for Intercellular Transfer of Cellular Contents in Human Malignant Pleural Mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Lou, E. A Ticket to Ride: The Implications of Direct Intercellular Communication via Tunneling Nanotubes in Peritoneal and Other Invasive Malignancies. Front. Oncol. 2020, 10, 559548. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Imparato, V.; Masone, S.; Masullo, M.; Nasso, R.; Montagnani, S.; Arcucci, A. Involvement of Breast Cancer-Associated Fibroblasts in Tumor Development, Therapy Resistance and Evaluation of Potential Therapeutic Strategies. Curr. Med. Chem. 2018, 25, 3414–3434. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein-Achiasaf, L.; Morein, D.; Ben-Yaakov, H.; Liubomirski, Y.; Meshel, T.; Elbaz, E.; Dorot, O.; Pichinuk, E.; Gershovits, M.; Weil, M.; et al. Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers 2021, 13, 1472. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Ruocco, M.R.; Martucci, N.; Vecchio, E.; Insabato, L.; Russo, D.; Accurso, A.; Masone, S.; Montagnani, S.; et al. Influence of Fibroblasts on Mammary Gland Development, Breast Cancer Microenvironment Remodeling, and Cancer Cell Dissemination. Cancers 2020, 12, 1697. [Google Scholar] [CrossRef]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.; Franzen, C.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1–TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2014, 34, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maishi, N.; Annan, D.A.; Kikuchi, H.; Hida, Y.; Hida, K. Tumor Endothelial Heterogeneity in Cancer Progression. Cancers 2019, 11, 1511. [Google Scholar] [CrossRef] [Green Version]
- Madu, C.O.; Wang, S.; Madu, C.O.; Lu, Y. Angiogenesis in Breast Cancer Progression, Diagnosis, and Treatment. J. Cancer 2020, 11, 4474–4494. [Google Scholar] [CrossRef]
- Nagl, L.; Horvath, L.; Pircher, A.; Wolf, D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironment—New Findings and Future Perspectives. Front. Cell Dev. Biol. 2020, 8, 766. [Google Scholar] [CrossRef]
- Singhal, M.; Augustin, H.G. Beyond Angiogenesis: Exploiting Angiocrine Factors to Restrict Tumor Progression and Metastasis. Cancer Res. 2019, 80, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Halabi, N.; Guerrouahen, B.S.; Rafii, S.; Rafii, A. Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J. Transl. Med. 2015, 13, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmura-Kakutani, H.; Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Kawamoto, T.; Iida, J.; Shindoh, M.; Tsuchiya, K.; Shinohara, N.; et al. Identification of Tumor Endothelial Cells with High Aldehyde Dehydrogenase Activity and a Highly Angiogenic Phenotype. PLoS ONE 2014, 9, e113910. [Google Scholar] [CrossRef] [PubMed]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al. Heterogeneity of Tumor Endothelial Cells. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef]
- Bussolati, B.; Assenzio, B.; Deregibus, M.C.; Camussi, G. The proangiogenic phenotype of human tumor-derived endothelial cells depends on thrombospondin-1 downregulation via phosphatidylinositol 3-kinase/Akt pathway. J. Mol. Med. 2006, 84, 852–863. [Google Scholar] [CrossRef]
- Fujisaki, K.; Fujimoto, H.; Sangai, T.; Nagashima, T.; Sakakibara, M.; Shiina, N.; Kuroda, M.; Aoyagi, Y.; Miyazaki, M. Cancer-Mediated adipose reversion promotes cancer cell migration via IL-6 and MCP-1. Breast Cancer Res. Treat. 2015, 150, 255–263. [Google Scholar] [CrossRef] [PubMed]
- D’Esposito, V.; Liguoro, D.; Ambrosio, M.R.; Collina, F.; Cantile, M.; Spinelli, R.; Raciti, G.; Miele, C.; Valentino, R.; Campiglia, P.; et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 2016, 7, 24495–24509. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Santander, A.M.; Lopez-Ocejo, O.; Casas, O.; Agostini, T.; Sanchez, L.; Lamas-Basulto, E.; Carrio, R.; Cleary, M.P.; Gonzalez-Perez, R.R.; Torroella-Kouri, M. Paracrine Interactions between Adipocytes and Tumor Cells Recruit and Modify Macrophages to the Mammary Tumor Microenvironment: The Role of Obesity and Inflammation in Breast Adipose Tissue. Cancers 2015, 7, 143–178. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2015, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lee, J.S.; Jie, C.; Park, M.H.; Iwakura, Y.; Patel, Y.; Soni, M.; Reisman, D.; Chen, H. HER2 Overexpression Triggers an IL1α Proinflammatory Circuit to Drive Tumorigenesis and Promote Chemotherapy Resistance. Cancer Res. 2018, 78, 2040–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Goto, H.; Shimono, Y.; Funakoshi, Y.; Imamura, Y.; Toyoda, M.; Kiyota, N.; Kono, S.; Takao, S.; Mukohara, T.; Minami, H. Adipose-Derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene 2018, 38, 767–779. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- Attané, C.; Milhas, D.; Hoy, A.; Muller, C. Metabolic Remodeling Induced by Adipocytes: A New Achilles’ Heel in Invasive Breast Cancer? Curr. Med. Chem. 2020, 27, 3984–4001. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Ebert, M.P.; Dooley, S.; Meyer, C. Caveolin-1 in the regulation of cell metabolism: A cancer perspective. Mol. Cancer 2016, 15, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Graney, P.L.; Tavakol, D.N.; Chramiec, A.; Ronaldson-Bouchard, K.; Vunjak-Novakovic, G. Engineered models of tumor metastasis with immune cell contributions. iScience 2021, 24, 102179. [Google Scholar] [CrossRef]
- Müller, L.; Tunger, A.; Plesca, I.; Wehner, R.; Temme, A.; Westphal, D.; Meier, F.; Bachmann, M.; Schmitz, M. Bidirectional Crosstalk Between Cancer Stem Cells and Immune Cell Subsets. Front. Immunol. 2020, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.-Q.; Waaijer, S.J.; Zwager, M.C.; de Vries, E.; van der Vegt, B.; Schröder, C.P. Tumor-Associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tan, W.; Wang, C. Tumor-Associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial–mesenchymal transition. OncoTargets Ther. 2018, 11, 3817–3826. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Fang, W.; Zhou, T.; Shi, H.; Yao, M.; Zhang, D.; Qian, H.; Zeng, Q.; Wang, Y.; Jin, F.; Chai, C.; et al. Progranulin induces immune escape in breast cancer via up-regulating PD-L1 expression on tumor-associated macrophages (TAMs) and promoting CD8+ T cell exclusion. J. Exp. Clin. Cancer Res. 2021, 40, 1–11. [Google Scholar] [CrossRef]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor Entrained Neutrophils Inhibit Seeding in the Premetastatic Lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güç, E.; Pollard, J.W. Redefining macrophage and neutrophil biology in the metastatic cascade. Immunity 2021, 54, 885–902. [Google Scholar] [CrossRef]
- Ma, X.; Wang, M.; Yin, T.; Zhao, Y.; Wei, X. Myeloid-Derived Suppressor Cells Promote Metastasis in Breast Cancer After the Stress of Operative Removal of the Primary Cancer. Front. Oncol. 2019, 9, 855. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Meng, M.; Wang, G.; Han, R.; Zhang, Y.; Jing, X.; Zhao, L.; Gu, S.; Zhao, X. Myeloid-Derived Suppressor Cells Recruited by Chemokine (C-C Motif) Ligand 3 Promote the Progression of Breast Cancer via Phosphoinositide 3-Kinase-Protein Kinase B-Mammalian Target of Rapamycin Signaling. J. Breast Cancer 2020, 23, 141–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, N.M.; Martinez, L.M.; Murdock, S.; Deligio, J.T.; Olex, A.L.; Effi, C.; Dozmorov, M.G.; Bos, P.D. Regulatory T Cells Support Breast Cancer Progression by Opposing IFN-γ-Dependent Functional Reprogramming of Myeloid Cells. Cell Rep. 2020, 33, 108482. [Google Scholar] [CrossRef]
- Yang, P.; Li, Q.-J.; Feng, Y.; Zhang, Y.; Markowitz, G.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-β-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karavitis, J.; Hix, L.M.; Shi, Y.H.; Schultz, R.F.; Khazaie, K.; Zhang, M. Regulation of COX2 Expression in Mouse Mammary Tumor Cells Controls Bone Metastasis and PGE2-Induction of Regulatory T Cell Migration. PLoS ONE 2012, 7, e46342. [Google Scholar] [CrossRef] [PubMed]
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4+ T Cells to T-Regulatory Cells. Cancer Res. 2011, 71, 3505–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edechi, C.A.; Ikeogu, N.; Uzonna, J.E.; Myal, Y. Regulation of Immunity in Breast Cancer. Cancers 2019, 11, 1080. [Google Scholar] [CrossRef] [Green Version]
- Batalha, S.; Ferreira, S.; Brito, C. The Peripheral Immune Landscape of Breast Cancer: Clinical Findings and In Vitro Models for Biomarker Discovery. Cancers 2021, 13, 1305. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.-C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Paget, S. The Distribution of Secondary Growths in Cancer of the Breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Cho, E.S.; Kim, N.H.; Yun, J.S.; Cho, S.B.; Kim, H.S.; Yook, J.I. Breast Cancer Subtypes Underlying EMT-Mediated Catabolic Metabolism. Cells 2020, 9, 2064. [Google Scholar] [CrossRef]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2018, 194, 161–184. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christo-fori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktas, B.; Tewes, M.; Fehm, T.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [Green Version]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Moodie, A.; Blanchard, A.A.A.; Leygue, E.; Myal, Y. Claudin 1 in Breast Cancer: New Insights. J. Clin. Med. 2015, 4, 1960–1976. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, D.G.; Duyverman, A.M.M.J.; Kohno, M.; Snuderl, M.; Steller, E.J.A.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J.; Obenauf, A. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Banyard, J.; Bielenberg, D.R. The role of EMT and MET in cancer dissemination. Connect. Tissue Res. 2015, 56, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Wu, Q.; Shepard, C.; Wells, A. Hepatocyte induced re-expression of E-cadherin in breast and prostate cancer cells increases chemoresistance. Clin. Exp. Metastasis 2011, 29, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Gunasinghe, N.P.A.D.; Wells, A.; Thompson, E.W.; Hugo, H. Mesenchymal–Epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Rodrigues, G.; Hoshino, A.; Kenific, C.M.; Matei, I.R.; Steiner, L.; Freitas, D.; Kim, H.S.; Oxley, P.R.; Scandariato, I.; Casanova-Salas, I.; et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nature 2019, 21, 1403–1412. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Eisenblaetter, M.; Flores-Borja, F.; Lee, J.J.; Wefers, C.; Smith, H.; Hueting, R.; Cooper, M.S.; Blower, P.J.; Patel, D.; Rodríguez-Justo, M.; et al. Visualization of Tumor-Immune Interaction—Target-Specific Imaging of S100A8/A9 Reveals Pre-Metastatic Niche Establishment. Theranostics 2017, 7, 2392–2401. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Ochi, H.; Sunamura, S.; Kosaka, N.; Mabuchi, Y.; Fukuda, T.; Yao, K.; Kanda, H.; Ae, K.; Okawa, A.; et al. Cancer-Secreted hsa-miR-940 induces an osteoblastic phenotype in the bone metastatic microenvironment via targeting ARHGAP1 and FAM134A. Proc. Natl. Acad. Sci. USA 2018, 115, 2204–2209. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-Secreted miR-105 Destroys Vascular Endothelial Barriers to Promote Metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer Exosomes Perform Cell-Independent MicroRNA Biogenesis and Promote Tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [Green Version]
- Di Modica, M.; Regondi, V.; Sandri, M.; Iorio, M.; Zanetti, A.; Tagliabue, E.; Casalini, P.; Triulzi, T. Breast cancer-secreted miR-939 downregulates VE-cadherin and destroys the barrier function of endothelial monolayers. Cancer Lett. 2016, 384, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Martínez, A.; De Miguel-Pérez, D.; Ortega, F.G.; García-Puche, J.L.; Robles-Fernández, I.; Exposito, J.; Martorell-Marugan, J.; Carmona-Sáez, P.; Garrido-Navas, M.D.C.; Rolfo, C.; et al. Exosomal miRNA profile as complementary tool in the diagnostic and prediction of treatment response in localized breast cancer under neoadjuvant chemotherapy. Breast Cancer Res. 2019, 21, 1–9. [Google Scholar] [CrossRef]
- Guo, L.; Zhu, Y.; Li, L.; Zhou, S.; Yin, G.; Yu, G.; Cui, H. Breast cancer cell-derived exosomal miR-20a-5p promotes the proliferation and differentiation of osteoclasts by targeting SRCIN1. Cancer Med. 2019, 8, 5687–5701. [Google Scholar] [CrossRef] [Green Version]
- Ekström, E.J.; Bergenfelz, C.; von Bülow, V.; Serifler, F.; Carlemalm, E.; Jönsson, G.; Andersson, T.; Leandersson, K. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Hajjar, K.A. The annexin A2 system and angiogenesis. Biol. Chem. 2016, 397, 1005–1016. [Google Scholar] [CrossRef]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, P.M.; Hare, R.J.; Gryczynski, I.; Vishwanatha, J. Exosomal Annexin II Promotes Angiogenesis and Breast Cancer Metastasis. Mol. Cancer Res. 2016, 15, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzi, S.; Hebda, J.K.; Gavard, J. Vascular Permeability and Drug Delivery in Cancers. Front. Oncol. 2013, 3, 211. [Google Scholar] [CrossRef] [Green Version]
- Theodoro, T.R.; Matos, L.L.; Cavalheiro, R.P.; Justo, G.Z.; Nader, H.; Pinhal, M.A.S. Crosstalk between tumor cells and lymphocytes modulates heparanase expression. J. Transl. Med. 2019, 17, 103. [Google Scholar] [CrossRef] [PubMed]
- Tiedemann, K.; Sadvakassova, G.; Mikolajewicz, N.; Juhas, M.; Sabirova, Z.; Tabariès, S.; Gettemans, J.; Siegel, P.M.; Komarova, S.V. Exosomal Release of L-Plastin by Breast Cancer Cells Facilitates Metastatic Bone Osteolysis. Transl. Oncol. 2018, 12, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Luo, X.; Lv, W.; Hu, W.; Zhao, C.; Xiong, M.; Yi, Y.; Wang, D.; Wang, Y.; Wang, H.; et al. Tumor-derived exosomal components: The multifaceted roles and mechanisms in breast cancer metastasis. Cell Death Dis. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-Metastatic niches: Organ-Specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061. [Google Scholar] [CrossRef]
- Pulido, C.; Vendrell, I.; Ferreira, A.R.; Casimiro, S.; Mansinho, A.; Alho, I.; Costa, L. Bone metastasis risk factors in breast cancer. Ecancermedicalscience 2017, 11, 715. [Google Scholar] [CrossRef] [Green Version]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Funari, A.; Alimandi, M.; Pierelli, L.; Pino, V.; Gentileschi, S.; Sacchetti, B. Human Sinusoidal Subendothelial Cells Regulate Homing and Invasion of Circulating Metastatic Prostate Cancer Cells to Bone Marrow. Cancers 2019, 11, 763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göbel, A.; Dell’Endice, S.; Jaschke, N.; Pählig, S.; Shahid, A.; Hofbauer, L.; Rachner, T. The Role of Inflammation in Breast and Prostate Cancer Metastasis to Bone. Int. J. Mol. Sci. 2021, 22, 5078. [Google Scholar] [CrossRef] [PubMed]
- Hinz, N.; Baranowsky, A.; Horn, M.; Kriegs, M.; Sibbertsen, F.; Smit, D.; Clezardin, P.; Lange, T.; Schinke, T.; Jücker, M. Knockdown of AKT3 Activates HER2 and DDR Kinases in Bone-Seeking Breast Cancer Cells, Promotes Metastasis In Vivo and Attenuates the TGFβ/CTGF Axis. Cells 2021, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Rixiati, Y.; Huang, H.; Shi, Y.; Huang, C.; Jiao, B. Asperolide A prevents bone metastatic breast cancer via the PI3K/AKT/mTOR/c-Fos/NFATc1 signaling pathway. Cancer Med. 2020, 9, 8173–8185. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Kang, Y. Metastatic niche functions and therapeutic opportunities. Nat. Cell Biol. 2018, 20, 868–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guise, T.A. The Vicious Cycle of Bone Metastases. J. Musculoskelet. Neuronal Interact. 2002, 2, 570–572. [Google Scholar]
- Cheng, J.; Frye, J.; Whitman, S.; Kunihiro, A.; Pandey, R.; Funk, J. A Role for TGFβ Signaling in Preclinical Osteolytic Estrogen Receptor-Positive Breast Cancer Bone Metastases Progression. Int. J. Mol. Sci. 2021, 22, 4463. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Qiao, H.; Tang, T. Engineering 3D approaches to model the dynamic microenvironments of cancer bone metastasis. Bone Res. 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersini, S.; Jeon, J.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials 2013, 35, 2454–2461. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.; Allan, A.L. Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int. J. Mol. Sci. 2019, 20, 2272. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, S.; Goel, S.; Kamoun, W.S.; Maru, Y.; Fukumura, D.; Duda, D.G.; Jain, R.K. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc. Natl. Acad. Sci. USA 2011, 108, 3725–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Khan, S.; Shukla, S.; Lakra, A.D.; Kumar, S.; Das, G.; Maurya, R.; Meeran, S.M. Cucurbitacin B inhibits breast cancer metastasis and angiogenesis through VEGF-mediated suppression of FAK/MMP-9 signaling axis. Int. J. Biochem. Cell Biol. 2016, 77, 41–56. [Google Scholar] [CrossRef]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.; Manova-Todorova, K.; Massague, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Zha, H.; Long, H.; Wang, X.; Yang, F.; Gao, J.; Hu, C.; Zhou, L.; Guo, B.; Zhu, B. C3a-C3aR signaling promotes breast cancer lung metastasis via modulating carcinoma associated fibroblasts. J. Exp. Clin. Cancer Res. 2020, 39, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Keklikoglou, I.; Cianciaruso, C.; Güç, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nature 2018, 21, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-Positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nat. Cell Biol. 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Kersten, K.; Coffelt, S.; Hoogstraat, M.; Verstegen, N.; Vrijland, K.; Ciampricotti, M.; Doornebal, C.W.; Hau, C.-S.; Wellenstein, M.D.; Salvagno, C.; et al. Mammary tumor-derived CCL2 enhances pro-metastatic systemic inflammation through upregulation of IL1β in tumor-associated macrophages. OncoImmunology 2017, 6, e1334744. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Nosaka, T.; Nakamoto, Y.; Baba, T. Lung Macrophages: Multifunctional Regulator Cells for Metastatic Cells. Int. J. Mol. Sci. 2018, 20, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bale, R.; Putzer, D.; Schullian, P. Local Treatment of Breast Cancer Liver Metastasis. Cancers 2019, 11, 1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Feng, Y.; Lin, S.; Chen, J.; Lin, H.; Liang, X.; Zheng, H.; Cai, X. Mechanisms involved in breast cancer liver metastasis. J. Transl. Med. 2015, 13, 64. [Google Scholar] [CrossRef] [Green Version]
- Furusato, B.; Mohamed, A.; Uhlén, M.; Rhim, J.S. CXCR4 and Cancer. Pathol. Int. 2010, 60, 497–505. [Google Scholar] [CrossRef]
- Hembruff, S.L.; Jokar, I.; Yang, L.; Cheng, N. Loss of Transforming Growth Factor-β Signaling in Mammary Fibroblasts Enhances CCL2 Secretion to Promote Mammary Tumor Progression through Macrophage-Dependent and -Independent Mechanisms. Neoplasia 2010, 12, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Van den Eynden, G.G.; Majeed, A.W.; Illemann, M.; Vermeulen, P.B.; Bird, N.C.; Høyer-Hansen, G.; Eefsen, R.L.; Reynolds, A.R.; Brodt, P. The Multifaceted Role of the Microenvironment in Liver Metastasis: Biology and Clinical Implications. Cancer Res. 2013, 73, 2031–2043. [Google Scholar] [CrossRef] [Green Version]
- Tabariès, S.; Dupuy, F.; Dong, Z.; Monast, A.; Annis, M.G.; Spicer, J.; Ferri, L.E.; Omeroglu, A.; Basik, M.; Amir, E.; et al. Claudin-2 Promotes Breast Cancer Liver Metastasis by Facilitating Tumor Cell Interactions with Hepatocytes. Mol. Cell. Biol. 2012, 32, 2979–2991. [Google Scholar] [CrossRef] [Green Version]
- Eichler, A.F.; Chung, E.; Kodack, D.P.; Loeffler, J.S.; Fukumura, D.; Jain, R.K. The biology of brain metastases—Translation to new therapies. Nat. Rev. Clin. Oncol. 2011, 8, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–Endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Pedrosa, R.M.S.M.; Mustafa, D.A.; Soffietti, R.; Kros, J.M. Breast cancer brain metastasis: Molecular mechanisms and directions for treatment. Neuro Oncol. 2018, 20, 1439–1449. [Google Scholar] [CrossRef]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. Br. J. Pharmacol. 2006, 27, 697–709. [Google Scholar] [CrossRef]
- Sevenich, L.; Bowman, R.L.; Mason, S.D.; Quail, D.F.; Rapaport, F.; Elie, B.T.; Brogi, E.; Brastianos, P.; Hahn, W.C.; Holsinger, L.J.; et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nature 2014, 16, 876–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obenauf, A.; Massagué, J. Surviving at a Distance: Organ-Specific Metastasis. Trends Cancer 2015, 1, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Hou, Z.; Endsley, M.P.; Gronseth, E.I.; Rarick, K.R.; Jorns, J.M.; Yang, Q.; Du, Z.; Yan, K.; Bordas, M.L.; et al. Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-β2/ANGPTL4 axes. NPJ Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- González-Angulo, A.M.; Ferrer-Lozano, J.; Stemke-Hale, K.; Sahin, A.; Liu, S.; Barrera, J.A.; Burgues, O.; Lluch, A.; Chen, H.; Hortobagyi, G.N.; et al. PI3K Pathway Mutations and PTEN Levels in Primary and Metastatic Breast Cancer. Mol. Cancer Ther. 2011, 10, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.; Zhang, Q.; Huang, W.-C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma–Astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Hoffmann, A.D.; Liu, H.; Liu, X. Organotropism: New insights into molecular mechanisms of breast cancer metastasis. NPJ Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pukrop, T.; Dehghani, F.; Chuang, H.-N.; Lohaus, R.; Bayanga, K.; Heermann, S.; Regen, T.; Van Rossum, D.; Klemm, F.; Schulz, M.; et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia 2010, 58, 1477–1489. [Google Scholar] [CrossRef]
- Friedman, G.; Levi-Galibov, O.; David, E.; Bornstein, C.; Giladi, A.; Dadiani, M.; Mayo, A.; Halperin, C.; Pevsner-Fischer, M.; Lavon, H.; et al. Cancer-Associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat. Rev. Cancer 2020, 1, 692–708. [Google Scholar] [CrossRef]
- Bonneau, C.; Eliès, A.; Kieffer, Y.; Bourachot, B.; Ladoire, S.; Pelon, F.; Hequet, D.; Guinebretière, J.-M.; Blanchet, C.; Vincent-Salomon, A.; et al. A subset of activated fibroblasts is associated with distant relapse in early luminal breast cancer. Breast Cancer Res. 2020, 22, 1–22. [Google Scholar] [CrossRef]
- Kim, H.M.; Jung, W.H.; Koo, J.S. Expression of cancer-associated fibroblast related proteins in metastatic breast cancer: An immunohistochemical analysis. J. Transl. Med. 2015, 13, 222. [Google Scholar] [CrossRef] [Green Version]
- Lopatina, T.; Grange, C.; Cavallari, C.; Navarro-Tableros, V.; Lombardo, G.; Rosso, A.; Cedrino, M.; Pomatto, M.A.C.; Koni, M.; Veneziano, F.; et al. Targeting IL-3Rα on tumor-derived endothelial cells blunts metastatic spread of triple-negative breast cancer via extracellular vesicle reprogramming. Oncogenesis 2020, 9, 1–14. [Google Scholar] [CrossRef]
- Vasseur, A.; Cabel, L.; Tredan, O.; Chevrier, M.; Dubot, C.; Lorgis, V.; Jacot, W.; Goncalves, A.; Debled, M.; Levy, C.; et al. Prognostic value of CEC count in HER2-negative metastatic breast cancer patients treated with bevacizumab and chemotherapy: A prospective validation study (UCBG COMET). Angiogenesis 2019, 23, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.L.; Danforth, D.N. The Role of Immune Cells in Breast Tissue and Immunotherapy for the Treatment of Breast Cancer. Clin. Breast Cancer 2020, 21, e63–e73. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Montero, C.M.; Salem, M.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2008, 58, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Roxå, A.; Mehmeti, M.; Leandersson, K.; Larsson, A.-M. Clinical relevance of systemic monocytic-MDSCs in patients with metastatic breast cancer. Cancer Immunol. Immunother. 2020, 69, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Montero, A.J.; Diaz-Montero, C.M.; Deutsch, Y.E.; Hurley, J.; Koniaris, L.G.; Rumboldt, T.; Yasir, S.; Jorda, M.; Garret-Mayer, E.; Avisar, E.; et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II–IIIc breast cancer. Breast Cancer Res. Treat. 2011, 132, 215–223. [Google Scholar] [CrossRef]
- Jeong, H.; Hwang, I.; Kang, S.H.; Shin, H.C.; Kwon, S.Y. Tumor-Associated Macrophages as Potential Prognostic Biomarkers of Invasive Breast Cancer. J. Breast Cancer 2019, 22, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, J.; Zheng, R.; Shao, Q.; Gao, W.; Song, B.; Chen, X.; Qu, X. Regulatory T cells are an important prognostic factor in breast cancer: A systematic review and meta-analysis. Neoplasma 2016, 63, 789–798. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Peng, F.; Ai, L.; Mu, S.; Li, Y.; Yang, C.; Hu, Y. Tumor-infiltrating B cells as a favorable prognostic biomarker in breast cancer: A systematic review and meta-analysis. Cancer Cell Int. 2021, 21, 1–8. [Google Scholar] [CrossRef]
- Zhou, Y.; Shao, N.; Aierken, N.; Xie, C.; Ye, R.; Qian, X.; Hu, Z.; Zhang, J.; Lin, Y. Prognostic value of tumor-infiltrating Foxp3+ regulatory T cells in patients with breast cancer: A meta-analysis. J. Cancer 2017, 8, 4098–4105. [Google Scholar] [CrossRef] [PubMed]
- Stenström, J.; Hedenfalk, I.; Hagerling, C. Regulatory T lymphocyte infiltration in metastatic breast cancer—an independent prognostic factor that changes with tumor progression. Breast Cancer Res. 2021, 23, 1–12. [Google Scholar] [CrossRef]
- Van Der Spek, Y.M.; Kroep, J.R.; Tollenaar, R.A.E.M.; Mesker, W.E. Chemotherapy resistance and stromal targets in breast cancer treatment: A review. Mol. Biol. Rep. 2020, 47, 8169–8177. [Google Scholar] [CrossRef]
- Truffi, M.; Mazzucchelli, S.; Bonizzi, A.; Sorrentino, L.; Allevi, R.; Vanna, R.; Morasso, C.; Corsi, F. Nano-Strategies to Target Breast Cancer-Associated Fibroblasts: Rearranging the Tumor Microenvironment to Achieve Antitumor Efficacy. Int. J. Mol. Sci. 2019, 20, 1263. [Google Scholar] [CrossRef] [Green Version]
- Meng, M.; Wang, W.; Yan, J.; Tan, J.; Liao, L.; Shi, J.; Wei, C.; Xie, Y.; Jin, X.; Yang, L.; et al. Immunization of stromal cell targeting fibroblast activation protein providing immunotherapy to breast cancer mouse model. Tumor Biol. 2016, 37, 10317–10327. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Guo, J.; Guo, Q.-Q.; Xie, Y.; Dong, L.; Zhou, Y.; Liu, C.-L.; Yu, B.; Wu, H.; Wu, J.-X.; et al. A DNA vaccine expressing an optimized secreted FAPα induces enhanced anti-tumor activity by altering the tumor microenvironment in a murine model of breast cancer. Vaccine 2019, 37, 4382–4391. [Google Scholar] [CrossRef]
- Sharma, M.; Turaga, R.C.; Yuan, Y.; Satyanarayana, G.; Mishra, F.; Bian, Z.; Liu, W.; Sun, L.; Yang, J.; Liu, Z.-R. Simultaneously targeting cancer-associated fibroblasts and angiogenic vessel as a treatment for TNBC. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Haugen, M.H.; Lingjærde, O.C.; Hedenfalk, I.; Garred, Ø.; Borgen, E.; Loman, N.; Hatschek, T.; Børresen-Dale, A.-L.; Naume, B.; Mills, G.B.; et al. Protein Signature Predicts Response to Neoadjuvant Treatment With Chemo-therapy and Bevacizumab in HER2-Negative Breast Cancers. JCO Precis. Oncol. 2021, 5, 286–306. [Google Scholar] [CrossRef]
- Robert, N.J.; Diéras, V.; Glaspy, J.; Brufsky, A.M.; Bondarenko, I.; Lipatov, O.N.; Perez, E.A.; Yardley, D.A.; Chan, S.Y.; Zhou, X.; et al. RIBBON-1: Randomized, Double-Blind, Placebo-Controlled, Phase III Trial of Chemotherapy with or Without Bevacizumab for First-Line Treatment of Human Epidermal Growth Factor Receptor 2–Negative, Locally Recurrent or Metastatic Breast Cancer. J. Clin. Oncol. 2011, 29, 1252–1260. [Google Scholar] [CrossRef]
- Rashid, M.H.; Borin, T.F.; Ara, R.; Alptekin, A.; Liu, Y.; Arbab, A.S. Generation of Novel Diagnostic and Therapeutic Exosomes to Detect and Deplete Protumorigenic M2 Macrophages. Adv. Ther. 2020, 3. [Google Scholar] [CrossRef]
- Kulkarni, A.; Chandrasekar, V.; Natarajan, S.K.; Ramesh, A.; Pandey, P.; Nirgud, J.; Bhatnagar, H.; Ashok, D.; Ajay, A.K.; Sengupta, S. A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat. Biomed. Eng. 2018, 2, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, A.; Brouillard, A.; Kumar, S.; Nandi, D.; Kulkarni, A. Dual inhibition of CSF1R and MAPK pathways using supramolecular nanoparticles enhances macrophage immunotherapy. Biomaterials 2019, 227, 119559. [Google Scholar] [CrossRef] [PubMed]
- Parveen, S.; Siddharth, S.; Cheung, L.S.; Kumar, A.; Shen, J.; Murphy, J.R.; Sharma, D.; Bishai, W.R. Therapeutic targeting with DABIL-4 depletes myeloid suppressor cells in 4T1 triple-negative breast cancer model. Mol. Oncol. 2021, 15, 1330–1344. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 Blockade Depletes and Selectively Reprograms Regulatory T Cells in Concert with Immunotherapy in Cancer Patients. Sci. Transl. Med. 2012, 4, 134. [Google Scholar] [CrossRef] [Green Version]
- Ali, K.; Soond, D.R.; Piñeiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, E.; Scurr, M.; Campbell, E.; Jones, E.; Godkin, A.; Gallimore, A. T-Cell modulation by cyclophosphamide for tumour therapy. Immunology 2018, 154, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Huijts, C.M.; Lougheed, S.M.; Bodalal, Z.; van Herpen, C.M.; Hamberg, P.; Tascilar, M.; Haanen, J.B.; Verheul, H.M.; de Gruijl, T.D.; van der Vliet, H.J.; et al. The Effect of Everolimus and Low-Dose Cyclophosphamide on Immune Cell Subsets in Patients with Metastatic Renal Cell Carcinoma: Results from a Phase I Clinical Trial. Cancer Immunol. Immunother. 2019, 68, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edechi, C.A.; Ikeogu, N.M.; Akaluka, G.N.; Terceiro, L.E.L.; Machado, M.; Salako, E.S.; Barazandeh, A.F.; Kung, S.K.P.; Uzonna, J.E.; Myal, Y. The Prolactin Inducible Protein Modulates Antitumor Immune Responses and Metastasis in a Mouse Model of Triple Negative Breast Cancer. Front. Oncol. 2021, 11, 639859. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Li, J.; Wang, C.; Lou, Z.; Gao, S.; Zhao, L.; Wang, S.; Chaulagain, A.; Zhang, M.; Li, X.; et al. Single cell RNA sequencing for breast cancer: Present and future. Cell Death Discov. 2021, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pal, B.; Chen, Y.; Vaillant, F.; Capaldo, B.D.; Joyce, R.; Song, X.; Bryant, V.L.; Penington, J.S.; Di Stefano, L.; Ribera, N.T.; et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021, 40, e107333. [Google Scholar] [CrossRef] [PubMed]
- McNamara, K.L.; Caswell-Jin, J.L.; Joshi, R.; Ma, Z.; Kotler, E.; Bean, G.R.; Kriner, M.; Zhou, Z.; Hoang, M.; Beechem, J.; et al. Spatial proteomic characterization of HER2-positive breast tumors through neoadjuvant therapy predicts response. Nat. Rev. Cancer 2021, 2, 400–413. [Google Scholar] [CrossRef]
- Estrada, M.; Rebelo, S.P.; Davies, E.J.; Pinto, M.; Pereira, H.A.; Santo, V.E.; Smalley, M.; Barry, S.T.; Gualda, E.J.; Alves, P.; et al. Modelling the tumour microenvironment in long-term microencapsulated 3D co-cultures recapitulates phenotypic features of disease progression. Biomaterials 2015, 78, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mejia, D.L.; Chiang, B.; Luker, K.E.; Luker, G.D. Hybrid collagen alginate hydrogel as a platform for 3D tumor spheroid invasion. Acta Biomater. 2018, 75, 213–225. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. Three-Dimensional Culture System of Cancer Cells Combined with Biomaterials for Drug Screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Nii, T.; Katayama, Y. Biomaterial-Assisted Regenerative Medicine. Int. J. Mol. Sci. 2021, 22, 8657. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terceiro, L.E.L.; Edechi, C.A.; Ikeogu, N.M.; Nickel, B.E.; Hombach-Klonisch, S.; Sharif, T.; Leygue, E.; Myal, Y. The Breast Tumor Microenvironment: A Key Player in Metastatic Spread. Cancers 2021, 13, 4798. https://doi.org/10.3390/cancers13194798
Terceiro LEL, Edechi CA, Ikeogu NM, Nickel BE, Hombach-Klonisch S, Sharif T, Leygue E, Myal Y. The Breast Tumor Microenvironment: A Key Player in Metastatic Spread. Cancers. 2021; 13(19):4798. https://doi.org/10.3390/cancers13194798
Chicago/Turabian StyleTerceiro, Lucas E. L., Chidalu A. Edechi, Nnamdi M. Ikeogu, Barbara E. Nickel, Sabine Hombach-Klonisch, Tanveer Sharif, Etienne Leygue, and Yvonne Myal. 2021. "The Breast Tumor Microenvironment: A Key Player in Metastatic Spread" Cancers 13, no. 19: 4798. https://doi.org/10.3390/cancers13194798
APA StyleTerceiro, L. E. L., Edechi, C. A., Ikeogu, N. M., Nickel, B. E., Hombach-Klonisch, S., Sharif, T., Leygue, E., & Myal, Y. (2021). The Breast Tumor Microenvironment: A Key Player in Metastatic Spread. Cancers, 13(19), 4798. https://doi.org/10.3390/cancers13194798