
Sulforaphane: A Broccoli Bioactive Phytocompound with Cancer Preventive Potential



Abstract
Simple Summary
Abstract
1. Introduction
2. Bioavailability and Pharmacokinetics of SFN
3. Toxicity Studies
4. Sulforaphane in Cancer Prevention and Intervention
4.1. Literature Search Methodology
4.2. Preclinical Studies (In Vitro and In Vivo)
4.2.1. Breast Cancer
4.2.2. Gastrointestinal Tract and Associate Cancers
Esophageal Cancer
Gastric Cancer
Small Intestine Cancer
Colon Cancer
Hepatocellular Cancer
Pancreatic Cancer
4.2.3. Gynecological Cancers
Cervical Cancer
Endometrial Cancer
Ovarian Cancer
4.2.4. Hematological Cancers
Leukemia
Lymphoma
4.2.5. Lung Cancer
4.2.6. Neural Cancer
4.2.7. Skin Cancer
4.2.8. Urogenital Cancers
Bladder Cancer
Prostate Cancer
4.3. Clinical Studies
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APAF-1 | apoptotic protease activating factor-1 |
| ARE | antioxidant response element |
| BSE | broccoli sprout extract |
| BSP | broccoli sprout powders |
| COX-2 | cyclooxygenase-2 |
| CYP | cytochrome P-450 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DCIS | ductal carcinoma in situ |
| DMH | dimethylhydrazine |
| DMNT | DNA methyltransferase |
| EMT | epithelial–mesenchymal transition |
| ER | estrogen receptor |
| ERK | extracellular signal-related kinase |
| FADD | Fas-associated death domain |
| GFN | glucoraphanin |
| GGT | γ-glutamyl transpeptidase |
| GSH | glutathione |
| GST | glutathione S-transferase |
| HDAC | histone deacytylase |
| HER | human growth factor receptor |
| HIF | hypoxia-inducible factor |
| HMOX1 | heme oxygenase 1 |
| HSP70 | heat shock protein 70 |
| IDC | invasive ductal carcinoma |
| IFN-γ | interferon-γ |
| IL | interleukin |
| i.p. | intraperitoneal |
| ITC | isothiocyanate |
| i.v. | intravenous |
| JNK | Jun NH2-terminal kinase |
| Keap1 | Kelch-like ECH-associated protein 1 |
| MAPK | mitogen-activated protein kinase |
| MEK1 | mitogen-activated protein kinase 1 |
| MMP | matrix metalloproteinase |
| NF-κB | nuclear factor-κB |
| NQO1 | NADPH-dependent oxidoreductase 1 |
| Nrf2 | nuclear factor erythroid-2-related factor 2 |
| NSCLCs | non-small cell lung cancer cells |
| PARP | poly (ADP-ribose) polymerase |
| PBMCs | peripheral blood mononuclear cells |
| PCNA | proliferating cell nuclear antigen |
| PR | progesterone receptor |
| PRIMSA | Preferred Reporting Item for Systemic Review and Meta-Analysis |
| PSA | prostate-specific antigen |
| PTEN | phosphatase and tensin homolog |
| PUMA | p53 upregulated modulator of apoptosis |
| ROS | reactive oxygen species |
| SF | sulforaphane |
| STAT3 | signal transducer and activator of transcription 3 |
| TGF-β | transforming growth factor-β |
| TNF-α | tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
| XRE | xenobiotic response element |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 68, 394–424. [Google Scholar] [CrossRef]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef]
- Sporn, M.B.; Dunlop, N.M.; Newton, D.L.; Smith, J.M. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids). Fed. Proc. 1976, 35, 1332–1338. [Google Scholar]
- Fahey, J.W.; Talalay, P.; Kensler, T.W. Notes from the field: “green” chemoprevention as frugal medicine. Cancer Prev. Res. 2012, 5, 179–188. [Google Scholar] [CrossRef]
- Aune, D.; Giovannucci, E.; Boffetta, P.; Fadnes, L.T.; Keum, N.; Norat, T.; Greenwood, D.C.; Riboli, E.; Vatten, L.J.; Tonstad, S. Fruit and vegetable intake and the risk of cardiovascular disease, total cancer and all-cause mortality-a systematic review and dose-response meta-analysis of prospective studies. Int. J. Epidemiol. 2017, 46, 1029–1056. [Google Scholar] [CrossRef]
- Amin, A.R.; Kucuk, O.; Khuri, F.R.; Shin, D.M. Perspectives for cancer prevention with natural compounds. J. Clin. Oncol. 2009, 27, 2712–2725. [Google Scholar] [CrossRef]
- Gullett, N.P.; Ruhul Amin, A.R.; Bayraktar, S.; Pezzuto, J.M.; Shin, D.M.; Khuri, F.R.; Aggarwal, B.B.; Surh, Y.J.; Kucuk, O. Cancer prevention with natural compounds. Semin. Oncol. 2010, 37, 258–281. [Google Scholar] [CrossRef]
- Liu, R.H. Health-promoting components of fruits and vegetables in the diet. Adv. Nutr. 2013, 4, 384S–392S. [Google Scholar] [CrossRef]
- Lee, K.W.; Bode, A.M.; Dong, Z. Molecular targets of phytochemicals for cancer prevention. Nat. Rev. Cancer. 2011, 11, 211–218. [Google Scholar] [CrossRef]
- Block, K.I.; Gyllenhaal, C.; Lowe, L.; Amedei, A.; Amin, A.R.M.R.; Amin, A.; Aquilano, K.; Arbiser, J.; Arreola, A.; Arzumanyan, A.; et al. Designing a broad-spectrum integrative approach for cancer prevention and treatment. Semin. Cancer Biol. 2015, 35, S276–S304. [Google Scholar] [CrossRef]
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2018, 128, 366–375. [Google Scholar] [CrossRef]
- Das, B.; Sarkar, N.; Bishayee, A.; Sinha, D. Dietary phytochemicals in the regulation of epithelial to mesenchymal transition and associated enzymes: A promising anticancer therapeutic approach. Semin. Cancer Biol. 2019, 56, 196–218. [Google Scholar] [CrossRef]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2019, S1044-579X, 30405–30455. [Google Scholar] [CrossRef]
- Fakhri, S.; Moradi, S.Z.; Farzaei, M.H.; Bishayee, A. Modulation of dysregulated cancer metabolism by plant secondary metabolites: A mechanistic review. Semin. Cancer Biol. 2020, S1044-579X, 30040–30047. [Google Scholar] [CrossRef]
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.R.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 9, 1451. [Google Scholar] [CrossRef]
- Tuli, H.S.; Tuorkey, M.J.; Thakral, F.; Sak, K.; Kumar, M.; Sharma, A.K.; Sharma, U.; Jain, A.; Aggarwal, V.; Bishayee, A. Molecular Mechanisms of Action of Genistein in Cancer: Recent Advances. Front. Pharmacol. 2019, 10, 1336. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Tania, M.; Srivastava, S.; Ritzer, E.E.; Pandey, A.; Aggarwal, D.; Barwal, T.S.; Jain, A.; Kaur, G.; et al. Molecular mechanisms of action of epigallocatechin gallate in cancer: Recent trends and advancement. Semin. Cancer Biol. 2020, 24, 30107. [Google Scholar] [CrossRef]
- Mirza, B.; Croley, C.R.; Ahmad, M.; Pumarol, J.; Das, N.; Sethi, G.; Bishayee, A. Mango (Mangifera indica L.): A magnificent plant with cancer preventive and anticancer therapeutic potential. Crit. Rev. Food 2020, 8, 2125–2151. [Google Scholar] [CrossRef]
- De Greef, D.; Barton, E.M.; Sandberg, E.N.; Croley, C.R.; Pumarol, J.; Wong, T.L.; Das, N.; Bishayee, A. Anticancer potential of garlic and its bioactive constituents: A systematic and comprehensive review. Semin. Cancer Biol. 2020, 73, 219–264. [Google Scholar] [CrossRef]
- Nouri, Z.; Fakhri, S.; Nouri, K.; Wallace, C.E.; Farzaei, M.H.; Bishayee, A. Targeting Multiple Signaling Pathways in Cancer: The Rutin Therapeutic Approach. Cancers 2020, 12, 2276. [Google Scholar] [CrossRef]
- Ghanbari-Movahed, M.; Jackson, G.; Farzaei, M.H.; Bishayee, A. A Sstematic Review of the Preventive and Therapeutic Effects of Naringin Against Human Malignancies. Front. Pharmacol. 2021, 12, 639840. [Google Scholar] [CrossRef]
- Wong, T.L.; Strandberg, K.R.; Croley, C.R.; Fraser, S.E.; Nagulapalli Venkata, K.C.; Fimognari, C.; Sethi, G.; Bishayee, A. Pomegranate bioactive constituents target multiple oncogenic and oncosuppressive signaling for cancer prevention and intervention. Semin. Cancer Biol. 2021, 73, 265–293. [Google Scholar] [CrossRef]
- Lin, H.J.; Probst-Hensch, N.M.; Louie, A.D.; Kau, I.H.; Witte, J.S.; Ingles, S.A.; Frankl, H.D.; Lee, E.R.; Haile, R.W. Glutathione transferase null genotype, broccoli, and lower prevalence of colorectal adenomas. Cancer Epidemiol. Biomark. Prev. 1998, 7, 647–652. [Google Scholar]
- Michaud, D.S.; Spiegelman, D.; Clinton, S.K.; Rimm, E.B.; Willett, W.C.; Giovannucci, E.L. Fruit and vegetable intake and incidence of bladder cancer in a male prospective cohort. J. Natl. Cancer Inst. 1999, 91, 605–613. [Google Scholar] [CrossRef]
- Cohen, J.H.; Kristal, A.R.; Stanford, J.L. Fruit and vegetable intakes and prostate cancer risk. J. Natl. Cancer Inst. 2000, 92, 61–68. [Google Scholar] [CrossRef]
- Ambrosone, C.B.; McCann, S.E.; Freudenheim, J.L.; Marshall, J.R.; Zhang, Y.; Shields, P.G. Breast cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. Nutrition 2004, 134, 1134–1138. [Google Scholar] [CrossRef]
- Mori, N.; Shimazu, T.; Sasazuki, S.; Nozue, M.; Mutoh, M.; Sawada, N.; Iwasaki, M.; Yamaji, T.; Inoue, M.; Takachi, R.; et al. Cruciferous Vegetable Intake Is Inversely Associated with Lung Cancer Risk among Current Nonsmoking Men in the Japan Public Health Center (JPHC) Study. Nutrients 2017, 147, 841–849. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: Metabolism and excretion in humans. Cancer Epidemiol. Biomark. Prev. 2001, 10, 501–508. [Google Scholar]
- Navarro, S.L.; Li, F.; Lampe, J.W. Mechanisms of action of isothiocyanates in cancer chemoprevention: An update. Food and Funct. 2011, 2, 579–587. [Google Scholar] [CrossRef]
- Zhang, Y. The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis 2012, 33, 2–9. [Google Scholar] [CrossRef]
- Gupta, P.; Kim, B.; Kim, S.H.; Srivastava, S.K. Molecular targets of isothiocyanates in cancer: Recent advances. Mol. Nutr. Food Res. 2014, 58, 1685–1707. [Google Scholar] [CrossRef]
- Kumar, G.; Tuli, H.S.; Mittal, S.; Shandilya, J.K.; Tiwari, A.; Sandhu, S.S. Isothiocyanates: A class of bioactive metabolites with chemopreventive potential. Tumor Biol. 2015, 36, 4005–4016. [Google Scholar] [CrossRef]
- Palliyaguru, D.L.; Yuan, J.M.; Kensler, T.W.; Fahey, J.W. Isothiocyanates: Translating the Power of Plants to People. Mol. Nutr. Food Res. 2018, 62, e1700965. [Google Scholar] [CrossRef]
- Singh, D.; Arora, R.; Bhatia, A.; Singh, H.; Singh, B.; Arora, S. Molecular targets in cancer prevention by 4-(methylthio)butyl isothiocyanate—A comprehensive review. Life Sci. 2020, 241, 117061. [Google Scholar] [CrossRef]
- Hudlikar, R.; Wang, L.; Wu, R.; Li, S.; Peter, R.; Shannar, A.; Chou, P.J.; Liu, X.; Liu, Z.; Kuo, H.D.; et al. Epigenetics/epigenomics and prevention of early stages of cancer by isothiocyanates. Cancer Prev. Res. 2021, 14, 151–164. [Google Scholar] [CrossRef]
- Xu, T.; Ren, D.; Sun, X.; Yang, G. Dual roles of sulforaphane in cancer treatment. Anticancer Agents Med. Chem. 2012, 12, 1132–1142. [Google Scholar] [CrossRef]
- Kumar, A.; Sabbioni, G. New biomarkers for monitoring the levels of isothiocyanates in humans. Chem. Res. Toxicol. 2010, 23, 756–765. [Google Scholar] [CrossRef]
- De Figueiredo, S.M.; Binda, N.S.; Nogueira-Machado, J.A.; Vieira-Filho, S.A.; Caligiorne, R.B. The antioxidant properties of organosulfur compounds (sulforaphane). Recent Pat. Endocr. Metab. Immune Drug Discov. 2015, 9, 24–39. [Google Scholar] [CrossRef]
- Houghton, C.A. Sulforaphane: Its “Coming of Age” as a Clinically Relevant Nutraceutical in the Prevention and Treatment of Chronic Disease. Oxid. Med. Cell. Longev. 2019, 2019, 2716870. [Google Scholar] [CrossRef]
- Mahn, A.; Castillo, A. Potential of Sulforaphane as a Natural Immune System Enhancer: A Review. Molecules 2021, 26, 752. [Google Scholar] [CrossRef]
- Zeren, S.; Bayhan, Z.; Kocak, F.E.; Kocak, C.; Akcılar, R.; Bayat, Z.; Simsek, H.; Duzgun, S.A. Gastroprotective effects of sulforaphane and thymoquinone against acetylsalicylic acid-induced gastric ulcer in rats. J. Surg. Res. 2016, 203, 348–359. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, X.; Zhao, S.; Ma, C.; Cui, J.; Zheng, Y. Sulforaphane Protects against Cardiovascular Disease via Nrf2 Activation. Oxid. Med. Cell. Longev. 2015, 2015, 407580. [Google Scholar] [CrossRef]
- Liebman, S.E.; Le, T.H. Eat Your Broccoli: Oxidative Stress, NRF2, and Sulforaphane in Chronic Kidney Disease. Nutrients 2021, 13, 266. [Google Scholar] [CrossRef] [PubMed]
- Santín-Márquez, R.; Alarcón-Aguilar, A.; López-Diazguerrero, N.E.; Chondrogianni, N.; Königsberg, M. Sulforaphane—Role in aging and neurodegeneration. GeroScience 2019, 41, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Schepici, G.; Bramanti, P.; Mazzon, E. Efficacy of Sulforaphane in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8637. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Kozumbo, W.J. The phytoprotective agent sulforaphane prevents inflammatory degenerative diseases and age-related pathologies via Nrf2-mediated hormesis. Pharmacol. Res. 2021, 163, 105283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Hrelia, P. Sulforaphane as a promising molecule for fighting cancer. Mutat. Res. 2007, 635, 90–104. [Google Scholar] [CrossRef]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, L. Discovery and development of sulforaphane as a cancer chemopreventive phytochemical. Acta Pharmacol. Sin. 2007, 28, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and Done? Targeting the NRF2 Pathway with Sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, Y.; Ma, L.; Ji, R.; Qu, Y.; Xin, Y.; Lv, G. Chemopreventive activity of sulforaphane. Drug Des. Dev. 2018, 12, 2905–2913. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Baluch, N.; Homayouni, T.S.; Morgatskaya, E.; Kumar, S.; Kazemi, P.; Yeger, H. The role of Sulforaphane in cancer chemoprevention and health benefits: A mini-review. Cell Commun. Signal. 2018, 12, 91–101. [Google Scholar] [CrossRef]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or Sulforaphane: Is It the Source or Dose That Matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef]
- Rafiei, H.; Ashrafizadeh, M.; Ahmadi, Z. MicroRNAs as novel targets of sulforaphane in cancer therapy: The beginning of a new tale? Phytother. Res. 2020, 34, 721–728. [Google Scholar] [CrossRef]
- Elkashty, O.A.; Tran, S.D. Sulforaphane as a Promising Natural Molecule for Cancer Prevention and Treatment. Curr. Med. Sci. 2021, 41, 250–269. [Google Scholar] [CrossRef]
- Kuran, D.; Pogorzelska, A.; Wiktorska, K. Breast Cancer Prevention-Is there a Future for Sulforaphane and Its Analogs? Nutrients 2020, 12, 1559. [Google Scholar] [CrossRef]
- Leone, A.; Diorio, G.; Sexton, W.; Schell, M.; Alexandrow, M.; Fahey, J.W.; Kumar, N.B. Sulforaphane for the chemoprevention of bladder cancer: Molecular mechanism targeted approach. Oncotarget 2017, 8, 35412–35424. [Google Scholar] [CrossRef] [PubMed]
- Amjad, A.I.; Parikh, R.A.; Appleman, L.J.; Hahm, E.R.; Singh, K.; Singh, S.V. Broccoli-Derived Sulforaphane and Chemoprevention of Prostate Cancer: From Bench to Bedside. Curr. Pharmacol. Rep. 2015, 1, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Calcabrini, C.; Maffei, F.; Turrini, E.; Fimognari, C. Sulforaphane Potentiates Anticancer Effects of Doxorubicin and Cisplatin and Mitigates Their Toxic Effects. Front. Pharmacol. 2020, 11, 567. [Google Scholar] [CrossRef]
- Kamal, M.M.; Akter, S.; Lin, C.; Nazzal, S. Sulforaphane as an anticancer molecule: Mechanisms of action, synergistic effects, enhancement of drug safety, and delivery systems. Arch. Pharm. Res. 2020, 43, 371–384. [Google Scholar] [CrossRef]
- Wu, G.; Yan, Y.; Zhou, Y.; Duan, Y.; Zeng, S.; Wang, X.; Lin, W.; Ou, C.; Zhou, J.; Xu, Z. Sulforaphane: Expected to Become a Novel Antitumor Compound. Oncol. Res. 2020, 28, 439–446. [Google Scholar] [CrossRef]
- Rathaur, P.; Johar, K.S.R. Metabolism and Pharmacokinetics of Phytochemicals in the Human Body. Curr. Drug Metab. 2019, 20, 1085–1102. [Google Scholar] [CrossRef]
- Fahey, J.W.; Holtzclaw, W.D.; Wehage, S.L.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Sulforaphane Bioavailability from Glucoraphanin-Rich Broccoli: Control by Active Endogenous Myrosinase. PLoS ONE 2015, 10, e0140963. [Google Scholar] [CrossRef]
- Sivapalan, T.; Melchini, A.; Saha, S.; Needs, P.W.; Traka, M.H.; Tapp, H.; Dainty, J.R.; Mithen, R.F. Bioavailability of Glucoraphanin and Sulforaphane from High-Glucoraphanin Broccoli. Mol. Nutr. Food Res. 2018, 62, e1700911. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Panjwani, A.A.; Liu, H.; Cornblatt, G.; Cornblatt, B.S.; Ownby, S.L.; Fuchs, E.; Holtzclaw, W.D.; et al. Bioavailability of Sulforaphane Following Ingestion of Glucoraphanin-Rich Broccoli Sprout and Seed Extracts with Active Myrosinase: A Pilot Study of the Effects of Proton Pump Inhibitor Administration. Nutrients 2019, 11, 1489. [Google Scholar] [CrossRef]
- Petri, N.; Tannergren, C.; Holst, B.; Mellon, F.A.; Bao, Y.; Plumb, G.W.; Bacon, J.; O’Leary, K.A.; Kroon, P.A.; Knutson, L.; et al. Absorption/metabolism of sulforaphane and quercetin, and regulation of phase II enzymes, in human jejunum in vivo. Drug Metab. Dispos. 2003, 31, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Hsu, A.; Riedl, K.; Bella, D.; Schwartz, S.J.; Stevens, J.F.; Ho, E. Bioavailability and inter-conversion of sulforaphane and erucin in human subjects consuming broccoli sprouts or broccoli supplement in a cross-over study design. Pharmacol. Res. 2011, 64, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Mennicke, W.H.; Görler, K.; Krumbiegel, G.; Lorenz, D.; Rittmann, N. Studies on the metabolism and excretion of benzyl isothiocyanate in man. Xenobiotica 1988, 18, 441–447. [Google Scholar] [CrossRef]
- Bricker, G.V.; Riedl, K.M.; Ralston, R.A.; Tober, K.L.; Oberyszyn, T.M.; Schwartz, S.J. Isothiocyanate metabolism, distribution, and interconversion in mice following consumption of thermally processed broccoli sprouts or purified sulforaphane. Mol. Nutr. Food Res. 2015, 58, 1991–2000. [Google Scholar] [CrossRef]
- Hanlon, N.; Coldham, N.; Gielbert, A.; Kuhnert, N.; Sauer, M.; King, L.; Ioannides, C. Absolute bioavailability and dose-dependent pharmacokinetic behaviour of dietary doses of the chemopreventive isothiocyanate sulforaphane in rat. Br. J. Nutr. 2008, 99, 559–564. [Google Scholar] [CrossRef]
- Zhang, Y.; Wade, K.L.; Prestera, T.; Talala, P. Quantitative determination of isothiocyanates, dithiocarbamates, carbon disulfide, and related thiocarbonyl compounds by cyclocondensation with 1,2-benzenedithiol. Anal. Biochem. 1996, 239, 160–167. [Google Scholar] [CrossRef]
- Cornblatt, B.; Ye, L.; Dinkova-Kostova, A.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef]
- Oliviero, T.; Lamers, S.; Capuano, E.; Dekker, M.; Verkerk, R. Bioavailability of Isothiocyanates from Broccoli Sprouts in Protein, Lipid, and Fiber Gels. Mol. Nutr. Food Res. 2018, 62, e1700837. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Atwell, L.L.; Hsu, A.; Wong, C.P.; Stevens, J.F.; Bella, D.; Yu, T.W.; Pereira, C.B.; Löhr, C.V.; Christensen, J.M.; Dashwood, R.H.; et al. Absorption and chemopreventive targets of sulforaphane in humans following consumption of broccoli sprouts or a myrosinase-treated broccoli sprout extract. Mol. Nutr. Food Res. 2015, 59, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Qian, Q.; Adaikalakoteswari, A.; Rabbani, N.; Babaei-Jadidi, R.; Thornalley, P.J. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes 2008, 57, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Gross-Steinmeyer, K.; Stapleton, P.L.; Tracy, J.H.; Bammler, T.K.; Strom, S.C.; Eaton, D.L. Sulforaphane- and phenethyl isothiocyanate-induced inhibition of aflatoxin B1-mediated genotoxicity in human hepatocytes: Role of GSTM1 genotype and CYP3A4 gene expression. Toxicol. Sci. 2010, 116, 422–432. [Google Scholar] [CrossRef]
- Abbaoui, B.; Riedl, K.M.; Ralston, R.A.; Thomas-Ahner, J.M.; Schwartz, S.J.; Clinton, S.K.; Mortazavi, A. Inhibition of bladder cancer by broccoli isothiocyanates sulforaphane and eruicin: Characterization, metabolism, and interconversion. Mol. Nutr. Food Res. 2012, 56, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, P.; Ragonese, F.; Stabile, A.; Pistilli, A.; Kuligina, E.; Rende, M.; Bottoni, U.; Calvieri, S.; Crisanti, A.; Spaccapelo, R. Antitumor activity and expression profiles of genes induced by sulforaphane in human melanoma cells. Eur. J. Nutr. 2018, 57, 2547–2569. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Tong, P.; Dashwood, W.M.; Dashwood, R.H.; Ho, E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp. Biol. Med. 2007, 232, 227–234. [Google Scholar]
- Socała, K.; Nieoczym, D.; Kowalczuk-Vasilev, E.; Wyska, E.; Wlaź, P. Increased seizure susceptibility and other toxicity symptoms following acute sulforaphane treatment in mice. Toxicol. Appl. Pharmacol. 2017, 326, 43–53. [Google Scholar] [CrossRef]
- Castro, N.P.; Rangel, M.C.; Merchant, A.S.; MacKinnon, G.; Cuttitta, F.; Salomon, D.S.; Kim, Y.S. Sulforaphane Suppresses the Growth of Triple-negative Breast Cancer Stem-like Cells In vitro and In vivo. Cancer Prev. Res. 2019, 12, 147–158. [Google Scholar] [CrossRef]
- Alumkal, J.J.; Slottke, R.; Schwartzman, J.; Cherala, G.; Munar, M.; Graff, J.N.; Beer, T.M.; Ryan, C.W.; Koop, D.R.; Gibbs, A.; et al. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Investig. New Drugs. 2015, 33, 480–489. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Stephenson, K.K.; Wade, K.L.; Ye, L.; Talalay, P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: A clinical phase I study. Nutr. Cancer. 2006, 55, 53–62. [Google Scholar] [CrossRef]
- Tahata, S.; Singh, S.V.; Lin, Y.; Hahm, E.; Beumer, J.H.; Christner, S.M.; Rao, U.N.; Sander, C.; Tarhini, A.A.; Tawbi, H.; et al. Evaluation of Biodistribution of Sulforaphane after Administration of Oral Broccoli Sprout Extract in Melanoma Patient with Multiple Atypical Nevi. Cancer Prev. Res. 2018, 11, 429–438. [Google Scholar] [CrossRef]
- Zhang, Z.; Garzotto, M.; Davis II, E.W.; Mori, M.; Stoller, W.A.; Farris, P.E.; Wong, C.P.; Beaver, L.M.; Thomas, G.V.; Williams, D.E.; et al. Sulforaphane Bioavailability and chemopreventive Activity in Men Presenting for Biopsy of the Prostate Gland: A Randomized Controlled Trial. Nutr. Cancer 2020, 72, 74–84. [Google Scholar] [CrossRef]
- Jeffery, E.H.; Keck, A.S. Translating knowledge generated by epidemiological and in vitro studies into dietary cancer prevention. Mol. Nutr. Food Res. 2008, 52, S7–S17. [Google Scholar]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic review and meta-analysis of studies that evaluate health care interventions: Explanation and elaboration. PLoS Med. 2009, 6, e1000100. [Google Scholar] [CrossRef]
- Tseng, E.; Scott-Ramsay, E.A.; Morris, M.E. Dietary organic isothiocyanates are cytotoxic in human breast cancer and mammary epithelial MCF-12A cell lines. Exp. Biol. Med. 2004, 229, 835–842. [Google Scholar] [CrossRef]
- Jackson, J.T.; Singletary, K.W. Sulforaphane inhibits human MCF-7 mammary cancer cell mitotic progression and tubulin polymerization. J. Nutr. 2004, 134, 2229–2236. [Google Scholar] [CrossRef]
- Jackson, J.T.; Singletary, K.W. Sulforaphane: A naturally occurring mammary carcinoma mitotic inhibitor, which disrupts tubulin polymerization. Carcinogenesis 2004, 25, 219–227. [Google Scholar] [CrossRef]
- Azarenko, O.; Okouneva, T.; Singletary, K.W.; Jordan, M.A.; Wilson, L. Suppression of microtubule dynamic instability and turnover in MCF7 breast cancer cells by sulforaphane. Carcinogenesis 2008, 29, 2360–2368. [Google Scholar] [CrossRef]
- Ahmed, Z.; Li, X.; Li, F.; Cheaito, H.A.; Patel, K.; Mosallam, E.M.; Elbargeesy, G.; Dou, Q.P. Computational and biochemical studies of isothiocyanates as inhibitors of proteasomal cysteine deubiquitinases in human cancer cells. J. Cell. Biochem. 2018, 119, 9006–9016. [Google Scholar] [CrossRef]
- Cao, C.; Wu, H.; Vasilatos, S.N.; Chandran, U.; Qin, Y.; Wan, Y.; Oesterreich, S.; Davidson, N.E.; Huang, Y. HDAC5-LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 2018, 143, 1388–1401. [Google Scholar] [CrossRef]
- Royston, K.J.; Paul, B.; Nozell, S.; Rajbhandari, R.; Tollefsbol, T. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp. Cell Res. 2018, 368, 67–74. [Google Scholar] [CrossRef]
- Pledgie-Tracey, A.; Sobolewski, M.; Davidson, N. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol. Cancer Ther. 2007, 6, 1013–1021. [Google Scholar] [CrossRef]
- Lewinska, A.; Bednarz, D.; Adamczyk-Grochala, J.; Wnuk, M. Phytochemical-induced nucleolar stress results in the inhibition of breast cancer cell proliferation. Redox. Biol. 2017, 12, 459–482. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Deregowska, A.; Wnuk, M. Sulforaphane-induced cell cycle arrest and senescence are accompanied by DNA hypomethylation and changes in microRNA profile in breast cancer cells. Theranostics 2017, 7, 3461–3477. [Google Scholar] [CrossRef]
- Kanematsu, S.; Uehara, N.; Miki, H.; Yoshizawa, K.; Kawanaka, A.; Yuri, T.; Tsubura, A. Autophagy inhibition enhances sulforaphane-induced apoptosis in human breast cancer cells. Anticancer Res. 2010, 30, 3381–3390. [Google Scholar]
- Pawlik, A.; Wiczk, A.; Kaczynska, A.; Antosiewica, J.; Herman-Antosiewicz, A. Sulforaphane inhibits growth of phenotypically different breast cancer cells. Eur. J. Nutr. 2013, 52, 1949–1958. [Google Scholar] [CrossRef]
- Yang, F.; Wang, F.; Liu, Y.; Wang, S.; Li, X.; Huang, Y.; Xia, Y.; Chunyu, C. Sulforaphane induces autophagy by inhibition of HDAC6-mediated PTEN activation in triple negative breast cancer cells. Life Sci. 2018, 213, 149–157. [Google Scholar] [CrossRef]
- Yang, M.; Teng, W.; Qu, Y.; Wang, H.; Yuan, Q. Sulforaphane inhibits triple negative breast cancer through activating tumor suppressor Egr1. Breast Cancer Res. Treat. 2016, 158, 277–286. [Google Scholar] [CrossRef]
- Cheng, A.; Shen, C.; Hung, C.; Hsu, Y. Sulforaphane decrease of SERTAD1 expression triggers G1/S arrest in breast cancer cells. J. Med. Food. 2019, 22, 444–450. [Google Scholar] [CrossRef]
- Li, Y.; Buckhaults, P.; Cui, X.; Tollefsbol, T. Combinatorial epigenetic mechanisms and efficacy of early breast cancer inhibition by nutritive botanicals. Epigenomics 2016, 8, 1019–1037. [Google Scholar] [CrossRef]
- Ramirez, M.C.; Singletary, K. Regulation of estrogen receptor alpha expression in human breast cancer cells by sulforaphane. J. Nutr. Biochem. 2009, 20, 195–201. [Google Scholar] [CrossRef]
- Pawlik, A.; Wojewodzka, M.S.; Herman-Antosiewicz, A. Sensitization of estrogen receptor-positive breast cancer cell lines to 4-hydroxytamoxifen by isothiocyanates present in cruciferous plants. Eur. J. Nutr. 2016, 55, 1165–1180. [Google Scholar] [CrossRef]
- Jo, E.; Kim, S.; Ahn, N.; Park, J.; Hwang, J.; Lee, Y.; Kang, K. Efficacy of sulforaphane is mediated by p38 MAP kinase and caspase-7 activations in ER-positive and COX-2-expressed human breast cancer cell. Eur. J. Cancer Prev. 2007, 16, 505–510. [Google Scholar] [CrossRef]
- Hussain, A.; Mohsin, J.; Prabhu, S.A.; Begum, S.; Nursi, Q.E.; Harish, G.; Javed, E.; Khan, M.A.; Sharma, C. Sulforaphane inhibits growth of human breast cancer cells and augments the therapeutic index of the chemotherapeutic drug, gemcitabine. Asian Pac. J. Cancer Prev. 2013, 14, 5855–5860. [Google Scholar] [CrossRef]
- Licznerska, B.; Szaefer, H.; Matuszak, I.; Murias, M.; Baer-Dubowska, W. Modulating potential of L-Sulforaphane in the expression of cytochrome P450 to identify potential targets for breast cancer chemoprevention and therapy using breast cell lines. Phytother. Res. 2015, 29, 93–99. [Google Scholar] [CrossRef]
- Lubecka-Pietruszewski, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Cebula-Obrzut, B.; Smolewski, P.; Fabianowska-Majewska, K. Sulforaphane alone and in combination with clofarabine epigenetically regulates the expression of DNA methylation-silenced tumour suppressor genes in human breast cancer cells. J. Nutr. Nutrigenom. 2015, 8, 91–101. [Google Scholar] [CrossRef]
- Meeran, S.; Patel, S.; Tollefsbol, T. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE 2010, 5, e11457. [Google Scholar] [CrossRef]
- Sarkar, R.; Mukherjee, S.; Biswas, J.; Roy, M. Sulphoraphane, a naturally occurring isothiocyanate induces apoptosis in breast cancer cells by targeting heat shock proteins. Biochem. Biophys. Res. Commun. 2012, 427, 80–85. [Google Scholar] [CrossRef]
- Lo, R.; Matthews, J. The aryl hydrocarbon receptor and estrogen receptor alpha differentially modulate nuclear factor erythroid-2-related factor 2 transactivation in MCF-7 breast cancer cells. Toxicol. Appl. Pharmacol. 2013, 270, 139–148. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Howie, F.; Beckett, G.; Mithen, R.; Bao, Y. Sulforaphane, erucin, and iberin up-regulate thioredoxin reductase 1 expression in human MCF-7 cells. J. Agric. Food Chem. 2005, 53, 1417–1421. [Google Scholar] [CrossRef]
- Thangasamy, A.; Rogge, J.; Krishnegowda, N.; Freeman, J.; Ammanamanchi, S. Novel function of transcription factor Nrf2 as an inhibitor of RON tyrosine kinase receptor-mediated cancer cell invasion. J. Biol. Chem. 2011, 286, 32115–32122. [Google Scholar] [CrossRef]
- Deb, M.; Sengupta, D.; Kar, S.; Rath, S.K.; Parbin, S.; Shilpi, A.; Roy, S.; Das, G.; Patra, S.K. Elucidation of caveolin 1 both as a tumor suppressor and metastasis promoter in light of epigenetic modulators. Tumor Biol. 2014, 35, 12031–12047. [Google Scholar] [CrossRef]
- Qazi, A.; Pal, J.; Maitah, M.; Fulciniti, M.; Pelluru, D.; Nanjappa, P.; Lee, S.; Batchu, R.; Prasad, M.; Bryant, C.; et al. Anticancer activity of a broccoli derivative, sulforaphane, in barrett adenocarcinoma: Potential use in chemoprevention and as adjuvant in chemotherapy. Transl. Oncol. 2010, 3, 389–399. [Google Scholar] [CrossRef]
- Lu, Z.; Ren, Y.; Yang, L.; Jia, A.; Hu, Y.; Zhao, Y.; Zhao, W.; Yu, B.; Zhao, W.; Zhang, J.; et al. Inhibiting autophagy enhances sulforaphane-induced apoptosis via targeting NRF2 in esophageal squamous cell carcinoma. Acta Pharm. Sin. B 2020, 11, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Biswas, R.; Rhee, Y.; Kim, J.; Ahn, J. Sulforaphene promotes Bax/Bcl2, MAPK-dependent human gastric cancer AGS cells apoptosis and inhibits migration via EGFR, p-ERK1/2 down regulation. Gen. Physiol. Biophys. 2016, 35, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H. ROS-mediated activation of AMPK plays a critical role in sulforaphane-induced apoptosis and mitotic arrest in AGS human gastric cancer cells. Gen. Physiol. Biophys. 2018, 37, 129–140. [Google Scholar] [CrossRef]
- Dong, Q.; Wang, Q.; Wang, L.; Jiang, Y.; Liu, M.; Hu, H.; Liu, Y.; Zhou, H.; He, H.; Zhang, T.; et al. SMYD3-associated pathway is involved in the anti-tumor effects of sulforaphane on gastric carcinoma cells. Food Sci. Biotechnol. 2018, 27, 1165–1173. [Google Scholar] [CrossRef]
- Kiani, S.; Akhavan-Niaki, H.; Fattahi, S.; Kavoosian, S.; Jelodar, N.; Bagheri, N.; Zarrini, H. Purified sulforaphane from broccoli (Brassica oleracea var. italica) leads to alterations of CDX1 and CDX2 expression and changes in miR-9 and miR-326 levels in human gastric cancer cells. Gene 2018, 678, 115–123. [Google Scholar] [CrossRef]
- Andělová, H.; Rudolf, E.; Červinka, M. In vitro antiproliferative effects of sulforaphane on human colon cancer cell line SW620. Acta Med. 2007, 50, 171–176. [Google Scholar] [CrossRef]
- Rudolf, E.; Andelová, H.; Cervinka, M. Activation of several concurrent proapoptic pathways by sulforaphane in human colon cancer cells SW620. Food Chem. Toxicol. 2009, 47, 2366–2373. [Google Scholar] [CrossRef]
- Lan, H.; Yuan, H.; Lin, C. Sulforaphane induces p53 deficient SW480 cell apoptosis via the ROS MAPK signaling pathway. Mol. Med. Rep. 2017, 16, 7796–7804. [Google Scholar] [CrossRef]
- Gamet-Payrastre, L.; Li, P.; Lumeau, S.; Cassar, G.; Dupont, M.A.; Chevolleau, S.; Gasc, N.; Tulliez, J.; Tercé, F. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000, 60, 1426–1433. [Google Scholar] [PubMed]
- Pappa, G.; Lichtenberg, M.; Iori, R.; Barillari, J.; Bartsch, H.; Gerhäuser, C. Comparison of growth inhibition profiles and mechanisms of apoptosis induction in human colon cancer cell lines by isothiocyanates and indoles from Brassicaceae. Mutat. Res. 2006, 599, 76–87. [Google Scholar] [CrossRef]
- Pappa, G.; Strathmann, J.; Löwinger, M.; Bartsch, H.; Gerhäuser, C. Quantitative combination effects between sulforaphane and 3,3′-diindolylmethane on proliferation of human colon cancer cells in vitro. Carcinogenesis 2007, 28, 1471–1477. [Google Scholar] [CrossRef]
- Rudolf, E.; Červinka, M. Sulforaphane induces cytotoxicity and lysosome- and mitochondria-dependent cell death in colon cancer cells with deleted p53. Toxicol. In Vitro 2011, 25, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Shin, S.H.; Park, J.; Lim, S.; Lee, E.; Lee, C.; Sung, D.; Farrand, L.; Lee, S.R.; Kim, K.H.; et al. Sulforaphene suppresses growth of colon cancer-derived tumors via induction of glutathione depletion and microtubule depolymerization. Mol. Nutr. Food Res. 2016, 60, 1068–1078. [Google Scholar] [CrossRef]
- Shen, G.; Xu, C.; Chen, C.; Hebbar, V.; Kong, A.T. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with inductionof p21CIP1 and inhibition of expression of cyclin D1. Cancer Chemother. Pharmacol. 2006, 57, 317–327. [Google Scholar] [CrossRef]
- Zeng, H.; Trujillo, O.N.; Moyer, M.P.; Botnen, J.H. Prolonged sulforaphane treatment activates survival signaling in nontumorigenic NCM460 colon cells but apoptotic signaling in tumorigenic HCT116 colon cells. Nutr. Cancer. 2011, 63, 248–255. [Google Scholar] [CrossRef]
- Pappa, G.; Bartsch, H.; Gerhäuser, C. Biphasic modulation of cell proliferation by sulforaphane at physiologically relevant exposure times in a human colon cancer cell line. Mol. Nutr. Food Res. 2007, 51, 977–984. [Google Scholar] [CrossRef]
- Parnaud, G.; Li, P.; Cassar, G.; Rouimi, P.; Tulliez, J.; Combaret, L.; Gamet-Payrastre, L. Mechanism of Sulforaphane-Induced Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells. Nutr. Cancer 2004, 48, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Tsuno, N.H.; Okaji, Y.; Shuno, Y.; Sasaki, K.; Hongo, K.; Sunami, E.; Kitayama, J.; Takahashi, K.; Nagawa, H. Inhibition of Autophagy Potentiates Sulforaphane-Induced Apoptosis in Human Colon Cancer Cells. Ann. Surg. Oncol. 2010, 17, 592–602. [Google Scholar] [CrossRef]
- Chung, Y.K.; Or, R.C.; Lu, C.H.; Ouyang, W.T.; Yang, S.Y.; Chang, C.C. Sulforaphane down-regulates SKP2 to stabilize p27KIP1 for inducing antiproliferation in human colon adenocarcinoma cells. J. Biosci. Bioeng. 2015, 119, 35–42. [Google Scholar] [CrossRef]
- Traka, M.; Gasper, A.V.; Smith, J.A.; Hawkey, C.J.; Bao, Y.; Mithen, R.F. Transcriptome Analysis of Human Colon Caco-2 Cells Exposed to Sulforaphane. J. Nutr. 2005, 135, 1865–1872. [Google Scholar] [CrossRef]
- Johnson, G.S.; Li, J.; Beaver, L.M.; Dashwood, W.M.; Sun, D.; Rajendran, P.; Williams, D.E.; Ho, E.; Dashwood, R.H. A functional pseudogene, NMRAL2P, is regulated by Nrf2 and serves as a coactivator of NQO1 in sulforaphane-treated colon cancer cells. Mol. Nutr. Food Res. 2017, 61, 1600769. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, J.; Chen, S.; Qing, Y.; Wu, D.; Lin, Y.; Luo, J.Z.; Han, W.; Li, Y. Effects of co-treatment with sulforaphane and autophagy modulators on uridine 5’-diphospho-glucuronosyltransferase 1A isoforms and cytochrome P450 3A4 expression in Caco-2 human colon cancer cells. Oncol. Lett. 2014, 8, 2407–2416. [Google Scholar] [CrossRef]
- Harris, K.E.; Jeffery, E.H. Sulforaphane and erucin increase MRP1 and MRP2 in human carcinoma cell lines. J. Nutr. Biochem. 2008, 19, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Kidane, A.I.; Yu, T.W.; Dashwood, W.M.; Bisson, W.H.; Löhr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef]
- Martin, S.L.; Kala, R.; Tollefsbol, T.O. Mechanisms for the Inhibition of Colon Cancer Cells by Sulforaphane through Epigenetic Modulation of MicroRNA-21 and Human Telomerase Reverse Transcriptase (hTERT) Down-regulation. Curr. Cancer Drug Targets 2018, 18, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, A.; Mitra, J.; Johnson, G.S.; Li, L.; Dashwood, W.M.; Hegde, M.L.; Yue, C.; Dashwood, R.H.; Rajendran, P. Heterocyclic Analogs of Sulforaphane Trigger DNA Damage and Impede DNA Repair in Colon Cancer Cells: Interplay of HATs and HDACs. Mol. Nutr. Food Res. 2018, 62, e1800228. [Google Scholar] [CrossRef]
- Bessler, H.; Djaldetti, M. Broccoli and human health: Immunomodulatory effect of sulforaphane in a model of colon cancer. Int. J. Food Sci. Nutr. 2018, 69, 946–953. [Google Scholar] [CrossRef]
- Tafakh, M.S.; Saidijam, M.; Ranjbarnejad, T.; Malih, S.; Mirzamohammadi, S.; Najafi, R. Sulforaphane, a Chemopreventive Compound, Inhibits Cyclooxygenase-2 and Microsomal Prostaglandin E Synthase-1 Expression in Human HT-29 Colon Cancer Cells. Cells Tissues Organs 2018, 206, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Jakubíková, J.; Sedlák, J.; Mithen, R.; Bao, Y. Role of PI3K/Akt and MEK/ERK signaling pathways in sulforaphane- and erucin-induced phase II enzymes and MRP2 transcription, G2/M arrest and cell death in Caco-2 cells. Biochem. Pharmacol. 2005, 69, 1543–1552. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, S.H.; Lim, S.J. Comparison of the apoptosis-inducing capability of sulforaphane analogues in human colon cancer cells. Anticancer Res. 2010, 30, 3611–3619. [Google Scholar] [PubMed]
- Kim, D.H.; Sung, B.; Kang, Y.J.; Hwang, S.Y.; Kim, M.J.; Yoon, J.H.; Im, E.; Kim, N.D. Sulforaphane inhibits hypoxia-induced HIF-1α and VEGF expression and migration of human colon cancer cells. Int. J. Oncol. 2015, 47, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lei, W.; Mandlekar, S.; Weber, M.J.; Der, C.J.; Wu, J.; Kong, A.N. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J. Biol. Chem. 1999, 274, 27545–27552. [Google Scholar] [CrossRef]
- Yeh, C.T.; Yen, G.C. Effect of sulforaphane on metallothionein expression and induction of apoptosis in human hepatoma HepG2 cells. Carcinogenesis 2005, 26, 2138–2148. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, G.Y.; Bae, S.; Yoo, Y.; Choi, Y.H. Induction of apoptosis by isothiocyanate sulforaphane in human cervical carcinoma HeLa and hepatocarcinoma HepG2 cells through activation of caspase-3. Oncol. Rep. 2007, 18, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Keum, Y.S.; Yu, S.; Chang, P.P.; Yuan, X.; Kim, J.H.; Xu, C.; Han, J.; Agarwal, A.; Kong, A.N. Mechanism of action of sulforaphane: Inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 2006, 66, 8804–8813. [Google Scholar] [CrossRef]
- Jeon, Y.K.; Yoo, D.R.; Jang, Y.H.; Jang, S.Y.; Nam, M.J. Sulforaphane induces apoptosis in human hepatic cancer cells through inhibition of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase4, mediated by hypoxia inducible factor-1-dependent pathway. Biochim. Biophys. Acta 2011, 1814, 1340–1348. [Google Scholar] [CrossRef]
- Liu, P.; Atkinson, S.J.; Akbareian, S.E.; Zhou, Z.; Munsterberg, A.; Robinson, S.D.; Bao, Y. Sulforaphane exerts anti-angiogenesis effects against hepatocellular carcinoma through inhibition of STAT3/HIF-1α/VEGF signaling. Sci. Rep. 2017, 7, 12651. [Google Scholar] [CrossRef]
- Moon, D.O.; Kang, S.H.; Kim, K.C.; Kim, M.O.; Choi, Y.H.; Kim, G.Y. Sulforaphane decreases viability and telomerase activity in hepatocellular carcinoma Hep3B cells through the reactive oxygen species-dependent pathway. Cancer Lett. 2010, 295, 260–266. [Google Scholar] [CrossRef]
- Wu, J.; Han, J.; Hou, B.; Deng, C.; Wu, H.; Shen, L. Sulforaphane inhibits TGF-β-induced epithelial-mesenchymal transition of hepatocellular carcinoma cells via the reactive oxygen species-dependent pathway. Oncol. Rep. 2016, 35, 2977–2983. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Qu, Z.; Fang, Y.; Shi, X.; Ji, Y. Endoplasmic reticulum stress mediates sulforaphane-induced apoptosis of HepG2 human hepatocellular carcinoma cells. Mol. Med. Rep. 2017, 15, 331–338. [Google Scholar] [CrossRef][Green Version]
- Anwar-Mohamed, A.; El-Kadi, A.O. Sulforaphane induces CYP1A1 mRNA, protein, and catalytic activity levels via an AhR-dependent pathway in murine hepatoma Hepa 1c1c7 and human HepG2 cells. Cancer Lett. 2009, 275, 93–101. [Google Scholar] [CrossRef]
- Pham, N.A.; Jacobberger, J.W.; Schimmer, A.D.; Cao, P.; Gronda, M.; Hedley, D.W. The dietary isothiocyanate sulforaphane targets pathways of apoptosis, cell cycle arrest, and oxidative stress in human pancreatic cancer cells and inhibits tumor growth in severe combined immunodeficient mice. Mol. Cancer Ther. 2004, 3, 1239–1248. [Google Scholar] [PubMed]
- Li, Y.; Karagöz, G.E.; Seo, Y.H.; Zhang, T.; Jiang, Y.; Yu, Y.; Duarte, A.M.; Schwartz, S.J.; Boelens, R.; Carroll, K.; et al. Sulforaphane inhibits pancreatic cancer through disrupting HSP90-p50(Cdc37) complex and direct interactions with amino acids residues of HSP90. J. Nutr. Biochem. 2012, 23, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Kallifatidis, G.; Rausch, V.; Baumann, B.; Apel, A.; Beckermann, B.M.; Groth, A.; Mattern, J.; Li, Z.; Kolb, A.; Moldenhauer, G.; et al. Sulforaphane targets pancreatic tumour-initiating cells by NF-kappaB-induced antiapoptotic signaling. Gut 2009, 58, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Rodova, M.; Fu, J.; Watkins, D.N.; Srivastava, R.K.; Shankar, S. Sonic hedgehogsignaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS ONE 2012, 7, e46084. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Xiao, X.; Georgikou, C.; Yin, Y.; Liu, L.; Karakhanova, S.; Luo, Y.; Gladkich, J.; Fellenberg, J.; Sticht, C.; et al. MicroRNA-365a-3p inhibits c-Rel-mediated NF-κB signaling and the progression of pancreatic cancer. Cancer Lett. 2019, 452, 203–212. [Google Scholar] [CrossRef]
- Yin, L.; Xiao, X.; Georgikou, C.; Luo, Y.; Liu, L.; Gladkich, J.; Gross, W.; Herr, I. Sulforaphane Induces miR135b-5p and Its Target Gene, RASAL2, thereby Inhibiting the Progression of Pancreatic Cancer. Mol. Ther. Oncolytics 2019, 14, 74–81. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Z.; Zhou, C.; Chen, K.; Li, X.; Wang, Z.; Wu, Z.; Ma, J.; Ma, Q.; Duan, W. Activation of Nrf2 by Sulforaphane Inhibits High Glucose-Induced Progression of Pancreatic Cancer via AMPK Dependent Signaling. Cell. Physiol. Biochem. 2018, 50, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Forster, T.; Rausch, V.; Zhang, Y.; Isayev, O.; Heilmann, K.; Schoensiegel, F.; Liu, L.; Nessling, M.; Richter, K.; Labsch, S.; et al. Sulforaphane counteracts aggressiveness of pancreatic cancer driven by dysregulated Cx43-mediated gap junctional intercellular communication. Oncotarget 2014, 5, 1621–1634. [Google Scholar] [CrossRef]
- Georgikou, C.; Yin, L.; Gladkich, J.; Xiao, X.; Sticht, C.; Torre, C.; Gretz, N.; Gross, W.; Schäfer, M.; Karakhanova, S.; et al. Inhibition of miR30a-3p by sulforaphane enhances gap junction intercellular communication in pancreatic cancer. Cancer Lett. 2020, 469, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Sadrieh, L.; Priyani, A.; Ahmed, M.; Hassan, A.H.; Hussain, A. Anti-carcinogenic effects of sulforaphane in association with its apoptosis-inducing and anti-inflammatory properties in human cervical cancer cells. Cancer Epidemiol. 2011, 33, 272–278. [Google Scholar] [CrossRef]
- Khan, M.A.; Sundaram, M.; Hamza, A.; Quraishi, U.; Gunasekera, D.; Ramesh, L.; Goala, P.; Al Alami, U.; Ansari, M.; Rizvi, T.A.; et al. Sulforaphane Reverses the Expression of Various Tumor Suppressor Genes by Targeting DNMT3B and HDAC1 in Human Cervical Cancer Cells. Evid. Based Complement. Alternat. Med. 2015, 2015, 412149. [Google Scholar] [CrossRef]
- Cheng, Y.; Tsai, C.; Hsu, Y. Sulforaphane, a Dietary Isothiocyanate, Induces G2/M Arrest in Cervical Cancer Cells through Cyclin B1 Downregulation and GADD45B/CDC2 Association. Int. J. Mol. Sci. 2016, 17, 1530. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Essel, K.G.; Benbrook, D.M.; Garland, J.; Zhao, Y.D.; Chandra, V. Preclinical efficacy and involvement of AKT, mTOR, and ERK kinases in the mechanism of sulforaphane against endometrial cancer. Cancers 2020, 12, 1273. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Orsulic, S.; Ashok, B.T. Antiproliferative activity of sulforaphane in Akt-overexpressing ovarian cancer cells. Mol. Cancer Ther. 2007, 6, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.T.; Moqattash, S.T.; Gretz, H.F.; Nezhat, F.; Rahaman, J.; Chiao, J. Sulforaphane induces growth arrest and apoptosis in human ovarian cancer cells. Acta Obstet. Gynecol. Scand. 2007, 86, 1263–1268. [Google Scholar] [CrossRef]
- Bryant, C.S.; Kumar, S.; Chamala, S.; Shah, J.; Pal, J.; Haider, M.; Seward, S.; Qazi, A.M.; Morris, R.; Semaan, A.; et al. Sulforaphane induces cell cycle arrest by protecting RB-E2F-1 complex in epithelial ovarian cancer cells. Mol. Cancer 2010, 9, 47. [Google Scholar] [CrossRef]
- Kim, S.C.; Choi, B.; Kwon, Y. Thiol-reducing agents prevent sulforaphane-induced growth inhibition in ovarian cancer cells. Food Nutr. Res. 2017, 6, 1–12. [Google Scholar] [CrossRef]
- Hudecova, S.; Markova, J.; Simko, V.; Csaderova, L.; Stracina, T.; Sirova, M.; Fojtu, M.; Savastova, E.; Gronesova, P.; Pastorek, M.; et al. Sulforaphane-induced apoptosis involves the type 1 IP3 receptor. Oncotarget 2016, 7, 61403–61418. [Google Scholar] [CrossRef][Green Version]
- Chang, C.; Hung, C.; Yang, Y.; Lee, M.; Hsu, Y. Sulforaphane induced cell cycle arrest in the G2/M phase via the blockade of cyclin B1/CDC2 in human ovarian cancer cells. J. Ovarian Res. 2013, 6, 41. [Google Scholar] [CrossRef]
- Fimognari, C.; Nusse, M.; Cesari, R.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis 2002, 23, 581–585. [Google Scholar] [CrossRef]
- Fimognari, C.; Nusse, M.; Berti, F.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Sulforaphane Modulates Cell Cycle and Apoptosis in Transformed Non-transformed Human T Lymphocytes. Ann. N. Y. Acad. Sci. 2003, 1010, 393–398. [Google Scholar] [CrossRef]
- Fimognari, C.; Lenzi, M.; Cantelli-forti, G.; Hrelia, P. Induction of Differentiation in Human Promyelocytic Cells by the Isothiocyanate Sulforaphane. In Vivo 2008, 22, 317–320. [Google Scholar] [PubMed]
- Choi, W.Y.; Choi, B.T.; Lee, W.H.; Choi, Y.H. Sulforaphane generates reactive oxygen species leading to mitochondrial perturbation for apoptosis in human leukemia U937 cells. Biomed. Pharmacother. 2008, 62, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Koolivand, M.; Ansari, M.; Piroozian, F.; Moein, S.; MalekZadeh, K. Alleviating the progression of acute myeloid leukemia (AML) by sulforaphane through controlling miR-155 levels. Mol. Biol. Rep. 2018, 45, 2491–2499. [Google Scholar] [CrossRef]
- Shang, H.; Shih, Y.; Lee, C.H.S.; Liu, J.; Liao, N.; Chen, Y.; Huang, Y.; Lu, H.; Chung, J. Sulforaphane-induced apoptosis in human leukemia HL-60 cells through extrinsic and intrinsic signal pathways and altering associated genes expression assayed by cDNA microarray. Environ. Toxicol. 2016, 32, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.M.; Oraee, A.; Doonan, B.B.; Pinto, J.T.; Hsieh, T. Activation of NQO1 in NQO1*2 polymorphic human leukemic HL-60 cells by diet-derived sulforaphane. Exp. Hematol. Oncol. 2016, 5, 27. [Google Scholar] [CrossRef]
- Suppipat, K.; Park, C.S.; Shen, Y.; Zhu, X.; Lacorazza, H.D. Sulforaphane Induces Cell Cycle Arrest and Apoptosis in Acute Lymphoblastic Leukemia Cells. PLoS ONE 2012, 7, e051251. [Google Scholar] [CrossRef]
- Prata, C.; Facchini, C.; Leoncini, E.; Lenzi, M.; Maraldi, T.; Angeloni, C.; Zambonin, L.; Hrelia, S.; Fiorentini, D. Sulforaphane Modulates AQP8-Linked Redox Signalling in Leukemia Cells. Oxid. Med. Cell. Longev. 2018, 2018, 4125297. [Google Scholar] [CrossRef]
- Misiewicz, I.; Skupinska, K.; Kasprzycka-Guttman, T. Sulforaphane and 2-oxohexyl isothiocyanate induce cell growth arrest and apoptosis in L-1210 leukemia and ME-18 melanoma cells. Oncol. Rep. 2003, 10, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Ishirua, Y.; Ishimaru, H.; Watanabe, T.; Fujimuro, M. Sulforaphane Exhibits Cytotoxic Effects against Primary Effusion Lymphoma Cells by suppressing p38 MAPK and AKT Phosphorylation. Biol. Pharm. Bull. 2019, 42, 2019–2112. [Google Scholar] [CrossRef]
- Mi, L.; Chung, F.L. Binding to protein by isothiocyanates: A potential mechanism for apoptosis induction in human non small lung cancer cells. Nutr. Cancer 2008, 60, 12–20. [Google Scholar] [CrossRef]
- Liang, H.; Lai, B.; Yuan, Q. Sulforaphane induces cell-cycle arrest and apoptosis in cultured human lung adenocarcinoma LTEP-A2 cells and retards growth of LTEP-A2 xenografts in vivo. J. Nat. Prod. 2008, 71, 1911–1914. [Google Scholar] [CrossRef]
- Żuryń, A.; Litwiniec, A.; Safiejko-Mroczka, B.; Klimaszewska-Wiśniewska, A.; Gagat, M.; Krajewski, A.; Gackowska, L.; Grzanka, D. The effect of sulforaphane on the cell cycle, apoptosis and expression of cyclin D1 and p21 in the A549 non-small cell lung cancer cell line. Int. J. Oncol. 2016, 48, 2521–2533. [Google Scholar] [CrossRef]
- Żuryń, A.; Krajewski, A.; Klimaszewska-Wiśniewska, A.; Grzanka, A.; Grzanka, D. Expression of cyclin B1, D1 and K in non-small cell lung cancer H1299 cells following treatment with sulforaphane. Oncol. Rep. 2019, 41, 1313–1323. [Google Scholar] [CrossRef]
- Jiang, L.L.; Zhou, S.J.; Zhang, X.M.; Chen, H.Q.; Liu, W. Sulforaphane suppresses in vitro and in vivo lung tumorigenesis through downregulation of HDAC activity. Biomed. Pharmacother. 2016, 78, 74–80. [Google Scholar] [CrossRef]
- Gao, L.; Cheng, D.; Yang, J.; Wu, R.; Li, W.; Kong, A.N. Sulforaphane epigenetically demethylates the CpG sites of the miR-9-3 promoter and reactivates miR-9-3 expression in human lung cancer A549 cells. J. Nutr. Biochem. 2018, 56, 109–115. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, S.; Chen, Y.; Li, X.; Jiang, Y.; Yang, X.; Li, Y.; Wang, X.; Meng, Y.; Zhu, M.; et al. miR-19 targeting of GSK3β mediates sulforaphane suppression of lung cancer stem cells. J. Nutr. Biochem. 2017, 44, 80–91. [Google Scholar] [CrossRef]
- Wang, D.X.; Zou, Y.J.; Zhuang, X.B.; Chen, S.X.; Lin, Y.; Li, W.L.; Lin, J.J.; Lin, Z.Q. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR-616-5p-mediated GSK3β/β-catenin signaling pathways. Acta Pharmacol. Sin. 2017, 38, 241–251. [Google Scholar] [CrossRef]
- Tsai, J.Y.; Tsai, S.H.; Wu, C.C. The chemopreventive isothiocyanate sulforaphane reduces anoikis resistance and anchorage-independent growth in non-small cell human lung cancer cells. Toxicol. Appl. Pharmacol. 2019, 362, 116–124. [Google Scholar] [CrossRef]
- Zhou, L.; Yao, Q.; Li, Y.; Huang, Y.C.; Jiang, H.; Wang, C.Q.; Fan, L. Sulforaphane-induced apoptosis in Xuanwei lung adenocarcinoma cell line XWLC-05. Thorac. Cancer 2017, 8, 16–25. [Google Scholar] [CrossRef]
- Wang, Y.; Mandal, A.K.; Son, Y.O.; Pratheeshkumar, P.; Wise, J.T.F.; Wang, L.; Zhang, Z.; Shi, X.; Chen, Z. Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol. Appl. Pharmacol. 2018, 353, 23–30. [Google Scholar] [CrossRef]
- Chen, C.Y.; Yu, Z.Y.; Chuang, Y.S.; Huang, R.M.; Wang, T.C. Sulforaphane attenuates EGFR signaling in NSCLC cells. J. Biomed. Sci. 2015, 22, 38. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.Q.; Ge, M.M.; Zhang, Q.; Wang, X.Q.; Zhu, J.Y.; Xie, C.F.; Li, X.T.; Zhong, C.Y.; Han, H.Y. Sulforaphane inhibits epithelial-mesenchymal transition by activating extracellular signal-regulated kinase 5 in lung cancer cells. J. Nutr. Biochem. 2019, 72, 108219. [Google Scholar] [CrossRef]
- Geng, Y.; Zhou, Y.; Wu, S.; Hu, Y.; Lin, K.; Wang, Y.; Zheng, Z.; Wu, W. Sulforaphane Induced Apoptosis via Promotion of Mitochondrial Fusion and ERK1/2-Mediated 26S Proteasome Degradation of Novel Pro-survival Bim and Upregulation of Bax in Human Non-Small Cell Lung Cancer Cells. J. Cancer 2017, 8, 2456–2470. [Google Scholar] [CrossRef]
- Xie, C.; Zhu, J.; Jiang, Y.; Chen, J.; Wang, X.; Geng, S.; Wu, J.; Zhong, C.; Li, X.; Meng, Z. Sulforaphane Inhibits the Acquisition of Tobacco Smoke-Induced Lung Cancer Stem Cell-Like Properties via the IL-6/ΔNp63α/Notch Axis. Theranostics 2019, 9, 4827–4840. [Google Scholar] [CrossRef]
- Karmakar, S.; Weinberg, M.S.; Banik, N.L.; Patel, S.J.; Ray, S.K. Activation of multiple molecular mechanisms for apoptosis in human malignant glioblastoma T98G and U87MG cells treated with sulforaphane. Neuroscience 2006, 141, 1265–1280. [Google Scholar] [CrossRef]
- Bijangi-Vishehsaraei, K.; Reza Saadatzadeh, M.; Wang, H.; Nguyen, A.; Kamocka, M.M.; Cai, W.; Cohen-Gadol, A.A.; Halum, S.L.; Sarkaria, J.N.; Pollok, K.E.; et al. Sulforaphane suppresses the growth of glioblastoma cells, glioblastoma stem cell-like spheroids, and tumor xenografts through multiple cell signaling pathways. J. Neurosurg. 2017, 127, 1219–1230. [Google Scholar] [CrossRef]
- Miao, Z.; Yu, F.; Ren, Y.; Yang, J. d,l-Sulforaphane Induces ROS-Dependent Apoptosis in Human Gliomablastoma Cells by Inactivating STAT3 Signaling Pathway. Int. J. Mol. Sci. 2017, 18, 72. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, C.; Shang, L.; Zhang, Y.; Zou, R.; Zhan, Y.; Bi, B. Sulforaphane induces apoptosis and inhibits invasion in U251MG glioblastoma cells. SpringerPlus 2016, 5, 235. [Google Scholar] [CrossRef]
- Li, C.; Zhou, Y.; Peng, X.; Du, L.; Tian, H.; Yang, G.; Niu, J.; Wu, W. Sulforaphane inhibits invasion via activating ERK1/2 signaling in human glioblastoma U87MG and U373MG cells. PLoS ONE 2014, 9, e90520. [Google Scholar] [CrossRef]
- Arcidiacono, P.; Stabile, A.M.; Ragonese, F.; Pistilli, A.; Calvieri, S.; Bottoni, U.; Crisanti, A.; Spaccapelo, R.; Rende, M. Anticarcinogenic activities of sulforaphane are influenced by Nerve Growth Factor in human melanoma A375 cells. Food Chem. Toxicol. 2018, 113, 154–161. [Google Scholar] [CrossRef]
- Mantso, T.; Sfakianos, A.P.; Atkinson, A.; Anestopoulos, I.; Mitsiogianni, M.; Botaitis, S.; Perente, S.; Simopoulos, C.; Vasileiadis, S.; Franco, R.; et al. Development of a Novel Experimental In Vitro Model of Isothiocyanate-induced Apoptosis in Human Malignant Melanoma Cells. Anticancer Res. 2016, 36, 6303–6309. [Google Scholar] [CrossRef]
- Fisher, M.L.; Adhikary, G.; Grun, D.; Kaetzel, D.M.; Eckert, R.L. The Ezh2 polycomb group protein drives an aggressive phenotype in melanoma cancer stem cells and is a target of diet derived sulforaphane. Mol. Carcinog. 2016, 55, 2024–2036. [Google Scholar] [CrossRef]
- Hamsa, T.P.; Thejass, P.; Kuttan, G. Induction of apoptosis by sulforaphane in highly metastatic B16F-10 melanoma cells. Drug Chem. Toxicol. 2011, 34, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Enriquez, G.G.; Rizvi, S.A.; D’Souza, M.J.; Do, D.P. Formulation and Evaluation of drug-loaded targeted magnetic microspheres for cancer therapy. Int. J. Nanomed. 2013, 8, 1393–1402. [Google Scholar] [CrossRef]
- Do, D.P.; Pai, S.B.; Rizvi, S.A.; D’Souza, M.J. Development of sulforaphane-encapsulated microspheres for cancer epigenetic therapy. Int. J. Pharm. 2010, 386, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, K.; Cervinka, M.; Rudolf, E. Sulforaphane-induced apoptosis involvesp53 and p38 in melanoma cells. Apoptosis 2014, 19, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Sun, C.; Wu, K.; Cassidy, A.; Bao, Y. Effect of sulforaphane on cell growth, G0/G1 phase cell progression and apoptosis in human bladder cancer T24 cells. Int. J. Oncol. 2006, 29, 883–888. [Google Scholar] [CrossRef]
- Shan, Y.; Wu, K.; Wang, W.; Wang, S.; Lin, N.; Zhao, R.; Cassidy, A.; Bao, Y. Sulforaphane down-regulates COX-2 expression by activating p38 and inhibiting NF-kappaB-DNA-binding activity in human bladder T24 cells. Int. J. Oncol. 2009, 34, 1129–1134. [Google Scholar] [CrossRef]
- Shan, Y.; Wang, X.; Wang, W.; He, C.; Bao, Y. P38 MAPK plays a distinct role in sulforaphane-induced up-regulation of ARE-dependent enzymes and down-regulation of COX-2 in human bladder cancer cells. Oncol. Rep. 2010, 23, 1133–1138. [Google Scholar] [CrossRef]
- Shan, S.; Zhang, L.; Bao, Y.; Li, B.; He, C.; Gao, M.; Feng, X.; Xu, W.; Zhang, X.; Wang, S. Epithelial-mesenchymal transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J. Nutr. Biochem. 2013, 24, 1062–1069. [Google Scholar] [CrossRef]
- Jo, G.H.; Kim, G.; Kim, W.; Park, K.; Choi, Y.H. Sulforaphane induces apoptosis in T24 human urinary bladder cancer cells through a reactive oxygen species-mediated mitochondrial pathway: The involvement of endoplasmic reticulum stress and the Nrf2 signaling pathway. Int. J. Oncol. 2014, 45, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Abbaoui, B.; Telu, K.H.; Lucas, C.R.; Thomas-Ahner, J.M.; Schwarts, S.J.; Clinton, S.K.; Freitas, M.A.; Mortazavi, A. The impact of cruciferous vegetable of isothiocyanate on histone acetylation and histone phosphorylation in bladder cancer. J. Proteom. 2017, 156, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Huang, G.; Chen, Y.; Dang, Z.; Chen, C.; Liu, F.; Guo, Y.; Xie, X. Sulforaphane inhibits the proliferation of the BIU87 bladder cancer cell line via IGFBP-3 elevation. Asian Pac. J. Cancer Prev. 2014, 15, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Han, M.H.; Kim, G.; Moon, S.; Kim, W.; Hwang, H.J.; Park, K.Y.; Choi, Y.H. Sulforaphane induces reactive oxygen species-mediated mitotic arrest and subsequent apoptosis in human bladder cancer 5637 cells. Food Chem. Toxicol. 2014, 64, 157–165. [Google Scholar] [CrossRef]
- Brooks, J.D.; Paton, V.G.; Vidanes, G. Potent induction of phase 2 enzymes in human prostate cells by sulforaphane. Cancer Epidemiol. Biomark. Prev. 2001, 10, 949–954. [Google Scholar]
- Hahm, E.R.; Singh, S.V. Sulforaphane inhibits constitutive and interleukin-6-induced activation of signal transducer and activator of transcription 3 in prostate cancer cells. Cancer Prev. Res. 2010, 3, 484–494. [Google Scholar] [CrossRef]
- Choi, S.; Lew, K.L.; Xiao, H.; Herman-Antosiewicz, A.; Xiao, D.; Brown, C.K.; Singh, S.V. D,L-Sulforaphane-induced cell death in human prostate cancer cells is regulated by inhibitor of apoptosis family proteins and Apaf-1. Carcinogenesis 2007, 28, 151–162. [Google Scholar] [CrossRef]
- Carrasco-Pozo, C.; Tan, K.N.; Rodriguez, T.; Avery, V.M. The Molecular Effects of Sulforaphane and Capsaicin on Metabolism upon Androgen and Tip60 Activation of Androgen Receptor. Int. J. Mol. Sci. 2019, 20, 5384. [Google Scholar] [CrossRef]
- Kim, S.H.; Singh, S.V. D,L-Sulforaphane causes transcriptional repression of androgen receptor in human prostate cancer cells. Mol. Cancer Ther. 2009, 8, 1946–1954. [Google Scholar] [CrossRef]
- Herman-Antosiewicz, A.; Johnson, D.E.; Singh, S.V. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006, 66, 5828–5835. [Google Scholar] [CrossRef]
- Xiao, D.; Powolny, A.A.; Antosiewicz, J.; Hahm, E.R.; Bommareddy, A.; Zeng, Y.; Desai, D.; Amin, S.; Herman-Antosiewicz, A.; Singh, S.V. Cellular responses to cancer chemopreventive agent D,L-sulforaphane in human prostate cancer cells are initiated by mitochondrial reactive oxygen species. Pharm. Res. 2009, 26, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.W.; Wickramasekara, S.; Fang, Y.; Palomera-Sanchez, Z.; Maier, C.S.; Williams, D.E.; Dashwood, R.H.; Perez, V.I.; Ho, E. Analysis of autophagic flux in response to sulforaphane in metastatic prostate cancer cells. Mol. Nutr. Food Res. 2015, 59, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.D.; Li, G.; Hu, H.; Jiang, C.; Kang, K.S.; Lee, Y.S.; Kim, S.H.; Lu, J. Involvement of c-Jun N-terminal kinase in G2/M arrest and caspase-mediated apoptosis induced by sulforaphane in DU145 prostate cancer cells. Nutr. Cancer 2005, 52, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Hać, A.; Brokowska, J.; Rintz, E.; Bartkowski, M.; Węgrzyn, G.; Herman-Antosiewicz, A. Mechanism of selective anticancer activity of isothiocyanates relies on differences in DNA damage repair between cancer and healthy cells. Eur. J. Nutr. 2020, 59, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Jeong, S.J.; Yun, S.M.; Kim, J.H.; Lee, H.J.; Ahn, K.S.; Won, S.H.; Kim, H.S.; Lee, H.J.; Ahn, K.S.; et al. Down-regulation of phosphoglucomutase 3 mediates sulforaphane-induced cell death in LNCaP prostate cancer cells. Proteome Sci. 2010, 8, 67. [Google Scholar] [CrossRef]
- Singh, A.V.; Xiao, D.; Lew, K.L.; Dhir, R.; Singh, S.V. Sulforaphane induces caspase-mediated apoptosis in cultured PC-3 human prostate cancer cells and retards growth of PC-3 xenografts in vivo. Carcinogenesis 2004, 25, 83–90. [Google Scholar] [CrossRef]
- Singh, S.V.; Srivastava, S.K.; Choi, S.; Lew, K.L.; Antosiewicz, J.; Xiao, D.; Zeng, Y.; Watkins, S.C.; Johnson, C.S.; Trump, D.L.; et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J. Biol. Chem. 2005, 280, 19911–19924. [Google Scholar] [CrossRef]
- Negrette-Guzmán, M.; Huerta-Yepez, S.; Vega, M.I.; León-Contreras, J.C.; Hernández-Pando, R.; Medina-Campos, O.N.; Rodríguez, E.; Tapia, E.; Pedraza-Chaverri, J. Sulforaphane induces differential modulation of mitochondrial biogenesis and dynamics in normal cells and tumor cells. Food Chem. Toxicol. 2017, 100, 90–102. [Google Scholar] [CrossRef]
- Beaver, L.M.; Buchanan, A.; Sokolowski, E.I.; Riscoe, A.N.; Wong, C.P.; Chang, J.H.; Löhr, C.V.; Williams, D.E.; Dashwood, R.H.; Ho, E. Transcriptome analysis reveals a dynamic and differential transcriptional response to sulforaphane in normal and prostate cancer cells and suggests a role for Sp1 in chemoprevention. Mol. Nutr. Food Res. 2014, 58, 2001–2013. [Google Scholar] [CrossRef]
- Bhamre, S.; Sahoo, D.; Tibshirani, R.; Dill, D.; Brooks, J.D. Temporal changes in gene expression induced by sulforaphane in human prostate cancer cells. Prostate 2009, 69, 181–190. [Google Scholar] [CrossRef]
- Gibbs, A.; Schwartzman, J.; Deng, V.; Alumkal, J. Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc. Natl. Acad. Sci. USA 2009, 106, 16663–16668. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Dashwood, W.M.; Orner, G.A.; Ho, E.; Dashwood, R.H. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-min mice. FASEB J. 2006, 20, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Su, Z.Y.; Khor, T.O.; Shu, L.; Kong, A.N. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Beaver, L.M.; Kuintzle, R.; Buchanan, A.; Wiley, M.; Glasser, S.T.; Wong, C.P.; Johnson, G.S.; Chang, J.H.; Löhr, C.V.; Williams, D.E.; et al. Long noncoding RNAs and sulforaphane: A target for chemoprevention and suppression of prostate cancer. J. Nutr. Biochem. 2017, 42, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.R.; Chandra-Kuntal, K.; Desai, D.; Amin, S.; Singh, S.V. Notch activation is dispensable for D, L-sulforaphane-mediated inhibition of human prostate cancer cell migration. PLoS ONE 2012, 7, e44957. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenetics 2011, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Hsu, A.; Buchanan, A.; Palomera-Sanchez, Z.; Beaver, L.M.; Houseman, E.A.; Williams, D.E.; Dashwood, R.H.; Ho, E. Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS ONE 2014, 9, e86787. [Google Scholar] [CrossRef]
- Vyas, A.R.; Moura, M.B.; Hahm, E.R.; Singh, K.; Singh, S.V. Sulforaphane Inhibits c-Myc-Mediated Prostate Cancer Stem-Like Traits. J. Cell. Biochem. 2016, 117, 2482–2495. [Google Scholar] [CrossRef]
- Watson, G.W.; Wickramasekara, S.; Palomera-Sanchez, Z.; Black, C.; Maier, C.S.; Williams, D.E.; Dashwood, R.H.; Ho, E. SUV39H1/H3K9me3 attenuates sulforaphane-induced apoptotic signaling in PC3 prostate cancer cells. Oncogenesis 2014, 3, e131. [Google Scholar] [CrossRef]
- Singh, K.B.; Hahm, E.R.; Alumkal, J.J.; Foley, L.M.; Hitchens, T.K.; Shiva, S.S.; Parikh, R.A.; Jacobs, B.L.; Singh, S.V. Reversal of the Warburg phenomenon in chemoprevention of prostate cancer by sulforaphane. Carcinogenesis 2019, 40, 1545–1556. [Google Scholar] [CrossRef]
- Peng, X.; Zhou, Y.; Tian, H.; Yang, G.; Li, C.; Geng, Y.; Wu, S.; Wu, W. Sulforaphane inhibits invasion by phosphorylating ERK1/2 to regulate E-cadherin and CD44v6 in human prostate cancer DU145 cells. Oncol. Rep. 2015, 34, 1565–1572. [Google Scholar] [CrossRef]
- Vyas, A.R.; Singh, S.V. Functional relevance of D,L-sulforaphane-mediated induction of vimentin and plasminogen activator inhibitor-1 in human prostate cancer cells. Eur. J. Nutr. 2014, 53, 843–852. [Google Scholar] [CrossRef]
- Hać, A.; Domachowska, A.; Narajczyk, M.; Cyske, K.; Pawlik, A.; Herman-Antosiewicz, A. S6K1 controls autophagosome maturation in autophagy induced by sulforaphane or serum deprivation. Eur. J. Cell. Biol. 2015, 94, 470–481. [Google Scholar] [CrossRef]
- Pei, Y.; Wu, B.; Cao, Q.; Wu, L.; Yang, G. Hydrogen sulfide mediates the anti-survival effect of sulforaphane on human prostate cancer cells. Toxicol. Appl. Pharmacol. 2011, 257, 420–428. [Google Scholar] [CrossRef]
- Wiczk, A.; Hofman, D.; Konopa, G.; Herman-Antosiewicz, A. Sulforaphane, a cruciferous vegetable-derived isothiocyanate, inhibits protein synthesis in human prostate cancer cells. Biochim. Biophys. Acta 2012, 1823, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shen, G.; Chen, C.; Gélinas, C.; Kong, A.N. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene 2005, 24, 4486–4495. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shen, G.; Yuan, X.; Kim, J.H.; Gopalkrishnan, A.; Keum, Y.S.; Nair, S.; Kong, A.N. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis 2006, 27, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wang, H.; Zhang, Z.; Jiang, B.H.; Luo, J.; Shi, X. Sulforaphane inhibited expression of hypoxia-inducible factor-1alpha in human tongue squamous cancer cells and prostate cancer cells. Int. J. Cancer 2008, 123, 1255–1261. [Google Scholar] [CrossRef]
- Singh, K.B.; Kim, S.H.; Hahm, E.R.; Pore, S.K.; Jacobs, B.L.; Singh, S.V. Prostate cancer chemoprevention by sulforaphane in a preclinical mouse model is associated with inhibition of fatty acid metabolism. Carcinogenesis 2018, 39, 826–837. [Google Scholar] [CrossRef]
- Herman-Antosiewicz, A.; Xiao, H.; Lew, K.L.; Singh, S.V. Induction of p21 protein protects against sulforaphane-induced mitotic arrest in LNCaP human prostate cancer cell line. Mol. Cancer Ther. 2007, 6, 1673–1681. [Google Scholar] [CrossRef]
- Abbas, A.; Hall, J.A.; Patterson, W.L.; Ho, E.; Hsu, A.; Al-Mulla, F.; Georgel, P.T. Sulforaphane modulates telomerase activity via epigenetic regulation in prostate cancer cell lines. Biochem. Cell Biol. 2016, 94, 71–81. [Google Scholar] [CrossRef]
- Kanematsu, S.; Yoshizawa, K.; Uehara, N.; Miki, H.; Sasaki, T.; Kuro, M.; Lai, Y.; Kimura, A.; Yuri, T.; Tsubura, A. Sulforaphane inhibits the growth of KPL-1 human breast cancer cells in vitro and suppresses the growth and metastasis of orthotopically transplanted KPL-1 cells in female athymic mice. Oncol. Rep. 2011, 26, 603–608. [Google Scholar] [CrossRef]
- Hu, R.; Khor, T.O.; Shen, G.; Jeong, W.; Hebbar, V.; Chen, C.; Xu, C.; Reddy, B.; Chada, K.; Kong, A.T. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis 2006, 27, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Khor, T.; Hu, R.; Yu, S.; Nair, S.; Ho, C.; Reddy, B.; Huang, M.; Newmark, H.; Kong, A.T. Chemoprevention of familial adenomatous polyposis by natural dietary compounds sulforaphane and dibenzoylmethane alone and in combination in ApcMin/+ mouse. Cancer Res. 2007, 67, 9937–9944. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Dashwood, W.M.; Li, L.; Kang, Y.; Kim, E.; Johnson, G.; Fischer, K.A.; Löhr, C.V.; Williams, D.E.; Ho, E.; et al. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin. Epigenet. 2015, 7, 102. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, Y.; Nishikawa, A.; Kitamura, Y.; Kanki, K.; Ishii, Y.; Umemura, T.; Hirose, M. Protective effects of benzyl isothiocyanate and sulforaphane but not resveratrol against initiation of pancreatic carcinogenesis in hamsters. Cancer Lett. 2006, 241, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Fu, J.; Watkins, D.N.; Srivastava, R.K.; Shankar, S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog-GLI pathway. Mol. Cell. Biochem. 2013, 373, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Conaway, C.C.; Wang, C.X.; Pittman, B.; Yang, Y.M.; Schwartz, J.E.; Tian, D.; McIntee, E.J.; Hecht, S.S.; Chung, F.L. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005, 65, 8548–8557. [Google Scholar] [CrossRef]
- Thejass, P.; Kuttan, G. Antimetastatic activity of Sulforaphane. Life Sci. 2006, 78, 3043–3050. [Google Scholar] [CrossRef]
- Thejass, P.; Kuttan, G. Modulation of cell-mediated immune response in B16F-10 melanoma-induced metastatic tumor-bearing C57BL/6 mice by sulforaphane. Immunopharmacol. Immunotoxicol. 2007, 29, 173–186. [Google Scholar] [CrossRef]
- Wang, F.; Shan, Y. Sulforaphane retards the growth of UM-UC-3 xenographs, induces apoptosis, and reduces survivin in athymic mice. Nutr. Res. 2012, 32, 374–380. [Google Scholar] [CrossRef]
- Singh, S.V.; Warin, R.; Xiao, D.; Powolny, A.A.; Stan, S.D.; Arlotti, J.A.; Zeng, Y.; Hahm, E.R.; Marynowski, S.W.; Bommareddy, A.; et al. Sulforaphane inhibits prostate carcinogenesis and pulmonary metastasis in TRAMP mice in association with increased cytotoxicity of natural killer cells. Cancer Res. 2009, 69, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.H.; Spinks, C.A.; Doleman, J.F.; Melchini, A.; Ball, R.Y.; Mills, R.D.; Mithen, R.F. The dietary isothiocyanate sulforaphane modulates gene expression and alternative gene splicing in a PTEN null preclinical murine model of prostate cancer. Mol. Cancer 2010, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Gerhardsson de Verdier, M.; Hagman, U.; Peters, R.K.; Steineck, G.; Overvik, E. Meat, cooking methods and colorectal cancer: A case-referent study in Stockholm. Int. J. Cancer 1991, 49, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.G.; Young, P.J.; Agus, C.; Knize, M.G.; Boobis, A.R.; Gooderham, N.J.; Lake, B.G. Cruciferous vegetable consumption alters the metabolism of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo [4,5,-b] pyridine (PhIP) in humans. Carcinogenesis 2004, 25, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Chen, J.; Egner, P.A.; Fahey, J.W.; Jacobson, L.P.; Stephenson, K.K.; Ye, L.; Coady, L.J.; Wang, J.; Wu, Y.; et al. Effects of Glucosinolate-Rich Broccoli Sprouts on Urinary Levels of Aflatoxin-DNA Adducts and Phenanthrene Tetraol in Randomized Clinical Trial in He Zuo Township, Qidong, People’s Republic of China. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2605–2613. [Google Scholar] [CrossRef]
- Kensler, T.W.; Ng, D.; Carmella, S.G.; Chen, M.; Jacobson, L.P.; Muñoz, A.; Egner, P.A.; Chen, J.G.; Qian, G.S.; Chen, T.Y.; et al. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis 2012, 33, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Chen, J.G.; Zarth, A.T.; Ng, D.K.; Wang, J.B.; Kensler, K.H.; Jacobson, L.P.; Muñoz, A.; Johnson, J.L.; Groopman, J.D.; et al. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: Results of a randomized clinical trial in China. Cancer Prev. Res. 2014, 7, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Riso, P.; Martini, D.; Visioli, F.; Martinetti, A.; Porrini, M. Effect of broccoli intake on markers related to oxidative stress and cancer risk in healthy smokers and nonsmokers. Nutr. Cancer 2009, 61, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Riso, P.; Martini, D.; Møller, P.; Loft, S.; Bonacina, G.; Moro, M.; Porrini, M. DNA damage and repair activity after broccoli intake in young healthy smokers. Mutagenesis 2010, 25, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.E.; Zang, Y.; Sen, M.; Li, C.; Wang, L.; Egner, P.A.; Fahey, J.W.; Normolle, D.P.; Grandis, J.R.; Kensler, T.W.; et al. Prevention of Carcinogen- Induced Oral Cancer by Sulforaphane. Cancer Prev. Res. 2016, 9, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Atwell, L.L.; Zhang, Z.; Mori, M.; Farris, P.; Vetto, J.T.; Naik, A.M.; Oh, K.Y.; Thuillier, P.; Ho, E.; Shannon, J. Sulforaphane bioavailability and chemopreventive activity in women scheduled for breast biopsy. Cancer Prev. Res. 2015, 8, 1184–1191. [Google Scholar] [CrossRef]
- Zhang, Z.; Atwell, L.; Farris, P.E.; Ho, E.; Shannon, J. Associations between cruciferous vegetable intake and selected biomarkers among women schedule for breast biopsy. Public Health Nutr. 2015, 19, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.J.; Campone, M.; Cortes, J.; Duhoux, F.P.; Ross, S.; Franklin, M.S. Final results of the STEM trial: SFX-01 in the treatment and evaluation of ER+ Her2- metastatic breast cancer (mBC). Ann. Oncol. 2019, 30 (Suppl. 5), v122. [Google Scholar] [CrossRef]
- Lozanovski, V.J.; Houben, P.; Hinz, U.; Hackert, T.; Herr, I.; Schemmer, P. Pilot study evaluating broccoli sprouts in advanced pancreatic cancer (POUDER trial)-study protocol for a randomized controlled trial. Trials 2014, 15, 204. [Google Scholar] [CrossRef]
- Traka, M.; Gasper, A.V.; Melchini, A.; Bacon, J.; Needs, P.W.; Frost, V.; Chantry, A.; Jones, A.M.E.; Ortori, C.A.; Barrett, D.A.; et al. Broccoli Consumption Interacts with GSTM1 to Perturb Oncogenic Signaling Pathways in the Prostate. PLoS ONE 2008, 3, e02568. [Google Scholar] [CrossRef]
- Cipolla, B.G.; Mandron, E.; Lefort, J.M.; Coadou, Y.; Negra, E.D.; Corbel, L.; Scodan, R.L.; Azzouzi, A.R.; Mottet, N. Effect of sulforaphane in Men with Biochemical Recurrence after Radical Prostatectomy. Cancer Prev. Res. 2015, 8, 712–719. [Google Scholar] [CrossRef]
- Traka, M.; Melchinin, A.; Coode-Beta, J.; Al Kadhi, O.; Saha, S.; Defernez, M.; Troncoso-Rey, P.; Kibblewhite, H.M.; O’Neill, C.; Bernuzzi, F.; et al. Transcriptional changes in prostate of men on active surveillance after a 12-mo glucoraphanin-rich broccoli intervention- results from the Effect of Sulforaphane on prostate CAncer PrEvention (ESCAPE) randomized controlled trial. Am. J. Clin. Nutr. 2019, 109, 1133–1144. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Singh, S.; Lin, Y.; Hahm, E.; Beumer, J.H.; Christner, S.M.; Rao, U.N.M.; Sander, C.; Tarhini, A.A.; Tawbi, H.A.; et al. Dose-response evaluation of broccoli sprout extract sulforaphane (BSE-SFN) in melanoma patients (Pts) with atypical/dysplastic nevi (A/DN). J. Clin. Oncol. 2016, 34, e21022. [Google Scholar] [CrossRef]
- Pavia, M.; Pileggi, C.; Nobile, C.G.; Angelillo, I.F. Association between fruit and vegetable consumption and oral cancer: A meta-analysis of observational studies. Am. J. Clin. Nutr. 2006, 83, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Bravi, F.; Bosetti, C.; Filomeno, M.; Levi, F.; Garavello, W.; Galimberti, S.; Negri, E.; La Vecchia, C. Foods, nutrients and the risk of oral and pharyngeal cancer. Br. J. Cancer 2013, 109, 2904–2910. [Google Scholar] [CrossRef] [PubMed]
- Joseph, M.A.; Moysich, K.B.; Freudenheim, J.L.; Shields, P.G.; Bowman, E.D.; Zhang, Y.; Marshall, J.R.; Ambrosone, C.B. Cruciferous vegetables, genetic polymorphisms in glutathione S-transferases M1 and T1, and prostate cancer risk. Nutr. Cancer 2004, 50, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Kirsh, V.A.; Peters, U.; Mayne, S.T.; Subar, A.F.; Chatterjee, N.; Johnson, C.C.; Hayes, R.B. Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. Prospective study of fruit and vegetable intake and risk of prostate cancer. J. Natl. Cancer Inst. 2007, 99, 1200–1209. [Google Scholar] [CrossRef]






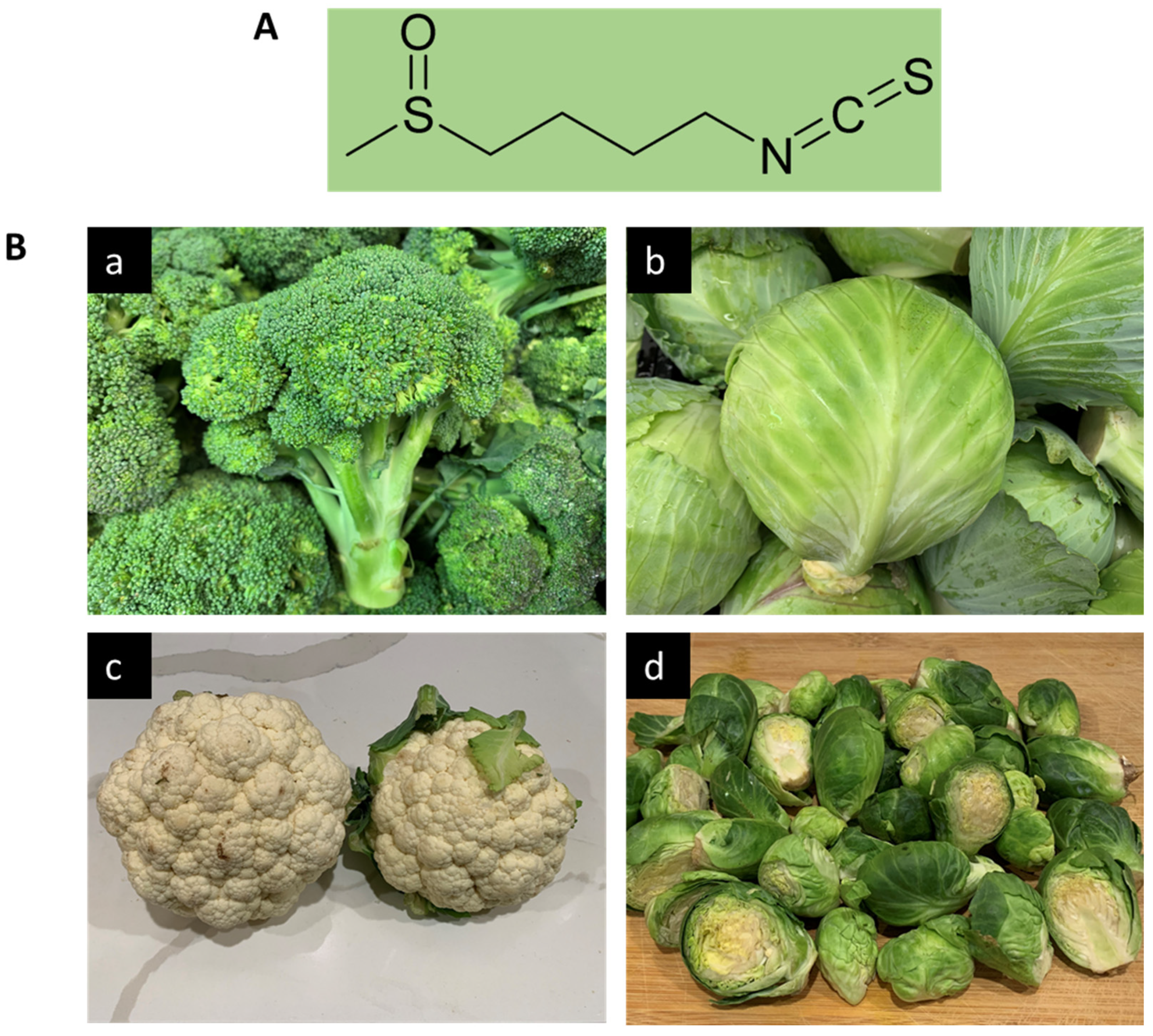{kind=link}
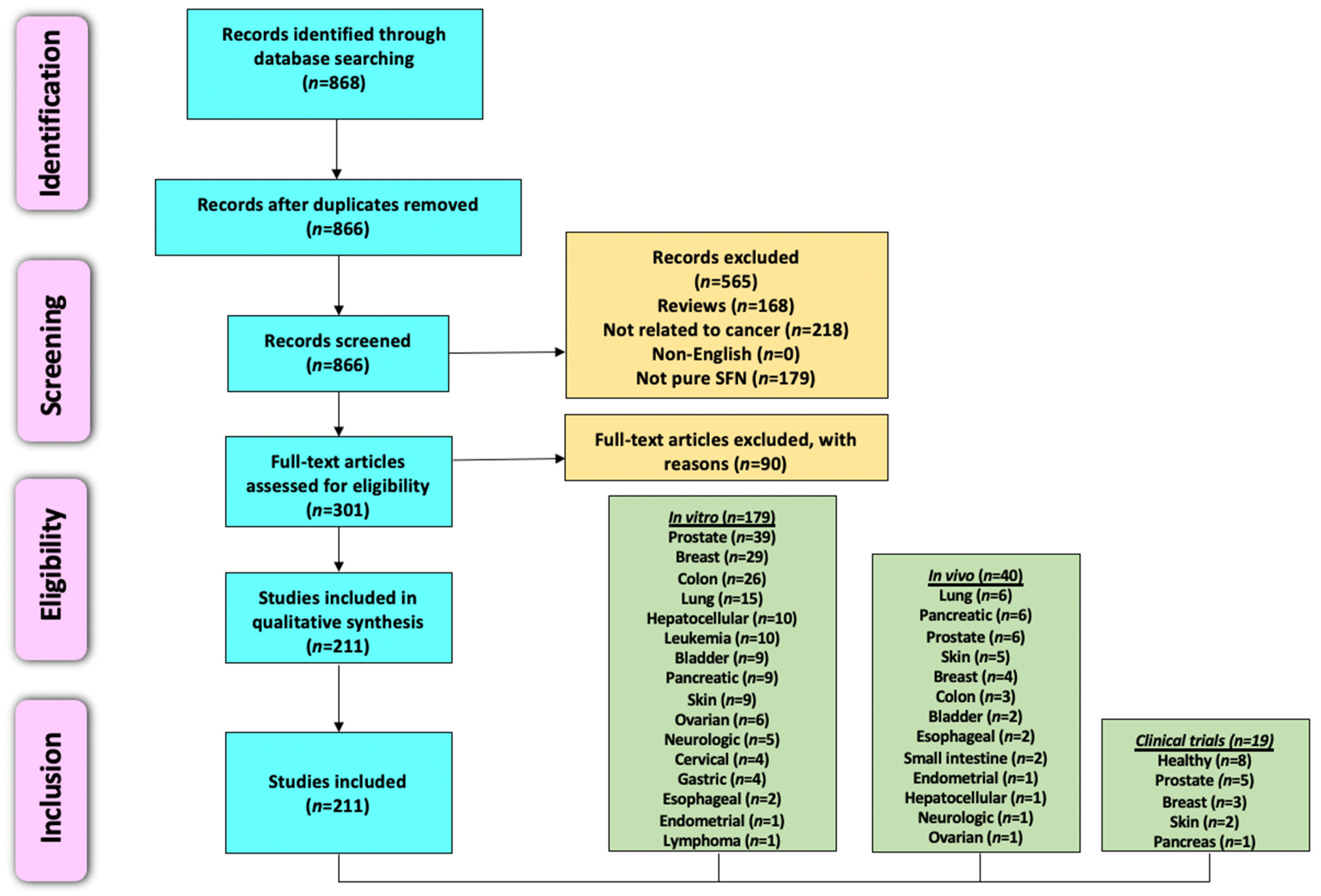{kind=link}
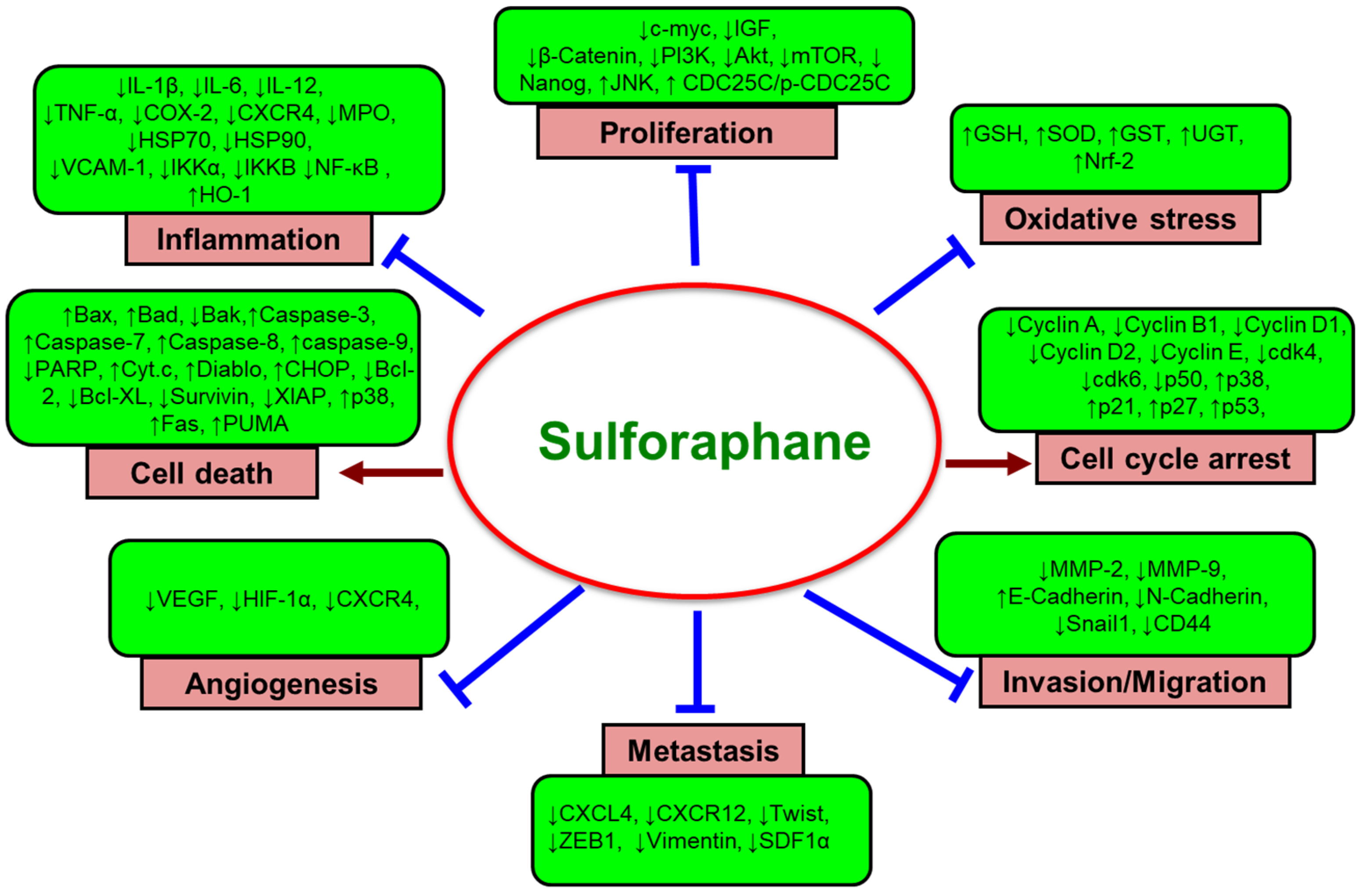{kind=link}
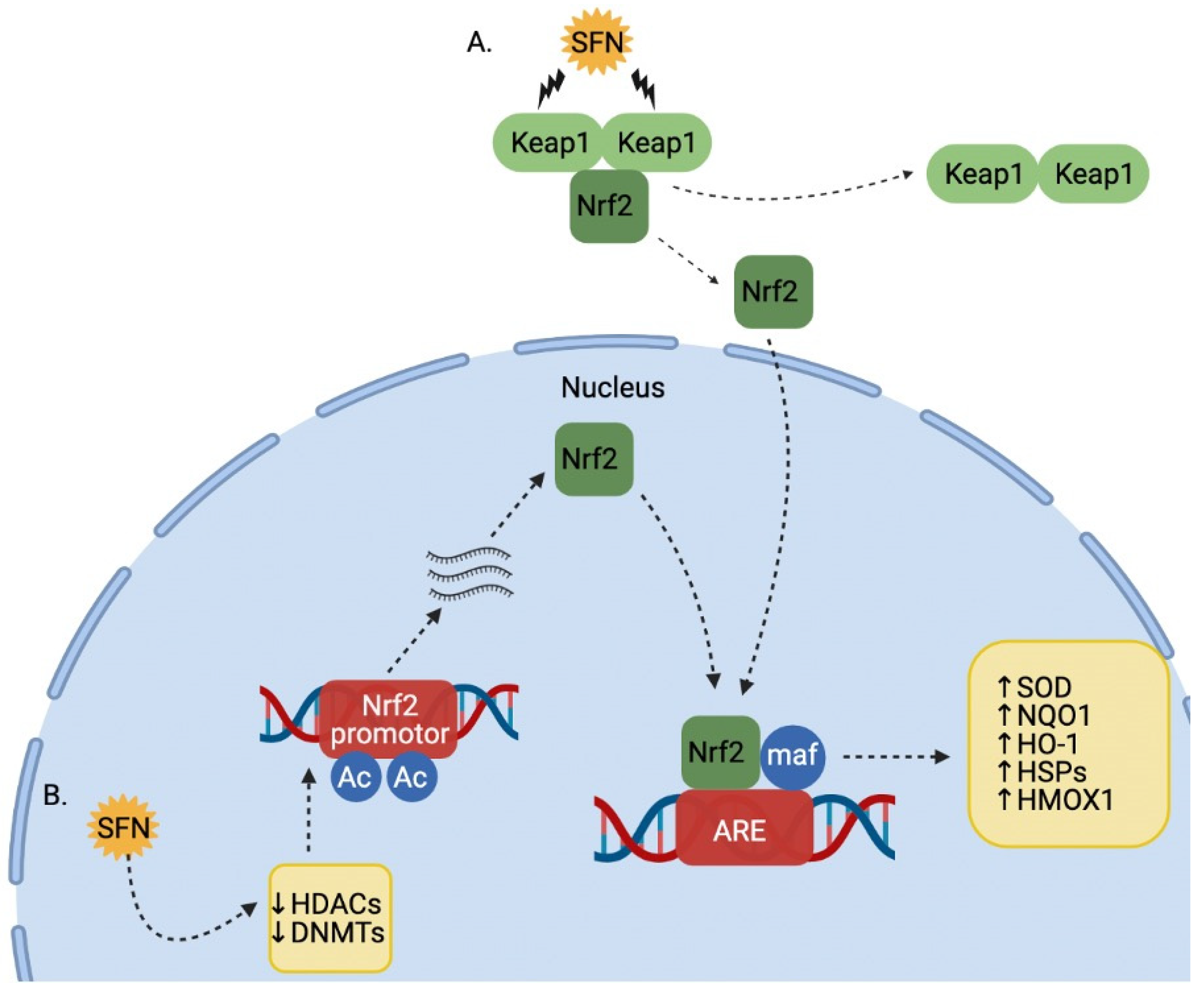{kind=link}
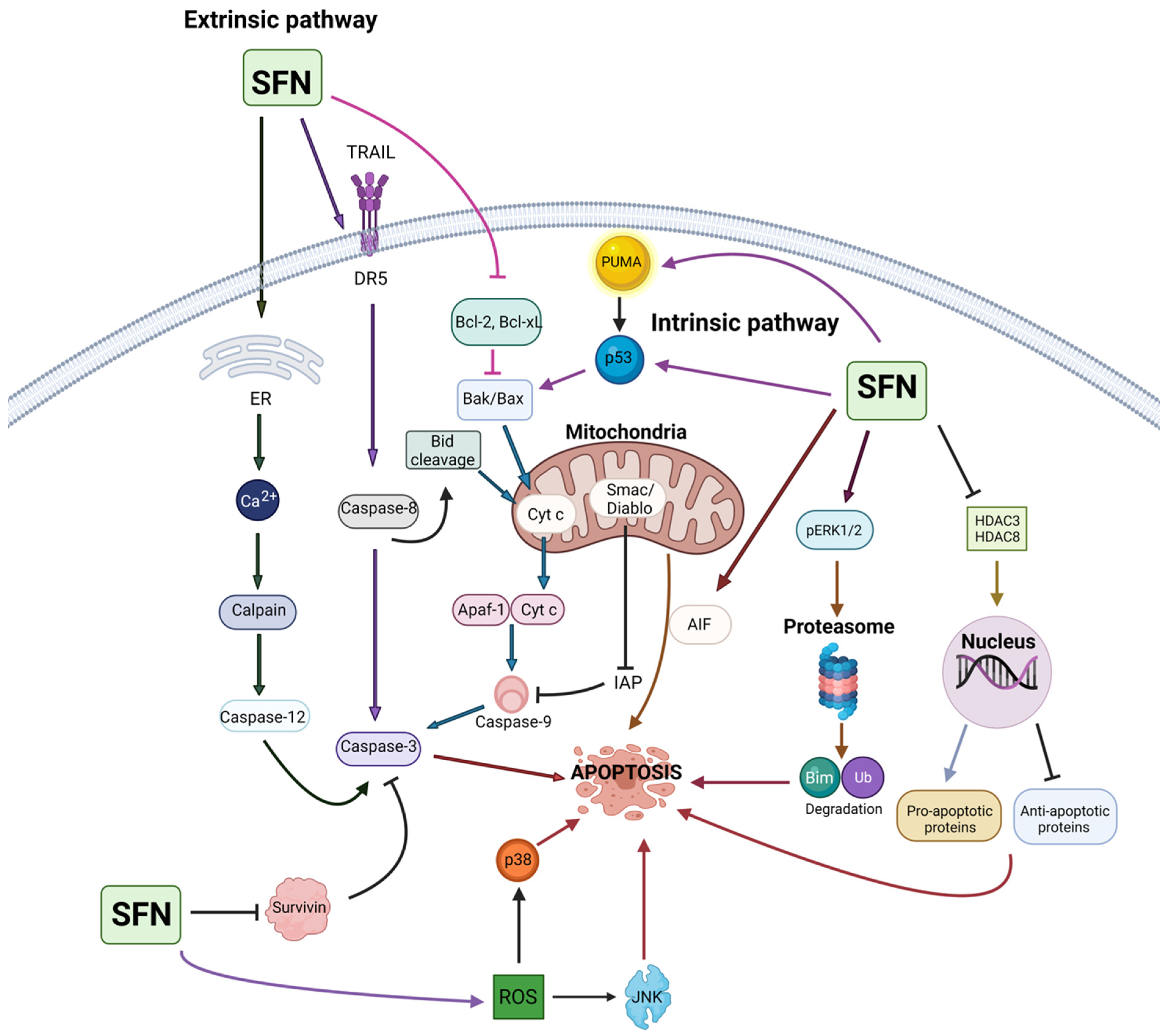{kind=link}
| Cell Lines Used | Conc. and Duration | Anticancer Effects | Mechanisms | References |
|---|---|---|---|---|
| Breast cancer | ||||
| MCF-7 | 0.1–100 µM (1–48 h) | Increased cytotoxicity | Not reported | Tseng et al., 2004 [93] |
| MCF-7 | 5–30 µM (6–24 h) | Suppressed cell proliferation | ↑G2/M phase arrest; ↑cyclin B1; ↑H1 phosphorylation; ↓tubulin polymerization | Jackson and Singletary, 2004 [94] |
| F3II | 5–30 µM (12–48 h) | Inhibited cell growth | ↑G2/M phase arrest; ↑cdc2 kinase activity; ↓tubulin polymerization; ↑apoptosis; ↓Bcl-2; ↓PARP; ↑caspase-3-like activity | Jackson and Singletary, 2004 [95] |
| MCF-7 | 2.5–50 µM (20–72 h) | Inhibited cell proliferation | ↑G2/M phase arrest; ↑microtubule dysfunction; ↑tubulin acetylation; ↓tubulin polymerization | Azarenko et al., 2008 [96] |
| MDA-MB-231, MDA-MB-468 | 5–50 µM (3–24 h) | Inhibited cell growth and invasion | ↑Apoptosis; ↑USP14; ↑UCHL5; ↑Ub-Prs | Ahmed et al., 2018 [97] |
| MDA-MB-231, MDA-MB-468, BT-474, MCF-7 | 1–25 µM (24, 72 h) | Decreased cell growth | ↓HDAC5; ↓HDAC5; ↓USF1; ↓USF1; ↓luciferase; ↓LSD1; ↑H3K4me1/2; ↑ AcH3K9; ↓USP28; ↑CTDSPL; ↑GLPR1; ↑CYLD; ↑TFP12; ↑PPP2R1B; ↑ISG15; ↑EGLN3 | Cao et al., 2018 [98] |
| MCF-7, MDA-MB-231 | 5 µM (24–72 h) | Inhibited cell growth | ↑G2-M phase arrest; ↓CCND1; ↓CDK4; ↓HDAC2; ↓HDAC3; ↓HMT; ↑p53; ↑p21; ↑H3K4Me3 | Royston et al., 2018 [99] |
| MDA-MB-231, MCF-7, T47D, MDA-MB-468 | 5–25 µM (24–72 h) | Inhibited cell growth | ↑G2-M phase arrest; ↓cyclin B1 (MDA-MB-231 and MCF-7); ↑apoptosis; ↓global HDAC; ↓EGFR; ↓HER-2 | Pledgie-Tracy et al., 2007 [100] |
| MCF-7, MDA-MB-231, SK-BR-3 | 5–20 µM (24 h) | Induced cytotoxicity | ↑p21; ↑oxidative stress; ↑carbonylation of lamin A/C; ↓lamin B1; ↓nucleolar RRN3; ↑nuclear RRN3; ↓NOP2; ↓WDR12 | Lewinska et al., 2017 [101] |
| MCF-7, MDA-MB-231, SK-BR-3 | 2.5–20 µM (24 h) | Inhibited cell proliferation | ↑Apoptosis; ↑G2/M phase arrest (MCF-7 and MDA-MB-231); ↑G0/G1 phase arrest (SK-BR-3); ↑p53; ↑p21; ↓CCNA2; ↓CCNB1; ↓CCNB2; ↓CCND3; ↓CCNE1, CCND1; ↓CCND2; ↓CCNH; ↓p-ERK1/2 (MDA-MB-231); ↑ROS; ↑DNA DSBs; ↑DNA SSBs; ↓Akt signaling; ↓ATP; ↓AMPK activation; ↓5-mdC; ↑HDAC5; ↓HDAC6-10; ↓DNMT1; ↓DNMT3B | Lewinska et al., 2017 [102] |
| MCF-7, MDA-MB-231 | 1–100 µM (24–72 h) | Induced cell death | ↑Apoptosis; ↑S phase cells; ↑G2/M phase cells; ↑p21; ↑p27; ↓cyclin A; ↓cyclin B1; ↓CDC2; ↑caspase-3; ↓Bcl-2; ↑autophagy; ↑LC3-I; ↑LC3-II | Kanematsu et al., 2010 [103] |
| MCF-7, SK-BR-3, MDA-MB-231, MDA-MB-468 | 5–30 µM (24 h) | Inhibited cell proliferation | ↑Autophagosomal vacuoles; ↓mTOR; ↓S6K1; ↓p-Akt | Pawlik et al., 2013 [104] |
| MDA-MB-231, BT549, MDA-MB-468 | 10, 25 µM (16–72 h) | Inhibited cell growth | ↑Autophagy; ↓P62; ↑Beclin1; ↑LC3-II; ↓HDAC6; ↑PTEN; ↓Akt | Yang et al., 2018 [105] |
| MDA-MB-231, MDA-MB-436, MDA-MB-468, MDA-MB-453 | 1–60 µM (24–72 h) | Inhibited cell proliferation | ↑Apoptosis; ↑G2/M phase arrest; ↑Egr1; ↑NQO1; ↑SL7A11; ↑G6PD; ↑GCLM; ↑SCD; ↑ID1; ↑IGFBP3; ↓cyclin B1; ↓Cdc2; ↓p-Cdc2; ↓Cdc25c | Yang et al., 2016 [106] |
| ZR-75-1 | 6.25–25 µM (4–72 h) | Inhibited cell growth | ↑G1/S phase arrest; ↓CDK2; ↓CDK4; ↓CDK6; ↓CDK2; ↓CDK4; ↓SERTAD1; ↓CCDN2; ↓HDAC3; ↓SERTAD; ↓CCDN2; ↓HDAC3 | Cheng et al., 2019 [107] |
| SH, SHR | 5–20 µM (72 h) | Decreased cell proliferation | ↑Apoptosis; ↑S-phase arrest (SH cells); ↑S-phase and G2/M-phase arrest (SHR cells); ↓HDAC1; ↑global histone H3 acetylation; ↑DCBLD2; ↓Septin 9 | Li et al., 2016 [108] |
| MCF-7, ZR-75-1 | 2.5–30 µM (24–72 h, 7 days) | Decreased cell proliferation | ↓ERα; ↓PR; ↑PSMB5 | Ramirez and Singletary, 2009 [109] |
| T47D, MCF-7, BT-474 | 2–50 µM (96 h) | Decreased cell viability | ↑Apoptosis; ↑PARP cleavage | Pawlik et al., 2016 [110] |
| MCF-7 | 25 µM 24–72 h | Inhibited cell proliferation | ↑Apoptosis; ↑caspase-7; ↑PARP cleavage; ↓Bcl-2; ↑Bax; ↓ERK1/2 MAPK; ↑p38 MAPK | Jo et al., 2007 [111] |
| MCF-7 | 0.01–75 µM (24 and 48 h) | Decreased cell viability | ↑Apoptosis; ↓Bcl-2; ↓COX-2 | Hussain et al., 2013 [112] |
| MCF-7, MDA-MB-231 | 5–20 µM (72 h) | Decreased cell viability | ↑Apoptosis; ↓CYP1A1 protein (all cell lines); ↓CYP19 (MCF-7); ↑CYP19; ↑aromatase (MDA-MB-231); ↑CYP1A2 (MDA-MB-231) | Licznerska et al., 2015 [113] |
| MCF-7, MDA-MB-231 | 10–70 µM (96 h) | Inhibited cell growth | ↑Apoptosis; ↑p53 (MCF-7); ↓PTEN methylation; ↑RARβ2; ↑p21 | Lubecka-Pietruszewska et al., 2015 [114] |
| MCF-7, MDA-MB-231 | 5–20 µM (3–15 days) | Inhibited cell proliferation | ↑Apoptosis; ↓hTERT; ↓telomerase activity; demethylation of CpGs of the CTCF binding site; ↑ac-H3; ↑ac-H3K9; ↓tri-me-H3K27; ↓tri-me-H3K9, ↓DNMT1; ↓DNMT3a | Meeran et al., 2010 [115] |
| MCF-7, MDA-MB-231 | 1–20 µM (24 h) | Inhibited cell proliferation | ↑Apoptosis; ↓HSP70; ↓HSP90; ↓HSF1; ↑p53; ↑p21; ↑AIF; ↑Bax; ↑Bad; ↓Bcl-2; ↑caspase-3; ↑caspase-8; ↑caspase-9 | Sarkar et al., 2012 [116] |
| MCF-7 | 10 µM (45 min, 6 h) | Inhibited cell growth | ↑Nrf2; ↑NQO1; ↑HMOX1; ↑H3K9Ac:H3 | Lo and Matthews, 2013 [117] |
| MCF-7 | 1–12 µM (4–24 h) | ↑TrxR1 | Wang et al., 2005 [118] | |
| MDA-MB-231, MDA-MB-468, BT-549, BT-474, SKBR3, HS578T | 20–60 µM (16, 24 h) | Decreased cell proliferation | ↑Nrf2; ↓RON | Thangasamy et al., 2011 [119] |
| MCF-7, MDA-MB-231 | 1–20 µM (24 h) | Decreased cell viability and migration | ↑CAV1; ↑CAV1; ↑condensed chromatin | Deb et al., 2014 [120] |
| MDA-MB-231-Luc-D3H1, JygMC(A) | 2.5–20 µM (48 h) | Decreased cell proliferation | ↓Primary tumorspheres; ↓secondary tumorspheres; ↓tertiary tumorspheres; ↓CR1; ↓CR3; ↓GRP78; ↓Alk4 | Castro et al., 2019 [86] |
| Gastrointestinal tract and associated cancers | ||||
| Esophageal cancer | ||||
| OE33, FLO-1 | 1–12.5 µM (0.5–5 h) | Inhibited cell growth | ↑Apoptosis; ↑G1 phase arrest; ↓HSP90; ↑p21 | Qazi et al., 2010 [121] |
| EC9706, ECa109 | 10–60 µM (3–72 h) | Inhibited cell proliferation and induced autophagy | ↑Apoptosis; ↑caspase-9; ↑LC3B-II; ↓p62; ↑Nrf2 | Lu et al., 2020 [122] |
| Gastric cancer | ||||
| AGS | 2.0–6.75 µM (3–24 h) | Decreased cell growth and migration | ↑Apoptosis; ↑ROS; ↑Bax; ↓Bcl-2; ↑cyt c; ↑caspase-8; ↑PARP-1 cleavage; ↑SFE; ↑p-JNK; ↑p-P-38; ↓p-ERK1/2 | Mondal et al., 2016 [123] |
| AGS | 2.5–20 µM (24, 48 h) | Inhibited cell viability | ↑Apoptosis; ↑G2/M phase arrest; ↑cyclin B1; ↑p53; ↑p21; ↑p-H3; ↑PARP cleavage; ↑p-AMPK; ↓cyt c; ↓MMP | Choi et al., 2018 [124] |
| MGC803, AGS | 2–32 µM (24–48 h) | Inhibited cell proliferation | ↑Apoptosis; ↑G2/M phase arrest; ↓SMYD2; ↓SMYD3 mRNA; ↓CYR61; ↓MYL9 | Dong et al., 2018 [125] |
| AGS, MKN45 | 31–250 µM (48 h) | Inhibited cell growth | ↑Apoptosis; ↑CDX1; ↑CDX2 | Kiani et al., 2018 [126] |
| Colon cancer | ||||
| SW620 | 5–100 µM (24–72 h) | Decreased cell proliferation | ↑Apoptosis; ↑caspase-3; ↑double-strand DNA breaks | Andělová et al., 2007 [127] |
| SW620 | 20 µM (12–48 h) | Inhibited cell proliferation | ↑Apoptosis; ↑caspase-9; ↑caspase-3; ↑caspase-7; ↑ATM kinase; ↑Chk2 kinase; ↑JNK | Rudolf et al., 2009 [128] |
| SW480 | 1–20 µM (3–48 h) | Inhibited cell proliferation | ↑Apoptosis; ↑caspase-3; ↑caspase-7; ↑caspase-9; ↑ERK; ↑p53; ↓Bcl-2; ↑Bax/Bcl-2 ratio; ↑ROS; ↑MDA | Lan et al., 2017 [129] |
| HT-29 | 5–30 µM (24–96 h) | Inhibited cell growth | ↑Apoptosis; ↑G2/M phase arrest; ↑cyclin A; ↑cyclin B1; ↑Bax; ↑PARP cleavage | Gamet-Payrastre et al., 2000 [130] |
| 40-16, 379.2 | 0.4–50 µM (10–72 h) | Decreased cell growth | ↑Apoptosis; ↑PARP cleavage; ↓Pro-C9; ↓Pro-C7; ↓Bax; ↓Bcl-xL | Pappa et al., 2006 [131] |
| 40-16 | 5, 10 µM (24–72 h) | Inhibited cell growth | ↑PARP cleavage; ↑subG1 phase arrest | Pappa et al., 2007 [132] |
| HCT-116 | 0.5–100 µM (16–48 h) | Suppressed cell growth | ↑Apoptosis; ↑histone H2A.X phosphorylation; ↑caspase-9; ↑caspase-3; ↑JNK; ↑Bid; ↑Bax; ↓Bcl-2 | Rudolf and Cervinka, 2011 [133] |
| HCT-116, HT-29, DLD1, KM12, SNU-1040 | 2.5, 5 µM (24–72 h) | Inhibited cell proliferation | ↑Apoptosis; ↑PARP cleavage; ↑G2/M phase arrest; ↑CDK1; ↑CDC25B; ↑MK2; ↑P38; ↑p-JNK; ↓microtubule polymerization | Byun et al., 2016 [134] |
| HT-29 | 6.25–100 µM (4–36 h) | Inhibited cell growth | ↑G1 phase arrest; ↓cyclin D1; ↓cyclin A; ↓c-myc; ↑P21; ↑ERK; ↑JNK; ↑p38 | Shen et al., 2006 [135] |
| CT116 | 5–15 µM (72 h) | Inhibited cell viability | ↑Apoptosis; ↑G2 phase arrest; ↑p-SAPK; ↓c-Myc | Zeng et al., 2011 [136] |
| 40-16 | 0.4–50 µM (3–48 h) | Inhibited cell proliferation | ↑G2/M phase arrest (6, 12, and 24 h); ↑subG1 phase arrest (48 h); ↓GSH | Pappa et al., 2007 [137] |
| HT-29 | 15 µM (48 h) | Inhibited cell growth | ↑Apoptosis; ↓p-cdc2; ↑p21; ↑G2/M phase arrest; ↑Rb phosphorylation; ↑Rb protein | Parnaud et al., 2004 [138] |
| WiDr | 2.5–80 µM (16 h) | Inhibited cell proliferation | ↑Apoptosis; ↑autophagy; ↑LC3-1; ↑LC3-II; ↓Bcl-2 | Nishikawa et al., 2010 [139] |
| DLD-1, HCT116, LoVo | 5–20 µM (24 h) | Inhibited cell proliferation | ↓SKP2 mRNA; ↓SKP2 protein; ↑p27KIP1; ↑Akt; ↑ERK | Chung et al., 2015 [140] |
| Caco-2 | 1–50 µM (2, 24 h) | Inhibited cell viability | ↑KLF4; ↑p21; ↓CDX-2; ↓KLF5; ↓AMACR | Traka et al., 2005 [141] |
| HCT-116, HT29 | 15 µM (6 h) | Inhibition of cell growth and migration | ↑p53; ↓Wnt/β-catenin; ↑Nrf2; ↑NMRAL2P | Johnson et al., 2017 [142] |
| Caco-2 | 5–25 µM (6–36 h) | Induced autophagy | ↑LC-II; ↑UGT1A1; ↑UGT1A8; ↑UGT1A10 mRNA; ↑Nrf2 | Wang et al., 2014 [143] |
| Caco-2 | 0.5–20 µM (48 h) | Decreased cell viability | ↑MRP2 | Harris and Jeffery, 2008 [144] |
| HTC-116, HT-29, SW48, SW480 | 15 µM (24 h) | Decreased cell viability | ↑G2/M phase arrest; ↓HDAC3; ↓HDAC6; ↑p-H2AX; ↑p-ATR; ↑CtIP acetylation | Rajendran et al., 2013 [145] |
| RKO, HCT-116 | 2.5–20 µM (72 h) | Inhibited cell growth | ↑Apoptosis; ↓miR-21; ↓HDAC mRNA; ↓hTERT mRNA | Martin et al., 2018 [146] |
| HCT-116, SW480 | 0.9–60 µM (24 h) | Decreased cell proliferation | ↑pH2AX; ↑pRPA32; ↑p300; ↑histone H4 acetylation | Okonkwo et al., 2018 [147] |
| HT-29 | 0.25–10 µM (24 h) | Inhibited cell growth | ↓TNF-α; ↓IL-1β; ↓IL-6; ↓IFN-γ; ↓IL-1β | Bessler and Djaldetti, 2018 [148] |
| HT-29 | 10–50 µM (24, 48 h) | Suppressed cell growth and migration | ↑Apoptosis; ↑subG1 phase arrest; ↑caspase-3; ↓COX-2; ↓HIF-1; ↓VEGF; ↓CXCR4; ↓PGE2 | Tafakh et al., 2018 [149] |
| Caco-2 | 5–100 µM (24, 72 h) | Inhibited cell proliferation | ↑p-ERK1/2; ↑p-Akt; ↑NQO1; ↑UGT1A1; ↓MRP2 | Jakubikova et al., 2005 [150] |
| HCT-116, LoVo, Caco-2, HT-29 | 1–15 µM (72 h) | Decreased cell growth | ↑Apoptosis; ↑ROS; ↓procaspase-8 | Kim et al., 2010 [151] |
| HCT-116 | 12.5–50 µM (1–24 h) | Inhibited cell migration | ↓HIF-1α; ↓VEGF; ↓HO-1; ↓GLUT1 | Kim et al., 2015 [152] |
| Hepatocellular cancer | ||||
| HepG2, Hepa1c1c7 | 5–100 µM (24 h) | Exhibited cell cytotoxicity | ↑ERK2; ↑MAPK pathway | Yu et al., 1999 [153] |
| HepG2 | 100 µM (24 h) | Reduced cell viability and promoted cell death | ↑Apoptosis; ↑MT-I RNA; ↑MT-II RNA, ↑MT protein expression; ↑Nrf2; ↑p38; ↑JNK/MAPK pathways; ↑caspase-3; ↑PARP cleavage; ↑Bax; ↓Bcl-2; ↓Bcl-xL | Yeh and Yen, 2005 [154] |
| HepG2 | 5–30 µM (48 h) | Inhibited cell viability and promotes cell death | ↑Apoptosis; ↑caspase-3; ↑Bax; ↓Bcl-2; ↓Bcl-xL; ↓PARP; ↓β-catenin | Park et al., 2007 [155] |
| HepG2 | 20 µM (24 h) | Reduced cellular proliferation | ↑HO-1; ↑ARE; ↑Nrf2; ↑ERK1/2; ↓Keap1; ↓p38 MAPK; ↓p-MKK3/6 | Keum et al., 2006 [156] |
| Huh-7, SNU-449, NCTC | 20–60 µM (24 h) | Reduced cell viability and promoted cell cycle arrest | ↑Apoptosis; ↑G2/M phase arrest; ↑caspase-3; ↑caspase-8; ↑caspase-9; ↓PFKFB4; ↓HIF-1α; ↓VEGF | Jeon et al., 2011 [157] |
| HepG2 | 1.25–20 µM (24 h) | Reduced cellular proliferation, adhesion, migration, and invasion | ↓STAT3; ↓HIF-1α; ↓VEGF | Liu et al., 2017 [158] |
| Hep3B | 5–20 µM (24, 48 h) | Decreased cell viability and promoted cell death | ↑Apoptosis; ↓telomerase; ↓hTERT; ↓Akt; ↑ROS | Moon et al., 2010 [159] |
| HepG2 | 10–80 µM (48 h) | Inhibited cell proliferation, migration, and invasion | ↑Apoptosis; ↓TGF-β–induced EMT; ↓Vimentin; ↑E-Cadherin; ↑GO/G1 arrest; ↑ROS | Wu et al., 2016 [160] |
| HepG2 | 80 µM (24–72 h) | Inhibited cell proliferation and promoted cell death | ↑Apoptosis; ↑Bip/GRP78; ↑XBP-1; ↑caspase-12; ↑Bid; ↑CHOP/GADD153 | Zou et al., 2017 [161] |
| Hepa 1c1c7, HepG2 | 1–40 µM (24 h) | Reduced cell viability | ↑CYP1A1; ↑AhR transformation; ↑AhR binding to XRE | Anwar-Mohamed and El-Kadi, 2009 [162] |
| Pancreatic cancer | ||||
| MIA PaCa-2, PANC-1 | 5–40 µM (24–72 h) | Promoted cell cycle arrest and death | ↑Apoptosis; ↑caspase-3, ↑caspase-8, ↑G2-M arrest, ↑ROS | Pham et al., 2004 [163] |
| AsPC-1, BxPc-3, MIA PaCa-2, PANC-1 | 0.1–100 µM (12–48 h) | Inhibited cell proliferation and promoted cell death | ↑Apoptosis; ↓Akt; ↓Cdk4; ↓p53; ↑proteasomal degradation of HSP90 client proteins; ↑caspase-3; ↓HSP90-p50Cdc37 complex | Li et al., 2012 [164] |
| AsPC-1, BxPc-3, Capan-1, MIA PaCa2 | 10 µM (24, 48 h) | Reduced cell viability | ↑Apoptosis; ↓NF-κB binding | Kallifatidis et al., 2009 [165] |
| ASPC, PANC-1, and human pancreatic CSCs | 5–20 µM (1–7 days) | Reduced cellular proliferation and promoted cell death | ↑Apoptosis; ↓Bcl-2, ↑caspase-3; ↓Nanog; ↓Oct-4; ↓PDGFα; ↓Smo; ↓Gli1; ↓Gli2 | Rodova et al., 2012 [166] |
| AsPC-1, BxPc-3, PANC-1, MIA PaCa-2 | 10 nM (24 h) | Promoted cell death | ↑Apoptosis; ↑miR-365a-3p | Yin et al., 2019 [167] |
| AsPC-1, BxPc-3, PANC-1 | 10 µM (24 h) | Promoted cell death | ↑miR-135b-5p; ↑RASAL2 | Yin et al., 2019 [168] |
| PANC-1, MIA PaCa-2 | 1–100 µM (24–72 h) | Inhibited cellular proliferation, invasion, and migration | ↑Apoptosis; ↑ROS; ↑AMPK; ↑E-Cadherin; ↓N-Cadherin; ↓Vimentin; ↑Nrf2; ↑HO-1 | Chen et al., 2018 [169] |
| BxPc-3, AsPC-1 | 10 µM (24 h) | Reduced cell viability | ↑E-Cadherin; ↑GJIC; ↑Cx43; ↓c-Met; ↓CD133 | Forster et al., 2014 [170] |
| PANC-1 | 10 µM (24 h) | Exhibited cytotoxicity | ↑Cx43; ↑GJA1 mRNA; ↑GJIC; ↓miR30a-3p | Georgikou et al., 2020 [171] |
| Gynecological cancers | ||||
| Cervical cancer | ||||
| HeLa | 5–30 µM (48 h) | Decreased cell viability | ↑Apoptosis; ↑sub-G1 phase arrest; ↑Bax; ↓Bcl-2; ↓Bcl-xL; ↓pro-caspase-3; ↓PARP; ↓β-catenin | Park et al., 2007 [156] |
| HeLa | 0.01–100 µM (24 h) | Inhibited cell growth | ↑Apoptosis; ↑caspase-3; ↓Bcl-2; ↓COX-2; ↓IL-1β | Sharma et al., 2011 [172] |
| HeLa | 2.5 µM (24–72 h) | Inhibited cell growth | ↓DNMT; ↓DNMT3B; ↓HDAC1; ↑RARβ; ↑CDH1; ↑DAPK1; ↑GSTP1 | Khan et al., 2015 [173] |
| HeLa | 6.25–25 µM (24, 72 h) | Decreased cell proliferation | ↑G2/M phase arrest; ↑MPM-2; ↓cyclin B1; ↓cyclin B1/CDC2 complex; ↑CDC25C/p-CDC25C ratio; ↑GADD45β | Cheng et al., 2016 [174] |
| Endometrial cancer | ||||
| MFE280, KLE, Ishikawa, Hec1B, Hec1A, MFE296, AN3CA | 1–32 µM (24–72 h) | Inhibited cell viability | ↑Apoptosis; ↑G2/M phase arrest; ↓ATP; ↑p21; ↑p27; ↑Cdc2 phosphorylation, ↑caspase-3; ↑Bax; ↓Bcl-2; ↓Cox IV; ↓MEK; ↓ERK | Rai et al., 2020 [175] |
| Ovarian cancer | ||||
| SKOV3 | 10–100 µM (12, 24 h) | Inhibited cell growth | ↑Apoptosis; ↓Akt; ↓p-Akt; ↓PI3K; ↓cyclin D1; ↓cdk4; ↓cdk6 | Chaudhuri et al., 2007 [176] |
| OVCAR3, SKOV3 | 2–50 µM (24–72 h) | Reduced cell proliferation | ↑Apoptosis; ↑G1 phase arrest | Chuang et al., 2007 [177] |
| MDAH 2274, SKOV3 | 5–20 µM (12–72 h) | Induced growth arrest and inhibited cell migration | ↑Apoptosis; ↑G1 phase arrest; ↓RB; ↓p130; ↑p107; ↓E2F-1; ↓E2F-2; ↓E2F-3; ↓G1 phase; ↓cyclins; ↓CDKs; ↑non-phosphorylated RB; ↓E2F-1 | Bryant et al., 2010 [178] |
| OVCAR3, OVCAR4, OVCAR5, SKOV3 | 100 µM (72 h) | Inhibited cell proliferation | ↑p38; ↑ERK; ↑JNK (OVCAR3 and SKOV3); ↑thioredoxin reductase (OVCAR3) | Kim et al., 2017 [179] |
| A2780, SKOV3 | 2–200 µM (6, 24 h) | Decreased cell growth | ↑Apoptosis; ↑HSP27; ↑JNK; ↑MEK1; ↑p38; ↑p90rsk phosphorylation; ↑IP3R2; ↑NFR2; ↑CHOP; ↑ATF4; ↑GCLC; ↑HMOX1; ↑NQO-1 | Hudecova et al., 2016 [180] |
| PA-1 | 6.25, 12.5 µM (24, 72 h) | Inhibited cell proliferation | ↑G2/M phase arrest; ↓CDC2; ↓cyclin B1/CDC2 complex | Chang et al., 2013 [181] |
| Hematological cancers | ||||
| Leukemia | ||||
| Jurkat T | 3–30 µM (24–72 h) | Decreased cell proliferation | ↑Apoptosis; ↑G2/M phase arrest | Fimognari et al., 2002 [182] |
| Jurkat T | 3–30 µM (24, 48 h) | Reduced cell viability | ↑Apoptosis; ↑G2/M phase arrest; ↑p53; ↑Bax; ↓cyclin D3; ↓CDK4; ↓CDK6 | Fimognari et al., 2003 [183] |
| HL60 | 10–110 µM (24–72 h) | Inhibited cell viability | ↑Apoptosis | Fimognari et al., 2008 [184] |
| U937 | 1–5 µM (48 h) | Reduced cell growth | ↑Apoptosis; ↑sub-G1 phase arrest; ↑Bax; ↓Bcl-2; ↑caspase-3; ↑PARP cleavage; ↑ROS; ↑MMP | Choi et al., 2008 [185] |
| U937, HL60, NB-4, KG-1 | 15–60 µM (24, 48 h) | Decreased cell proliferation | ↑Apoptosis; ↓miR-155 | Koolivand et al., 2018 [186] |
| HL60 | 6–10 µM (10–48 h) | Decreased cell viability | ↑G2/M phase arrest; ↑ROS ↑intracellular Ca2+; ↑caspase-3; ↑caspase-8; ↑caspase-9; ↑Bax; ↑Bid; ↑Fas; ↑Fas-L; ↑Endo G; ↑AIF; ↑cyt c; ↓Bcl-xL; ↑FADD | Shang et al., 2016 [187] |
| HL60 | 1–25 µM (24, 48 h) | Inhibited cell viability | ↑Apoptosis; ↑NQO1; ↓NQO1; ↓Keap1; ↑Nrf2; ↓PARP; ↓pro-caspase-2; ↓pro-caspase-3; ↓p50; ↑Bax; ↓Bcl-2; ↓NF-κB | Wu et al., 2016 [188] |
| Nalm-6, REH, RS4 | 2–40 µM (24, 48 h) | Reduced cell growth | ↑Caspase-3; ↑caspase-8; ↑caspase-9; ↑PARP cleavage; ↑G2/M phase; ↑S phase arrest; ↑p21; ↑cyclin B1; ↓Akt; ↓p-mTOR | Suppipat et al., 2012 [189] |
| B1647 | 10, 30 µM (24 h) | Inhibited cell proliferation | ↓AQP8; ↓ROS; ↑Nox4 | Prata et al., 2018 [190] |
| L-1210 | 1–5 µM (24, 48 h) | Induced cell growth arrest | ↑Apoptosis; ↑DNA strand breaks; ↑PS externalization | Misiewicz et al., 2003 [191] |
| Lymphoma | ||||
| B-lymphoma cells | 1–10 µM (24 h) | Inhibited cell growth | ↑Caspase-3; ↑caspase-7; ↑caspase- 9; ↑PARP cleavage; ↓p38 MAPK; ↓Akt | Ishiura et al., 2019 [192] |
| Lung cancer | ||||
| A549 | 1–100 µM (24 h) | Induced mitotic arrest and promoted cell death | ↑Apoptosis; ↑G1/S arrest; ↑G2/M arrest; ↓tubulin polymerization; ↑ROS; ↓GSH | Mi and Chung, 2008 [193] |
| LTEP-A2 | 6.25–50 µM (3–72 h) | Inhibited cellular proliferation | ↑Apoptosis; ↑G2/M arrest | Liang et al., 2008 [194] |
| A549 | 30–90 µM (24 h) | Decreased cell proliferation | ↑G2/M phase; ↓G0/S phase; ↑p21; ↓cyclin D1 | Zuryn et al., 2016 [195] |
| H1299 | 5–15 µM (24, 48 h) | Promoted cell cycle arrest and decreased cell viability | ↑Apoptosis; ↑necrosis; ↑G2/M phase; ↓G0/S phase; ↓cyclin B1; ↑cyclin D1; ↑cyclin K | Zuryn et al., 2019 [196] |
| A549, H1299 | 5–15 µM (48 h) | Promoted cell cycle arrest and cell death | ↑Apoptosis; ↑H3 acetylation; ↑H4 acetylation; ↑p53; ↑p21; ↑Bax; ↑G0/G1 arrest; ↑G2/M arrest; ↓HDAC | Jiang et al., 2016 [197] |
| A549 | 2.5, 5 µM (5 days) | Decreased cell viability | ↑H3K4me1; ↓miR-9-3; ↓DNMT3a; ↓HDAC1; ↓HDAC3; ↓HDAC6; ↓CDH1; ↓CpG methylation | Gao et al., 2018 [198] |
| A549, H1299 | 1–15 µM (7 days) | Inhibited cell proliferation and the formation of tumorspheres | ↑Apoptosis; ↓miR-19a; ↓miR-19b; ↓Wnt/β-catenin pathway; ↑Bax; ↑caspase-3; ↑caspase-8; ↑caspase-9; ↓CD133; ↓CD44; ↓ALDH1A1; ↓nanog; ↓oct4; ↓PCNA; ↓cyclin D1 | Zhu et al., 2017 [199] |
| H1299, 95C, 95D | 0.5–100 µM (24, 48 h) | Inhibited cell proliferation, migration, and invasion | ↓miRNA-616-5p; ↓β-catenin; ↓N-cadherin; ↓vimentin | Wang et al., 2017 [200] |
| A549, CL1-5 | 10–40 µM (72 h) | Reduced cell viability and aggregation | ↑Apoptosis; ↑chromatin condensation; ↑anoikis, ↑annexin V binding; ↑PS externalization; ↑p53; ↑p21; ↑Bad; ↑Bax; ↑cleaved PARP, ↓procaspase-3; ↓procaspase-7; ↓procaspase-9; ↓p-FAK; ↓p-Akt; ↓β-catenin | Tsai et al., 2019 [201] |
| XWLC-05 | 0.5–5 µg/L (24, 48, 72 h) | Promoted cell cycle arrest and death | ↑Apoptosis; ↑G2/M phase; ↓G0/S phase; ↑p73; ↑PUMA; ↑Bax; ↑caspase-9; ↓Bcl-2; ↓p53 | Zhou et al., 2017 [202] |
| Cadmium-transformed BEAS-2BR | 2.5, 5, 10 µM (24 h) | Exhibited cytotoxicity | ↑Apoptosis; ↓apoptosis resistance; ↑autophagy; ↑caspase-3; ↑C-PARP; ↓constitutive Nrf2; ↓Bcl-2 | Wang et al., 2018 [203] |
| PC9/gef, H1975, A549, CL1-5, H3255 | 5–20 µM (48 h) | Reduced cell proliferation | ↓pEGFR; ↓p-Akt; ↓p-STAT3; ↑proteasome activity | Chen et al., 2015 [204] |
| A549, H1299 | 0.5–5 µM (72 h) | Reduces cellular proliferation, migration, and invasion | ↑ERK5; ↑p-ERK5 ↑E-Cadherin; ↑ZO-1; ↓pc-jun; ↓pc-Fos; ↓N-Cadherin; ↓Snail1; ↓MMP-2 | Chen et al., 2019 [205] |
| SK-1, A549 | 5–30 µM (24 h) | Decreased cell viability and promoted cell death | ↑Apoptosis; ↑ERK1/2; ↑Bax; ↑caspase-3; ↑26S proteasome activity; ↓Bim | Geng et al., 2017 [206] |
| HBE exposed to 2% TS and A549 | 1–40 µM (1–7 days) | Inhibited TS-induced, CSC-like properties | ↓CD133; ↓ALDH1A1; ↓Oct4; ↓Nanog; ↓IL-6; ↓NICD; ↓Hes1; ↓ΔNp63α | Xie et al., 2019 [207] |
| Neurological cancer | ||||
| T98G and U87MG | 20, 40 µM (24, 48 h) | Decreased cell viability and promoted cell death | ↑Apoptosis; ↑intracellular Ca+2; ↑Bax:Bcl2; ↑caspase-3; ↑caspase-9; ↑caspase-12; ↑cyt. c; ↑calpain; ↑α-spectrin degradation; ↑ICAD cleavage; ↑AIF; ↑Smac; ↑Diablo; ↓IAPs; ↓NF-κB | Karmakar et al., 2006 [208] |
| U87, U373, U118, SF767 | 5–50 µM (24, 48 h) | Inhibited cell survival and promoted cell death | ↑Apoptosis; ↑ROS; ↑DNA double-strand breaks; ↑γ-H2AX ↑caspase-3; ↑caspase-7; ↑caspase-9 | Bijangi-Visheshsaraei et al., 2017 [209] |
| U87 and U251 | 1–50 µM (24, 48 h) | Reduced cell viability and promoted cell death | ↑Apoptosis; ↑caspase-3; ↑Bax; ↑ROS; ↓Bcl-2; ↓p-STAT3 | Miao et al., 2017 [210] |
| U251MG | 10–40 µM (24 h) | Reduced cell viability and invasion | ↑Apoptosis; ↑Bad; ↑Bax; ↑cyt. c; ↑Annexin V-binding capacity; ↓Bcl-2; ↓survivin; ↓invasion; ↓MMP-2; ↓MMP-9; ↓Galectin-3 | Zhang et al., 2016 [211] |
| U87MG, U373MG | 10–90 µM (24 h) | Decreased cell proliferation, migration, and invasion | ↑ERK1/2; ↑CD44v6; ↓MMP-2 | Li et al., 2014 [212] |
| Skin cancer | ||||
| ME-18 | 1–5 µM (24, 48 h) | Induced cell growth arrest | ↑Apoptosis; ↑DNA strand breaks; ↑PS externalization | Misiewicz et al., 2003 [191] |
| A375, 501MEL | 1–5 µg/mL (2–48 h) | Suppressed cell growth, invasion, and metastasis | ↑Apoptosis; ↑MDM2; ↑BAX; ↑PUMA; ↑GADD45A; ↓CDKN1A; ↑FAS; ↑caspase-3; ↑caspase-8; ↑caspase-9; ↓Bcl2; ↑BBC3; ↓ADORA1; ↑HMOX1; ↑TXNRD1; ↑GGLC; ↑GCLM;↑AKR1B10; ↑G6PD; ↑HTRA3; ↓FST; ↓ITGB4; ↓PLAT; ↓ITGB2; ↓G2/M phase; ↑CDKN1A; ↑EGR1; ↑GADD45B; ↑ATF3 | Arcidiacono et al., 2018 [83] |
| A375 | 2 µg/mL (24–72 h) | Shifted growth factor receptor ratio from prosurvival to proapoptotic | ↑Apoptosis | Arcidiacono et al., 2018 [213] |
| A375 | 0.1–100 µM (24, 48 h) | Decreased cell survival | ↑Apoptosis; ↑caspase-3; ↑caspase-4; ↑caspase-6; ↑caspase-7; ↑caspase-8; ↑caspase-9 | Mantso et al., 2016 [214] |
| A375 and WM793 | 1–20 µM (24, 48 h) | Reduced spheroid formation, migration, and invasion | ↑Apoptosis; ↓Ezh2; ↓H3K27me3; ↓Bmi-1; ↓Suz12 | Fisher et al., 2016 [215] |
| B16F-10 | 1–5 µg/mL | Reduced cell viability and proliferation | ↑Apoptosis; ↑caspase-3; ↑caspase-9; ↑Bax; ↑p53; ↓caspase-8; ↓Bcl-2; ↓Bid; ↓NF-κB; ↓IL-1β; ↓IL-6; ↓TNF-α; ↓IL-12p40; ↓GM-CSF; ↓p65; ↓p50; ↓c-Fos; ↓ATF-2; ↓CREB; ↓c-Rel | Hamsa et al., 2011 [216] |
| B16 | 20–50 µM (24–72 h) | Reduced cell viability | ↓HDAC | Enriquez et al., 2013 [217] |
| B16 and S91 | 20–50 µM (24–72 h) | Inhibited cell growth and proliferation | ↓HDAC | Do et al., 2010 [218] |
| Bowes and SK-MEL-28 | 5–100 µM (2–48 h) | Decreased cellular proliferation | ↑Apoptosis; ↑p-p38 kinase; ↑p53; ↑PUMA; ↑Bax; ↑ROS | Rudolf et al., 2014 [219] |
| Urogenital cancers | ||||
| Bladder cancer | ||||
| T24 | 5–20 µM (24, 48 h) | Inhibited cell proliferation | ↑Apoptosis; ↓S and G2/M phase cells; ↑p27 | Shan et al., 2006 [220] |
| T24 | 50–20 µM (4–24 h) | Decreased cell growth | ↓COX-2; ↑nuclear NF-κB translocation; ↑p38 | Shan et al., 2009 [221] |
| T24 | 5–20 µM (10, 24 h) | Inhibited cell growth | ↑TR-1 mRNA; ↑GSTA1 mRNA; ↓COX-2 | Shan et al., 2010 [222] |
| T24 | 5–20 µM (24 h) | Decreased cell invasion and migration | ↑E-cadherin; ↓Snail; ↓ZEB1; ↑miR200c | Shan et al., 2013 [223] |
| T24 | 10, 20 µM (24 h) | Inhibited cell growth | ↑Apoptosis; ↑caspase-3; ↑caspase-9; ↑PARP cleavage; ↓XIAP; ↓cIAP-1; ↓cIAP-2; ↑Bax; ↑cyt. c; ↑ER stress; ↑GRP78; ↑CHOP; ↑ROS; ↑Nrf2; ↓Keap1; ↑HO-1 | Jo et al., 2014 [224] |
| RT4, J82, UMUC3 | 5–100 µM (48 h) | Inhibited cell proliferation | ↑Apoptosis; ↓NHU; ↑G2/M phase arrest; ↑caspase-3; ↑caspase-7 activity; ↑PARP cleavage; ↓survivin; ↓EGFR; ↓HER2/neu | Abbaoui et al., 2012 [82] |
| RT4, J82, UMUC3 | 4–20 µM (3–48 h) | Decreased cell growth | ↓HDAC; ↑p21 (RT4 cells); ↓thymidylate synthase; ↓histone H1 phosphorylation; ↑PP1β; ↑PP2A | Abbaoui et al., 2017 [225] |
| BIU87 | 10–80 µM (24 h) | Decreased cell proliferation | ↑Apoptosis; ↑G2/M phase arrest; ↑IGFBP-3 mRNA; ↓NF-κB | Dang et al., 2014 [226] |
| 5637 | 20 µM (4–48 h) | Suppressed cell growth | ↑Apoptosis; ↑G2/M phase arrest; ↑histone H3 phosphorylation; ↑cyclin B1; ↑Cdk1; ↑caspase-3; ↑PARP cleavage; ↑MMP loss; ↑ROS | Park et al., 2014 [227] |
| Prostate cancer | ||||
| LNCaP, MDA PCa 2a, MDA PCa 2b, PC-3, TSU-Pr1 | 0.1–0.15 µM (1–72 h) | Reduced cellular proliferation | ↑NQO1; ↑QR; ↑γ-GCS-L; ↑GSH; ↑microsomal GST; ↑α-class GSTs | Brooks et al., 2001 [228] |
| DU145, LNCaP, PC-3, and CWR22Rv1 | 20, 40 µM (12, 24 h) | Reduced cell viability | ↑Apoptosis; ↓p-STAT3; ↓IL-6-induced STAT3 phosphorylation; ↓JAK2; ↓pSTAT3 nuclear translocation; ↓STAT3 dimerization; ↓Bcl-2; ↓cyclin D1; ↓survivin; ↓Mcl-1 | Hahm et al., 2010 [229] |
| LNCaP, PC-3 | 10–40 µM (2–24 h) | Reduced cell growth and proliferation | ↑Apoptosis; ↑p53; ↑Bax; ↑E2F1; ↑Apaf-1; ↓Bak; ↓Bcl-xL; ↓NF-κB; ↓cIAP1; ↓cIAP2; ↓XIAP | Choi et al., 2007 [230] |
| LNCaP | 1, 10 µM (24–72 h) | Reduced cell viability and growth | ↑Apoptosis; ↓Bcl-xL; ↓glycolysis; ↓HIF-1α; ↓nuclear AR; ↓PSA | Carrasco-Pozo et al., 2019 [231] |
| LNCap, C4-2 | 1–40 µM (24, 48 h) | Inhibited androgen-stimulated cell growth and proliferation | ↑Transcriptional repression of AR; ↓total AR; ↓Ser210/213 phosphorylated AR; ↓intracellular PSA; ↓secreted PSA | Kim and Singh, 2009 [232] |
| PC-3, LNCaP | 40 µM (16 h) | Reduced cell viability and promoted cell death | ↑Apoptosis; ↑autophagy; ↑LC3; ↑cyt. c | Herman-Antosiewicz et al., 2006 [233] |
| LNCaP, PC-3 | 20 µM (24 h) | Inhibited cell growth and proliferation | ↑Apoptosis; ↑autophagy; ↑LC3 cleavage; ↑ROS; ↑G2/M phase arrest; ↑cyt. c; ↑Bax; ↓Bcl-2; ↓respiratory chain activity | Xiao et al., 2009 [234] |
| LNCaP, PC-3 | 150, 300 µM (4 h) | Decreased cell proliferation | ↑Apoptosis; ↑autophagy; ↑LC3-II; ↓p62 | Watson et al., 2015 [235] |
| DU145 | 5–20 µM (24, 48 h) | Inhibited cell viability | ↑Apoptosis; ↑G2/M phase arrest; ↑PARP cleavage; ↑ROS; ↑JNK | Cho et al., 2005 [236] |
| PC-3 | 10–40 µM (24 h) | Reduced cell viabilityand proliferation | ↑Apoptosis; ↑DNA double-strand breaks; ↑S-phase arrest | Hac et al., 2020 [237] |
| LNCaP | 20–100 µM (24 h) | Decreased cell viability and growth | ↑Apoptosis; ↑PARP cleavage; ↑caspase-3; ↓PGM3 | Lee et al., 2010 [238] |
| PC-3 | 20–100 µM (24–72 h) | Reduced cell survival and proliferation | ↑Apoptosis; ↑G0/G1 arrest; ↑caspase-3; ↑caspase-8; ↑caspase-9; ↑Bax; ↑PARP cleavage; ↓Bcl-2 | Singh et al., 2004 [239] |
| PC-3, DU145 | 10–40 µM (1–24 h) | Reduced cell viability | ↑Apoptosis; ↑caspase-3; ↑caspase-9; ↑Bid cleavage; ↑PARP cleavage; ↑Fas; ↑cyt. c; ↑disruption of mitochondrial membrane potential; ↑ROS; ↓GSH | Singh et al., 2005 [240] |
| PC-3 | 5–20 µM (24–96 h) | Reduced cell viability | ↑Apoptosis; ↑NRF1; ↑mitochondrial fission; ↑Bax; ↑PGC1α; ↓HIF-1α | Negrette-Guzmán et al., 2017 [241] |
| 22Rv1 | 5–50 µM (3–24 h) | Inhibited cell growth | ↑Apoptosis; ↓USP14 and UCHL5 active sites; ↑USP14; ↑UCHL5 protein; ↑Ub-Prs | Ahmed et al., 2018 [97] |
| LNCaP, PC-3 | 15 µM (6, 24 h) | Inhibited cell proliferation | ↑HO-1; ↑NQO1; ↓BMX; ↓CDK2; ↓PLK1; ↓Sp1 | Beaver et al., 2014 [242] |
| LNCaP | 10, 25 µM (2–72h) | Reduced cell growth | ↑Apoptosis; ↑NQO1; ↑LTB4DH; ↑ME1; ↑TXNRD1; ↑GSTM1; ↑MGST1; ↑SOD1; ↑PRDX1; ↑GCLM; ↓Jun; ↑G2/M arrest | Bhamre et al., 2009 [243] |
| LNCaP, PC-3 | 15 µM (24, 48 h) | Inhibited cellular growth | ↑Ac-H3 at P21 promoter; ↑p21; ↑G2/M phase arrest; ↑HO-1; ↑NQO1; ↓HDAC3; ↓HDAC4; ↓HDAC6 | Clarke et al., 2011 [71] |
| LNCaP, VCaP | 10–20 µM (12, 24 h) | Decreased cell viability | ↑HSP90 acetylation; ↓AR; ↓HDAC6; ↓ERG | Gibbs et al., 2009 [244] |
| LNCaP, PC-3 | 15 µM (48 h) | Promoted cell cycle arrest and death | ↓HDAC activity; ↑Ac-H3; ↑Ac-H4; ↑caspase-3; ↑G2/M arrest | Myzak et al., 2006 [245] |
| TRAMP C1 | Exhibited cytotoxicity | ↑Nrf2; ↑NQO-1; ↑Ac-H3; ↓DNMT1; ↓DNMT3a; ↓HDAC1; ↓HDAC4; ↓HDAC5; ↓HDAC7 | Zhang et al., 2013 [246] | |
| LNCaP, PC-3 | 15 µM (3–24 h) | Decreased cellular proliferation | Altered ~100 lncRNA’s expression | Beaver et al., 2017 [247] |
| PC-3, LNCaP | 10, 20 µM (8–24 h) | Reduced cell proliferation and migration | ↓Notch1; ↓Notch2; ↓Notch4; ↑DNA fragmentation | Hahm et al., 2012 [248] |
| PC-3, LNCaP | 15, 30 µM (24, 48 h) | Decreased cellular proliferation | ↓DNMT1; ↓DNMT3b; ↓cyclin-D2-promoter methylation; ↑cyclin D2 | Hsu et al., 2011 [249] |
| LNCaP, PC-3 | 15 µM (48 h) | Exhibited cytotoxicity | ↓DNMT1; ↓DNMT3b; ↑CCR4; ↑TGFBR1 | Wong et al., 2014 [250] |
| LNCaP, PC-3 | 2.5–20 µM (24 h) | Decreased cell viability and proliferation | ↑Apoptosis; ↓pCSC; ↓CD24; ↓ITGA6; ↓ZEB2; ↓c-Myc | Vyas et al., 2016 [251] |
| LNCaP, PC-3 | 15 µM (6–24 h) | Decreased cell viability | ↑SUV39H1 post-translational modification; ↓H3K9me3; ↓chromatin-associated SUV39H1 | Watson et al., 2014 [252] |
| LNCaP, 22Rv1, PC-3 | 5, 10 µM (24 h) | Decreased cell viability | ↓Glycolysis; ↓HKII; ↓LDHA; ↓PMK2 | Singh et al., 2019 [253] |
| DU145 | 5–40 µM (24 h) | Decreased cell viability, migration, and invasion | ↓Pseudopodia; ↓MMP-2; ↑p-ERK1/2; ↑E-Cadherin; ↓CD44v6 | Peng et al., 2015 [254] |
| PC-3, DU145 | 10, 20 µM (24 h) | Decreased cell proliferation and migration | ↑Apoptosis; ↑Vimentin; ↑PAI-1; ↓E-cadherin | Vyas and Singh, 2014 [255] |
| PC-3 | 40 µM (3–24 h) | Inhibited cell viability | ↑Autophagy; ↓S6K1 phosphorylation; ↑LC3 | Hac et al., 2015 [256] |
| PC-3 | 5–50 µM (24 h) | Decreased cell viability | ↑H2S; ↑p38; ↑JNK | Pei et al., 2011 [257] |
| PC-3 | 10–40 µM (2–24 h) | Decreased cell survival | ↓Protein synthesis; ↓[3H]-leucine incorporation; ↓mTOR signaling; ↑S6K1 dephosphorylation; ↓survivin | Wiczk et al., 2012 [258] |
| PC-3 | 1–40 µM (24 h) | Decreased cell viability | ↓NF-κB; ↓p65 nuclear translocation; ↓VEGF; ↓cyclin-D1; ↓Bcl-xL; ↓IKKα phosphorylation; ↓IKKβ phosphorylation | Xu et al., 2005 [259] |
| PC-3 | 5–40 µM (6–24 h) | Reduced cell viability | ↑AP-1; ↑p-ERK1/2; ↑p-JNK1/2; ↑p-Elk-1; ↑p-c-Jun | Xu et al., 2006 [260] |
| DU145 | 5–40 µM (24 h) | Inhibited angiogenesis | ↓HIF-1α; ↑JNK signaling; ↑ERK signaling; ↓VEGF | Yao et al., 2008 [261] |
| LNCaP, 22Rv1 | 5, 10 µM (8–24 h) | Inhibited cell proliferation | ↓ACC1; ↓FASN; ↓CPT1A; ↓ACADVL; ↓ACADM; ↓HADHA; ↓SREBP1 | Singh et al., 2018 [262] |
| LNCaP | 10–60 µM (24 h) | Promoted cell cycle arrest and death | ↑G2/M arrest; ↑S phase arrest; ↑mitotic arrest; ↓cyclin D1; ↓cyclin E1; ↓Cdk4; ↓Cdk6; ↓Cdk1; ↓Cdc25C; ↑cyclin B1; ↑p53; ↑p21 | Herman-Antosiewicz et al., 2007 [263] |
| LNCaP, DU-145 | 15 µM (24 h) | Reduced cellular proliferation | ↓hTERT; ↓G0/G1 transition; ↓S phase; ↓NF-κB; ↓HDAC inhibitor activity; ↓H3K4me2 signal; ↓MeCP2; ↑H3K18Ac signal ↑DNMT1; ↑DNMT3a; ↑Pan-acetylated H3; ↑Pan-acetylated H4 | Abbas et al., 2016 [264] |
| Animal Tumor Models | Anticancer Effects | Mechanisms | Dose (Route) | Duration | References |
|---|---|---|---|---|---|
| Breast cancer | |||||
| BALB/c mice injected with F3II cells | Suppressed tumor development | ↓PCNA; ↑PARP fragment | 15 nmol, daily (i.v.) | 13 days | Jackson and Singletary, 2004 [95] |
| Nude female BALB/c mice xenografted with MDA-MB-231-Luc-D3H1 cells | Inhibited tumor growth | ↓ALDH1A1; ↓NANOG; ↓CR1; ↓GDF3; ↓FOXD3; ↓NOTCH4; ↓WNT3 | 50 mg/kg (i.p.) | 3, 5 weeks | Castro et al., 2019 [86] |
| Female athymic BALB/c mice transplanted with KPL-1 cells | Suppressed tumor growth | ↑Apoptotic ratio | 25, 50 mg/kg (i.p.) | 26 days | Kanematsu et al., 2011 [265] |
| Nude mice xenografted with MDA-MB-453 cells | Reduced tumor size | ↑Egr1; ↓cyclin B1; ↓CDC25c | 100 mg/kg (i.v.) | 15 days | Yang et al., 2016 [106] |
| Gastrointestinal tract and associated cancers | |||||
| Esophageal cancer | |||||
| SCID mice inoculated with BEAC and FLO-1 cells | Reduced tumor size | Not reported | 0.75 mg/day (s.c.) | 2 weeks | Qazi et al., 2010 [121] |
| Male BALB/c mice inoculated with ECa109 cells | Decreased tumor size | ↑LC3B-II; ↓P62 | 5 mg/kg, every other day (i.p.) | 2 weeks | Lu et al., 2020 [122] |
| Small intestine | |||||
| Male ApcMin/+ mice | Decreased tumor number and size | ↑Apoptosis; ↓p-JNK; ↓p-ERK; ↓p-Akt | 300 and 600 ppm/day (via diet) | 3 weeks | Hu et al., 2006 [266] |
| Male ApcMin/+ mice | Reduced tumor size | ↑Apoptosis; ↑p21; ↑caspase-3; ↑caspase-9; ↑COX-2; ↓p-Akt | 300 and 600 ppm/day (via diet) | 3, 10 weeks | Shen et al., 2007 [267] |
| Colon cancer | |||||
| Nude male mice xenografted with HCT116 cells | Suppressed tumor growth; decreased tumor size | ↑CDK1; ↑MK2; ↑p38 phosphorylation | 1 and 5 mg/kg/day (i.p.) | 13 days | Byun et al., 2016 [134] |
| Male C57BL/6J+/Min mice | Inhibited tumor growth | ↓HDAC; ↑acetylated histone H4; ↑p21; ↑Bax | ~6 µmol/day (via diet) | 10 weeks | Myzak et al., 2006 [245] |
| Male WT and Nrf2 mice induced tumors with DMT | Reduced tumor size | ↓HDAC; ↓HDAC3 protein; ↑global histone H4 acetylation | 400 ppm/day or alternate days (via diet) | 25, 35 weeks | Rajendran et al., 2015 [268] |
| Hepatocellular cancer | |||||
| Female BALB/c athymic mice inoculated with HepG2 cells | Reduced tumor growth and volume | Not reported | 50 mg/kg, every 2 days (i.p.) | 13 days | Wu et al., 2016 [160] |
| Pancreatic cancer | |||||
| Male Syrian Hamster injected with BOP to initiate carcinogenesis | Prevented pancreatic carcinogenesis | Not reported | 80 ppm/day (p.o.) | 3 weeks | Kuroiwa et al., 2006 [269] |
| Male SCID mice inoculated with PANC-1 | Decreased tumor growth | Not reported | 250–500 µmol/kg/d (i.p.) | 3 weeks | Pham et al., 2004 [163] |
| Female athymic (nu/nu) mice inoculated with Mia Paca-2 | Inhibited tumor growth | Not reported | 25 or 50 mg/kg (5× per week i.p.) | 4 weeks | Li et al., 2012 [164] |
| Male NOD/SCID/IL2Rγ mice inoculated with human pancreatic CSCs | Reduced tumor growth | ↓Smo; ↓Gli 1; ↓Gli 2; ↓Oct-4; ↓VEGF; ↓PDGFα; ↓Bcl-2; ↓XIAP; ↑E-Cadherin | 20 mg/kg/day (5× per week p.o.) | 6 weeks | Li et al., 2013 [270] |
| Nude mice inoculated with MIA-PaCa2 | Blocked tumor growth and angiogenesis | ↑Apoptosis; ↓NK-κB binding | 4.4 mg/kg (i.p.) on days 4, 5, and 6 after tumor transplant | 1 week | Kallifatidis et al., 2009 [165] |
| BALB/c nude mice (transgenic pancreatic cancer mice) | Reduced tumor volume and weight | ↑Nrf2; ↓Ki-67; ↑p-AMPK | 50 mg/kg, every other day (i.p.) | 120 days | Chen et al., 2018 [169] |
| Gynecological cancers | |||||
| Endometrial cancer | |||||
| Female SCID mice inoculated with Ishikawa cells | Reduced tumor volume | ↑Apoptosis | 50 mg/kg once a day (i.p.) | 30 days | Rai et al., 2020 [175] |
| Ovarian cancer | |||||
| Athymic mice inoculated with A2780 cells | Inhibited tumor growth | ↑IP3R | 40 mg/kg, once a day (i.p.) | 7 days | Hudecova et al., 2016 [180] |
| Lung cancer | |||||
| A/J mice treated with benzopyrene and NNK | Inhibited cellular proliferation. Reduced tumor size and weight | ↑Apoptosis; ↑casspase-3; ↓PCNA | 1.5 and 5 µmol/g (p.o.) | 42 weeks | Conaway et al., 2005 [271] |
| Nude mice inoculated with LTEP-A2 cells | Reduced tumor weight | ↑Apoptosis; ↑G2/M arrest | 25–100 mg/kg, 3 doses/week (i.p.) | 9 days | Liang et al., 2008 [194] |
| NOD/SCID mice inoculated with A549 cells | Reduced tumor volume and weight | ↑Apoptosis; ↑H3 acetylation, ↑H4 acetylation; ↑p53; ↑p21; ↑Bax; ↑G0/G1 arrest; ↑G2/M arrest; ↓HDAC | 9 µM/mice/day on alternate days (p.o.) | 28 days | Jiang et al., 2016 [197] |
| BALB/c nu/nu male mice inoculated with NSCLC | Reduced tumor volume | ↓EGFR | 10 µmol/kg, 5 doses/week (i.t.) | 21 days | Chen et al., 2015 [204] |
| BALB/c nude female inoculated with H1299 | Reduced tumor weight and volume and inhibited cell migration and invasion | ↑ERK5; ↑pERK5; ↑E-Cadherin; ↑ZO-1; ↓pc-jun; ↓pc-Fos; ↓N-Cadherin; ↓Snail1 | 25 and 50 mg/kg every 3 days (i.p.) | 21 days | Chen et al., 2019 [205] |
| Nude male BALB/c mice inoculated with H1299 and 95D cells | Decreased the incidence of lung metastasis | ↓miRNA-616-5p; ↓β-catenin; ↓N-cadherin; ↓Vimentin | 25 or 50 mg/kg, every 3 days (i.v.) | 4 weeks | Wang et al., 2017 [200] |
| Neurological cancer | |||||
| Female NSG mice inoculated with GBM10 cells | Inhibited tumor growth | Not reported | 100 mg/kg for 5-day cycles (p.o.) | 3 weeks | Bijangi-Visheshsaraei et al., 2017 [209] |
| Skin cancer | |||||
| C57BL/6 mice injected with B16F-10 melanoma cells | Inhibited tumor growth and lung metastasis | ↓Lung hydroxyproline; ↓lung uronic acid; ↓lung hexosamine; ↓serum sialic acid; ↓serum GGT; ↑IL-2; ↑IFN-γ; ↓IL-1β; ↓IL-6; ↓TNF-α | 500 µg/kg (i.p.) | 10 days | Thejass and Kuttan, 2006 [272]; Thejass and Kuttan, 2007 [273] |
| C57BL/6 mice inoculated with B16 cells | Reduced tumor volume | ↓HDAC | 500 µmol/kg, 3 doses/week (i.p.) | 4 weeks | Do et al., 2010 [218] |
| C57Bl/6 mice inoculated with B16 cells | Inhibited tumor growth and reduced volume | ↓HDAC | 500 µmol/kg, 3 doses/week (i.p.) | 4 weeks | Enriquez et al., 2013 [217] |
| NSG mice inoculated with A375 | Reduced tumor formation and volume | ↑Apoptosis; ↓Ezh2; ↓H3K27me3; ↓MMP-9; ↓MMP-2; ↑TIMP3; ↑PARP cleavage; ↑procaspase-8; ↑procaspase-9 | 10 µmol/kg, 3 doses/week (p.o.) | 6 weeks | Fisher et al., 2016 [215] |
| Urogenital cancers | |||||
| Bladder cancer | |||||
| Nude female athymic mice xenografted with UMUC3 cells | Inhibited tumor growth | Not reported | 295 µmol/kg (p.o.) | 2 weeks | Abbaoui et al., 2012 [82] |
| Male athymic mice xenografted with UMUC3 cells | Suppressed tumor growth | ↑Apoptosis; ↑caspase-3; ↑cyt. c; ↓survivin | 12 mg/kg (p.o.) | 5 weeks | Wang and Shan, 2012 [274] |
| Prostate cancer | |||||
| Nude male athymic BALB/c (nu/nu) mice xenografted with PC-3 cells | Reduced tumor growth | ↑Apoptosis; ↑Bax; ↑Ac-H3; ↑Ac-H4; ↓HDAC | 443 mg/kg/day (p.o.) | 3 weeks | Myzak et al., 2007 [84] |
| Male and female athymic mice PC-3 xenograft | Inhibited tumor growth | ↑Apoptosis; ↑Bax | 5.6 µmol, 3 times/week (p.o.) | 3 weeks | Singh et al., 2004 [240] |
| Male TRAMP [C57BL/6xFVB]F1 hybrid | Decreased cell proliferation and pulmonary metastasis | ↑Apoptosis; ↑E-Cadherin; ↑Bad; ↑Bak; ↑Bid; ↑Bax; ↑NK cell cytotoxicity; ↑PARP cleavage; ↓Mcl-1; ↑T-cell infiltration | 6 µmol, 3 times/week (p.o.) | 17–19 weeks | Singh et al., 2009 [275] |
| PTEN^ L/L;PB-Cre4 mice | Inhibited cell viability and proliferation | ↑Apoptosis; ↑cell cycle arrest; ↑caspase-3; ↑caspase-7; ↑cyclin B1; ↓cyclin D2 | 0.1, 1 µmol/g/day (p.o.) | 8 weeks | Traka et al., 2010 [276] |
| TRAMP mice | Inhibited tumor growth | ↓ACC1; ↓FASN; ↓acetyl-coA; ↓total FFA; ↓phospholipids | Not specified | Not specified | Singh et al., 2018 [262] |
| TRAMP and Hi-Myc mice with prostate adenocarcinoma | Decreased tumor size | ↓Glycolysis; ↓HKII; ↓PKM2; ↓LDHA; ↓lactate | 1 mg, 3 times/week (p.o.) | 5 weeks | Singh et al., 2019 [253] |
| Study Subjects | Study Type | Study Population | No. of Patients/Control Subjects | Intervention | Status | Main Findings/Objectives | References |
|---|---|---|---|---|---|---|---|
| Healthy males | Dietary intervention study | United Kingdom | 20 | Brussels sprouts and broccoli (250 g each) | Completed | Increased urinary excretion of PhIP metabolites in group that did not consume broccoli | Walters et al., 2004 [278] |
| Healthy adults | Randomized placebo-controlled clinical trial | China | 200 | Broccoli sprout infusion (400 μmol GFN) | Completed | Showed an inverse association of dithiocarbamate and aflatoxin-DNA adducts | Kensler et al., 2005 [279] |
| Healthy adults | Crossover clinical trail | China | 50 | Broccoli sprout beverages (800 μmol GFN or 150 μmol SFN) | Completed | Enhanced urinary excretion of mercapturic acids of acrolein, benzene, and crotonaldehyde | Kensler et al., 2012 [280] |
| Healthy adults | Randomized, placebo-controlled clinical trial | China | 148/143 | Broccoli sprout beverages (600 μmol GFN and 40 μmol SFN) | Completed | Increased the excretion of benzene-derived mercapturic acid in individuals with positive GSTT1 genotype compared to null genotype | Egner et al., 2014 [281] |
| Healthy adults (smokers and non-smokers) | Randomized crossover study | Italy | 20 | Broccoli (200 g/day) | Completed | Decreased DNA strand break in both smoker and non-smoker and reduced oxidative purine in the smoker group | Riso et al., 2009 [282] |
| Health adults (smokers) | Placebo-controlled intervention study | Italy | 27 | Broccoli (250 g/day, 110 μmol ITC/day) | Completed | Decreased oxidized DNA lesions and suppressed DNA strand breaks in the PBMCs of smokers with higher protections with GSTM1-null genotype | Riso et al., 2010 [283] |
| Healthy volunteers | Pilot clinical trial | United State | 10 | GFN-rich (600 μmol/L GFN) or SFN-rich (150 μmol/L SFN) BSE | Completed | Upregulated NQO1 mRNA in oral mucosa | Buamen et al., 2016 [284] |
| Healthy adults | Randomized clinical trial | United States | 20 | Fresh broccoli sprouts or BSE (200 µmol SFN) | Completed | Decreased PBMC HDAC activity, especially in higher dose group or following repeated intake | Atwell et al., 2015 [79] |
| Breast cancer patients | Double-blinded clinical trial | United States | 27/27 | Broccoli seed extract with GFN (224 mg or 512 µmol GFN) | Completed | Decreased PBMC HDAC activity and Ki-67 and HDAC3 serum levels | Atwell et al., 2015 [285] |
| Breast cancer patients | Double-blinded clinical trial | United States | 27/27 | Cruciferous vegetables | Completed | Increased consumption led to significantly lower Ki-67 levels in breast cancer samples | Zhang et al., 2015 [286] |
| Breast cancer patients | Non-randomized interventional clinical trial | Belgium, France, Spain, United Kingdom | 60 | α-cyclodextrin complex of SFN (SFX-01) | Not completed | Showed potential to reverse resistance to endocrine therapy in metastatic breast cancer | Howell et al., 2019 [287] |
| Pancreatic cancer patients | Double-blinded, randomized pilot trial | Germany | 40 | Freeze-dried broccoli sprout (90 mg SFN/day, 507.64 µmol/day) | Not completed | Primary goal is to increase the survival of patients with pancreatic ductal adenocarcinoma | Lozanovski et al., 2014 [288] |
| Prostate cancer patients | Non-randomized trial | United Kingdom | 20 | Broccoli (400 g/week) | Completed | Broccoli consumption interacted with GSTM1 genotype and resulted in alterations of TGFβ1 and EGF signaling pathways | Traka et. al., 2008 [289] |
| Prostate cancer patients | Single-arm trial | United States | 20 | SFN-rich extract (200 µmol/day) | Completed | Reduced PSA by more than 50% in one patient and registered a smaller decline (<50%) in PSA in seven patients; prolonged PSA doubling time | Alumkal et al., 2015 [87] |
| Prostate cancer patients | Double-blinded, randomized, placebo-controlled trial | France | 38/40 | SFN extracted from broccoli seeds (60 mg SFN/day, 338.42 µmol SFN/day) | Completed | Lowered PSA level at months 0, 1, 3, and 6 as well as prolonged PSA doubling time | Cipolla et al., 2015 [290] |
| Prostate cancer patients | Randomized double-blinded, controlled trial | United Kingdom | 41/20 | Broccoli soup (300 mL/week) | Completed | Attenuated the transcriptional changes in the prostate in line with a reduction in the risk of cancer progression | Traka et al., 2019 [291] |
| At-risk prostate cancer patients | Double-blinded, randomized, controlled trial | United States | 48/48 | BSE (200 μmol SFN/day) | Completed | Increased HDAC activity in prostate cancer patients; did not alter tissue biomarkers; downregulated AMACR and ARLNC1 genes | Zhang et al., 2020 [90] |
| Skin cancer patients | Double-blinded, randomized clinical trial | United State | 17 | BSE (50–200 μmol SFN/day) | Not completed | To evaluate the effect on biomarkers Ki-67, Bcl-2, and STAT3 | Kirkwoood et. al., 2016 [292] |
| Skin cancer patients | Double-blinded, randomized clinical trial | United States | 17 | BSE (50–200 μmol SFN/day) | Completed | Decreased the levels of pro-inflammatory cytokines (IP-10, MCP-1, MIG, and MIP-1β) and increased tumor suppressor decorin on day 28 | Tahata et al., 2018 [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, A.E.; Baniasadi, M.; Giansiracusa, D.; Giansiracusa, M.; Garcia, M.; Fryda, Z.; Wong, T.L.; Bishayee, A. Sulforaphane: A Broccoli Bioactive Phytocompound with Cancer Preventive Potential. Cancers 2021, 13, 4796. https://doi.org/10.3390/cancers13194796
Kaiser AE, Baniasadi M, Giansiracusa D, Giansiracusa M, Garcia M, Fryda Z, Wong TL, Bishayee A. Sulforaphane: A Broccoli Bioactive Phytocompound with Cancer Preventive Potential. Cancers. 2021; 13(19):4796. https://doi.org/10.3390/cancers13194796
Chicago/Turabian StyleKaiser, Anna E., Mojdeh Baniasadi, Derrek Giansiracusa, Matthew Giansiracusa, Michael Garcia, Zachary Fryda, Tin Lok Wong, and Anupam Bishayee. 2021. "Sulforaphane: A Broccoli Bioactive Phytocompound with Cancer Preventive Potential" Cancers 13, no. 19: 4796. https://doi.org/10.3390/cancers13194796
APA StyleKaiser, A. E., Baniasadi, M., Giansiracusa, D., Giansiracusa, M., Garcia, M., Fryda, Z., Wong, T. L., & Bishayee, A. (2021). Sulforaphane: A Broccoli Bioactive Phytocompound with Cancer Preventive Potential. Cancers, 13(19), 4796. https://doi.org/10.3390/cancers13194796